
Abstract
Purpose
Superoxide is produced by activated neutrophils during the inflammatory response to stimuli such as endotoxin, can directly or indirectly injure host cells, and has been implicated in the pathogenesis of acute lung injury (ALI)/acute respiratory distress syndrome (ARDS). We wished to determine the potential for pulmonary overexpression of the extracellular isoform of superoxide dismutase (EC-SOD) to reduce the severity of endotoxin-induced lung injury.
Methods
Animals were randomly allocated to undergo intratracheal instillation of (1) surfactant alone (vehicle); (2) adeno-associated virus (AAV) vectors containing a null transgene (AAV-null); and (3) adeno-associated virus vectors containing the EC-SOD transgene (AAV-EC-SOD) and endotoxin was subsequently administered intratracheally. Two additional groups were randomized to receive (1) vehicle or (2) AAV-EC-SOD, and to undergo sham (vehicle) injury. The severity of the lung injury was assessed in all animals 24 h later.
Results
Endotoxin produced a severe lung injury compared to sham injury. The AAV vector encoding EC-SOD increased lung EC-SOD concentrations, and enhanced the antioxidant capacity of the lung. EC-SOD overexpression decreased the severity of endotoxin-induced ALI, reducing the decrement in systemic oxygenation and lung compliance, decreasing lung permeability and decreasing histologic injury. EC-SOD attenuated pulmonary inflammation, decreased bronchoalveolar lavage neutrophil counts, and reduced interleukin-6 and CINC-1 concentrations. The AAV vector itself did not contribute to inflammation or to lung injury.
Conclusions
Pulmonary overexpression of EC-SOD protects the lung against endotoxin-induced ALI.
Similar content being viewed by others


Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are life-threatening clinical conditions, for which there is no specific therapy [1]. When ARDS occurs in the setting of multisystem organ failure, mortality rates over 60% have been reported, with significant morbidity in 50% of survivors [1–3]. Endotoxin is produced by gram negative bacteria and has been implicated in the pathogenesis of sepsis-induced ALI/ARDS, which is the commonest cause of ALI/ARDS, and has the worst outcome [4–6]. Endotoxin may also contribute to ALI/ARDS from other causes, such as major surgery, where alterations in mucosal permeability may permit access of gut-derived endotoxins to the systemic circulation [7]. Endotoxin binds the Toll-like receptor-4 (TLR-4), which activates the innate immune response [8].
Neutrophil-derived reactive oxygen species such as superoxide contribute to lung damage and dysfunction in patients with ALI/ARDS [9, 10]. Superoxide dismutase (SOD) catalyses the dismutation of superoxide and is a key component of the antioxidant defences in the lung. Pharmacologic strategies to directly augment the lung antioxidant defences via exogenous SOD administration have demonstrated efficacy in preclinical models [11–13], but have not translated to the clinical setting. An alternative approach is to overexpress the gene encoding for superoxide in the lung epithelium, using intrapulmonary delivery of viral vectors encoding the gene.
We wished to test the hypothesis that pulmonary overexpression of EC-SOD would attenuate endotoxin-induced ALI. We used this model because of the role of endotoxin in the pathogenesis of ALI/ARDS, and because this is a well-characterized model that closely mimics the clinical development of ARDS [14–16].
Materials and methods
A more detailed description of the methods and materials can be found in the online supplementary material.
Preparation of AAV vectors
The EC-SOD transgene was first ligated into the multiple cloning site of a p-AAVMCS vector (Agilent Technologies Inc., Santa Clara, CA, USA), product size was confirmed by gel electrophoresis, and the plasmid was sequenced to validate insert sequence integrity [17]. Plasmid DNA and adeno-associated virus envelope for AAV serotype 6 was transfected into 293 T cells. Cells were harvested and lysed 24 h later, the lysate was centrifuged and the clear lysate fraction loaded on an iodixanol gradient. Following further purification of the viral particles by affinity column chromatography, particles were concentrated and desalted. Viral vector particle titres were determined by quantitative real-time polymerase chain reaction (qRT-PCR). The process was then repeated for the synthesis of an empty AAV6 vector with no gene product, termed AAV-null.
The AAV particles were divided into aliquots of 125 μl containing 3.0 × 108 DNAase-resistant particles (drp) per microlitre and stored at −70°C. As required, an aliquot was thawed and added to 125 μl of the porcine surfactant Curosurf® (120 mg/ml) (Trinity-Chiesi Pharmaceuticals Limited, Cheadle, UK), to create a final instillate volume of 250 μl. For those animals receiving vehicle only, the instillate was 125 μl of Curosurf® mixed with 125 μl of phosphate-buffered saline (PBS).
Endotoxin-induced ALI model
Specific-pathogen-free adult male Sprague–Dawley rats (350–450 g) were used. The experimental model was based on those previously reported, with several modifications [14, 18]. All work was approved by the National University of Ireland, Galway Research Ethics Committee and conducted under licence from the Department of Health, Ireland.
Vector instillation
Animals were anaesthetised by inhalational induction with isoflurane and an intraperitoneal injection of 40 mg kg−1 of ketamine (Pfizer, Kent, UK) [19, 20], and the trachea intubated with a size 14 intravenous catheter (BD Insyte®; Becton–Dickinson Ltd., Oxford, UK). Animals were randomized to intratracheal instillation of the surfactant/PBS mixture containing (a) vehicle alone; (b) 3.5 × 1010 drp AAV-null; or (c) 3.5 × 1010 drp AAV-EC-SOD. Subsequently, two additional groups were randomized to receive (d) vehicle or (e) AAV-EC-SOD instillation. The dose of AAV-EC-SOD used was determined to produce effective transgene expression in preliminary studies. Following instillation in two aliquots, the animals were extubated, and allowed to recover from anaesthesia.
Endotoxin/vehicle instillation
Five days following vector/vehicle instillation, the animals were re-anaesthetised and 5 mg kg−1 of endotoxin derived from Escherichica coli serotype 055:B5 (Fluka, Poole, UK) or vehicle was instilled intratracheally [14].
Assessment of lung injury
Twenty-four hours following endotoxin instillation, all animals were again anaesthetized as described above, intravenous access was secured via the dorsal penile vein and anaesthesia maintained with repeated intravenous boli of Saffan® (alfaxadone 0.9% and alfadadolone acetate 0.3%; Schering Plough, Welwyn Garden City, UK) [19, 20]. A tracheostomy tube (1-mm internal diameter) was then inserted and intra-arterial access (22- or 24-gauge cannulae; Becton–Dickinson, Franklin Lakes, NJ, USA) was sited in the carotid artery. Cis-atracurium besylate 0.5 mg kg−1 (GlaxoSmithKline, Dublin, Ireland) was administered intravenously and the lungs were mechanically ventilated (Model 683; Harvard Apparatus, Holliston, MA, USA) at a respiratory rate of 80 min−1, tidal volume 6 ml kg−1 and positive end-expiratory pressure of 2 cmH2O. To minimize lung derecruitment, a recruitment manoeuvre consisting of positive end-expiratory pressure 15 cmH2O for 20 breaths was applied at the start of the protocol. All animals were ventilated with an inspired gas mixture of FiO2 = 0.3, and FiN2 = 0.7, for 20 min. Systemic arterial pressure, peak airway pressures and temperature were continuously measured, arterial blood samples were drawn for analysis (ABL 710; Radiometer, Copenhagen, Denmark) and static inflation lung compliance measured [14]. Heparin (400 IU kg−1) was administered intravenously and the animals were then killed by exsanguination under anaesthesia [19, 20].
Sampling and assay protocol
Bronchoalveolar lavage analysis
Immediately post-mortem, the heart–lung block was dissected from the thorax and bronchoalveolar lavage (BAL) collection performed [19, 20]. Total and differential cell counts were performed and the concentrations of cytokine-induced neutrophil chemoattractant-1 (CINC-1) and interleukin-6 (IL-6) in the BAL were determined using a quantitative sandwich ELISA (R&D Systems Europe Ltd., Abingdon, UK) [21, 22]. The Micro BCA™ protein assay kit (Pierce, Rockford, IL, USA), was utilized to determine total BAL protein levels [23].
Histologic analysis
The left lung was isolated and fixed [14, 24], and the extent of histologic lung damage determined using quantitative stereological techniques [25].
Assessment of gene transfer
SOD transgene expression was determined in lung homogenates by real-time rtPCR and Western blotting as previously described [11, 26, 27]. Briefly, RNA was extracted from lung tissue and cDNA synthesis performed using the ImProm-II™ reverse transcription system (Promega, Madison, WI, USA). Quantitative PCR was performed for EC-SOD gene, normalised against a GAPDH control product. Primer sequences (from MWG-Biotech) used were as follows: GAPDH sense: 5′-TTGTGAAGCTCATTTCCTGG-3′, antisense: 5′-CATGTAGGCCATGAGGTCCA-3′. EC-SOD sense: 5′-ATGCTGGCGCTACTGTGTTC-3′, antisense: 5′-ACTCCGCCGAGTCAGAGTT-3′.
Tissue Western blot analysis for EC-SOD
Lung tissue Western blot analysis for EC-SOD was carried out as previously described [28]. Briefly, total cell protein was extracted, protein concentration was determined and samples were electrophoresed on an SDS-PAGE gel and transferred to nitrocellulose. Primary goat anti-human EC-SOD polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used, with anti-goat antibody conjugated to horseradish peroxidase (Cell Signaling Technology, Danvers, MA, USA) as the secondary antibody. The membrane was subsequently incubated with a chemiluminescent substrate (SuperSignal West Pico; Pierce) and EC-SOD protein detected at size 32.5 kDa.
SOD activity assays
Lung homogenate SOD activity was assessed utilizing a colorimetric SOD activity assay kit (Sigma Aldrich, Dorset, UK) in which the degradation of a tetrazolium salt produces a formazan dye upon reduction with a superoxide anion. The rates of reduction are inhibited by superoxide dismutase activity.
Immunohistochemistry
Lung tissue was flash-frozen in liquid nitrogen, cut into 4-μm-thick sections, fixed in acetone, washed, and non-specific sites blocked with 10% (v/v) goat blocking serum. Sections were then incubated with EC-SOD primary antibody raised in goat (Santa Cruz), then incubated in rhodamine-conjugated donkey-anti-goat secondary antibody (Santa Cruz). Fluorescence was subsequently imaged on an Olympus IX71 microscope using Cell^P® software (Olympus Europa GmbH, Germany).
Statistical analysis
Results are expressed as mean ± standard deviation (SD) for normally distributed data, and as median (interquartile range, IQR) if non-normally distributed. Data were analysed by repeated measures two-way ANOVA or one-way ANOVA as appropriate, followed by Student–Newman–Keuls, t test or Kruskal–Wallis followed by Mann–Whitney U test with the Bonferroni correction for multiple comparisons, as appropriate. A p value of less than 0.05 was considered statistically significant.
Results
Forty-two animals were entered into this study. Two animals were excluded prior to assessment of injury due to technical failures of the protocol, namely failure to deliver endotoxin intratracheally. The remaining 40 animals were randomized to receive: (1) sham injury plus vehicle treatment (sham-vehicle, n = 5); (2) sham injury plus EC-SOD (sham-EC-SOD, n = 5); (3) endotoxin injury plus vehicle (LPS-vehicle, n = 10); (4) endotoxin injury plus null vector (LPS-null, n = 10); (5) endotoxin injury plus EC-SOD (LPS-EC-SOD, n = 10). All animals survived vector and endotoxin instillation. One animal in each endotoxin injury group died following induction of anaesthesia for final injury assessment, immediately prior to assessment of physiologic indices of lung injury, due to the severity of their lung injury. All other data were collected from these animals.
There were no differences between the groups in regard to arterial pH, PaCO2, serum bicarbonate, serum lactate, base excess or mean arterial pressure (Table 1).
EC-SOD was transferred and expressed
Endotoxin-induced injury significantly decreased lung tissue EC-SOD protein, and this decrease was fully restored by EC-SOD overexpression (Fig. 1a–b). EC-SOD protein was expressed throughout the lung tissue by immunohistochemical staining, with greater SOD protein evident in animals that received the EC-SOD transgene (Fig. 1c–d). These animals also demonstrated substantially greater lung expression of the EC-SOD transgene (online Fig. A). There was no difference in lung EC-SOD gene expression between the null vector and vehicle groups. Lung tissue SOD activity was significantly enhanced in the animals that received EC-SOD in comparison to non-transduced animals and animals that received the null vector (online Fig. A). Endotoxin-induced lung injury did not appear to reduce EC-SOD expression or activity.

a Densitometry of Western blots demonstrating relative EC-SOD protein concentrations in lung homogenates from each group. b Representative Western blots demonstrating relative EC-SOD protein concentrations in lung homogenates from each group. *Significantly different from sham-vehicle group (P < 0.05, ANOVA). Photomicrographs of representative confocal microscopy images of sections from lung tissue that received EC-SOD (c) and null vector (d) respectively exposed to DAPI nuclear stain and a stain for EC-SOD. The extent of staining for EC-SOD is greater in the lung sections from the animal that received EC-SOD. The scale bar equals 500 μm
EC-SOD attenuates lung injury
Endotoxin instillation produced a severe lung injury, which was attenuated by EC-SOD overexpression in comparison to animals that received vehicle or null vector. EC-SOD abolished the fall in arterial oxygenation seen in vehicle and null gene transfected animals following endotoxin instillation (Fig. 2a). EC-SOD also abrogated the decrement in alveolar–arterial oxygen gradient following endotoxin-induced injury (Table 1). EC-SOD completely attenuated the decrease in static lung compliance (Fig. 2b) and the increase in peak airway pressure (Fig. 2c) following endotoxin-induced injury.

a Histogram representing mean (SD) arterial oxygen partial pressures following endotoxin injury in each group. b Line graph representing mean (SD) static lung compliance following endotoxin injury in each group. c Histogram representing mean (SD) peak airway pressures following endotoxin injury in each group. Vehicle, animals that received intratracheal surfactant alone; Null vector, animals that received intratracheal AAV6 encoding no transgene; EC-SOD, animals that received intratracheal AAV6 encoding the EC-SOD transgene. *Significantly different from sham-vehicle group (P < 0.05, ANOVA). †Significantly different from LPS-vehicle and LPS-null groups (P < 0.05, ANOVA)
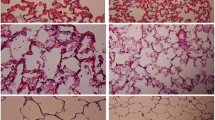
EC-SOD attenuated the endotoxin-induced increase in pulmonary permeability, as assessed by protein leak into the BAL fluid. BAL protein concentrations were significantly greater in all endotoxin-injured animals, but were significantly lower in EC-SOD animals (Table 1). EC-SOD decreased the degree of histologic injury compared to both endotoxin-injured groups (online Fig. B). Quantitative stereological analysis demonstrated that EC-SOD overexpression attenuated the increase in acinar tissue volume fraction and the decrease in acinar airspace volume fraction induced by endotoxin (Fig. 3a).

a Histogram representing mean (SD) alveolar tissue and airspace in the lung following endotoxin injury in each group. b Histogram representing mean (SD) bronchoalveolar lavage neutrophil counts from each group. Vehicle, animals that received intratracheal surfactant alone; Null vector, animals that received intratracheal AAV6 encoding no transgene; EC-SOD, animals that received intratracheal AAV6 encoding the EC-SOD transgene. *Significantly different from sham-vehicle group (P < 0.05, ANOVA). †Significantly different from LPS-vehicle and LPS-null groups (P < 0.05, ANOVA)
EC-SOD reduces pulmonary inflammation
EC-SOD overexpression attenuated the endotoxin-induced inflammatory response. The endotoxin-induced BAL neutrophil influx was attenuated by EC-SOD overexpression (Fig. 3b). EC-SOD overexpression also decreased endotoxin-induced BAL interleukin-6 concentrations, although this was not statistically significant (Table 1). EC-SOD abolished the endotoxin-induced increase in BAL CINC-1 concentrations (Table 1).
There was no evidence to suggest that the viral vector itself worsened the inflammatory response to endotoxin. Specifically, animals that received empty vector did not demonstrate increases in BAL IL-6, CINC-1, or neutrophil counts compared to animals that received vehicle alone (Fig. 3b, Table 1).
Discussion
These findings demonstrate the potential of antioxidant gene therapy to attenuate endotoxin-induced lung injury. Intratracheal delivery of AAV6 vectors encoding the EC-SOD transgene resulted in expression of the EC-SOD transgene, augmenting the antioxidant capacity of the lung. EC-SOD overexpression reduced the severity of subsequent endotoxin-induced lung injury, reducing pulmonary inflammation and reducing physiologic and histologic indices of lung injury and damage.
Superoxide is produced by activated neutrophils during the immune response, and may cause injury directly, or via conversion to more damaging oxidant species, such as the hydroxyl radical and hypochlorous acid [9, 29]. Superoxide can also react with nitric oxide to produce the potent nitrating agent peroxynitrite [10]. Evidence of superoxide-mediated lung damage has been demonstrated in patients with ARDS [9, 29]. Strategies to augment the lung antioxidant potential have included exogenous SOD administration [11–13]; however, limitations regarding direct pharmacologic administration of SOD have limited its therapeutic potential. In contrast, the extracellular SOD isoform (EC-SOD) may be a suitable candidate for gene therapy because it is a secreted product and therefore may protect adjacent cells against injury [26].
In these studies, endotoxin-induced injury decreased pulmonary SOD protein concentrations. Intrapulmonary delivery of AAV6 encoding EC-SOD increased pulmonary EC-SOD expression, and restored lung EC-SOD protein concentrations, to the levels seen in uninjured lungs. Of importance, the efficacy of the EC-SOD vector was not affected by the subsequent endotoxin-induced lung injury. We chose AAV serotype 6 on the basis of preliminary data from our group demonstrating superior transduction with this serotype compared to AAV serotypes 2 or 5. Increased total superoxide dismutase activity was detected in lung homogenates of those animals who were treated with EC-SOD using SOD activity assays. Whilst this assay is not specific for EC-SOD, the relative increase in overall antioxidant activity in the EC-SOD group is likely to be due to the increased EC-SOD present in these animals. Immunohistochemistry demonstrated that the EC-SOD protein was expressed throughout the pulmonary tissue. This strategy significantly reduced endotoxin-induced lung injury, which is characterized by increased oxidant-induced injury.
EC-SOD overexpression significantly reduced endotoxin-induced lung inflammation, and reduced the increase in alveolar neutrophil counts and IL-6 and CINC-1 concentrations. The reduction in alveolar neutrophil infiltration by EC-SOD overexpression may be due, at least in part, to the reduction in alveolar concentrations of the potent neutrophil chemoattractant CINC-1. Of importance, there was no evidence to suggest that the viral vector itself worsened the inflammatory response to endotoxin. This is significant given concerns regarding the potential of viral vectors to produce a host immune response, a potentially deleterious effect in the setting of a generalized pro-inflammatory state such as ALI/ARDS.
EC-SOD overexpression reduced endotoxin-induced lung injury, as evidenced by improved systemic arterial oxygenation, and lung static and dynamic compliance compared to untreated animals. EC-SOD almost completely attenuated the endotoxin-induced decrement in these indices as evidence by the fact that these indices were not different from those in uninjured animals. In contrast, administration of empty vector did not exert any beneficial effects compared to administration of vehicle alone. EC-SOD overexpression also reduced the extent of histologic injury, as evidenced by a greater preservation of alveolar airspace, and reduced alveolar tissue, compared to animals that received empty vector or vehicle alone.
There are a number of limitations in regard to this study. Firstly, this study involved the administration of vector prior to the onset of lung injury. In the clinical setting, lung injury is generally well established at presentation. It is not clear what effect EC-SOD overexpression might have if introduced after the onset of lung injury. However, there are certain settings, such as major cardiovascular or intra-abdominal surgery, where an insult, such as the development of endotoxemia, is a frequent, and relatively predictable occurrence [7]. A safe, low toxicity intervention, if proven effective in these settings, might confer substantial clinical benefit. Secondly, intratracheal delivery of transgene following the development of ALI/ARDS may be difficult, particularly where lung units are collapsed or filled with exudate, reducing the therapeutic utility of intrapulmonary delivered therapies. It is reassuring in these studies that endotoxin injury did not adversely affect transgene expression or activity. Lastly, ALI and ARDS generally have time courses significantly longer than the 24 h assessed by this model. Differences between groups that were not apparent at 24 h might be very clear after a number of days. Further experiments with a longer observation period, and which assess outcome, might be useful in giving insights into the likely clinical impact of pulmonary gene therapy for ARDS.
In conclusion, intrapulmonary delivery of EC-SOD decreased the severity of endotoxin-induced lung injury, demonstrating the potential beneficial effects of EC-SOD overexpression in the setting of ALI. More broadly, these findings suggest that gene therapy may have therapeutic potential for ALI/ARDS. However, additional experimental work is needed over longer time periods to further clarify the therapeutic potential of pulmonary EC-SOD overexpression in ALI/ARDS.
References
Angus DC, Musthafa AA, Clermont G, Griffin MF, Linde-Zwirble WT, Dremsizov TT, Pinsky MR (2001) Quality-adjusted survival in the first year after the acute respiratory distress syndrome. Am J Respir Crit Care Med 163:1389–1394
Rubenfeld GD (2003) Epidemiology of acute lung injury. Crit Care Med 31:S276–S284
Herridge MS, Cheung AM, Tansey CM, Matte-Martyn A, Diaz-Granados N, Al-Saidi F, Cooper AB, Guest CB, Mazer CD, Mehta S, Stewart TE, Barr A, Cook D, Slutsky AS (2003) One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 348:683–693
Valta P, Uusaro A, Nunes S, Ruokonen E, Takala J (1999) Acute respiratory distress syndrome: frequency, clinical course, and costs of care. Crit Care Med 27:2367–2374
TenHoor T, Mannino DM, Moss M (2001) Risk factors for ARDS in the United States: analysis of the 1993 National Mortality Followback Study. Chest 119:1178–1179
Zilberberg MD, Epstein SK (1998) Acute lung injury in the medical ICU: comorbid conditions, age, etiology, and hospital outcome. Am J Resp Crit Care Med 157:1159–1164
Laffey JG, Boylan JF, Cheng DC (2002) The systemic inflammatory response to cardiac surgery: implications for the anesthesiologist. Anesthesiology 97:215–252
Beutler B (2000) Endotoxin, toll-like receptor 4, and the afferent limb of innate immunity. Curr Opin Microbiol 3:23–28
Lamb NJ, Gutteridge JM, Baker C, Evans TW, Quinlan GJ (1999) Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Crit Care Med 27:1738–1744
Zhu S, Manuel M, Tanaka S, Choe N, Kagan E, Matalon S (1998) Contribution of reactive oxygen and nitrogen species to particulate-induced lung injury. Environ Health Perspect 106(Suppl 5):1157–1163
Abunasra HJ, Smolenski RT, Morrison K, Yap J, Sheppard MN, O’Brien T, Suzuki K, Jayakumar J, Yacoub MH (2001) Efficacy of adenoviral gene transfer with manganese superoxide dismutase and endothelial nitric oxide synthase in reducing ischemia and reperfusion injury. Eur J Cardiothorac Surg 20:153–158
Barnard ML, Baker RR, Matalon S (1993) Mitigation of oxidant injury to lung microvasculature by intratracheal instillation of antioxidant enzymes. Am J Physiol 265:L340–L345
Mossman BT, Surinrut P, Brinton BT, Marsh JP, Heintz NH, Lindau-Shepard B, Shaffer JB (1996) Transfection of a manganese-containing superoxide dismutase gene into hamster tracheal epithelial cells ameliorates asbestos-mediated cytotoxicity. Free Radic Biol Med 21:125–131
Laffey JG, Honan D, Hopkins N, Hyvelin JM, Boylan JF, McLoughlin P (2004) Hypercapnic acidosis attenuates endotoxin-induced acute lung injury. Am J Respir Crit Care Med 169:46–56
Brigham KL, Meyrick B (1986) Endotoxin and lung injury. Am Rev Respir Dis 133:913–927
van Helden HP, Kuijpers WC, Steenvoorden D, Go C, Bruijnzeel PL, van Eijk M, Haagsman HP (1997) Intratracheal aerosolization of endotoxin (LPS) in the rat: a comprehensive animal model to study adult (acute) respiratory distress syndrome. Exp Lung Res 23(4):297–316
Sen S, Conroy S, Hynes SO, McMahon J, O’Doherty A, Bartlett JS, Akhtar Y, Adegbola T, Connolly CE, Sultan S, Barry F, Katusic ZS, O’Brien T (2008) Gene delivery to the vasculature mediated by low-titre adeno-associated virus serotypes 1 and 5. J Gene Med 10:143–151
Ni Chonghaile M, Higgins BD, Costello JF, Laffey JG (2008) Hypercapnic acidosis attenuates severe acute bacterial pneumonia-induced lung injury by a neutrophil-independent mechanism. Crit Care Med 36:3135–3144
Costello J, Higgins B, Contreras M, Chonghaile MN, Hassett P, O’Toole D, Laffey JG (2009) Hypercapnic acidosis attenuates shock and lung injury in early and prolonged systemic sepsis. Crit Care Med 37:2412–2420
Higgins BD, Costello J, Contreras M, Hassett P, OT D, Laffey JG (2009) Differential effects of buffered hypercapnia versus hypercapnic acidosis on shock and lung injury induced by systemic sepsis. Anesthesiology 111:1317–1326
Calkins CM, Bensard DD, Heimbach JK, Meng X, Shames BD, Pulido EJ, McIntyre RC Jr (2001) l-Arginine attenuates lipopolysaccharide-induced lung chemokine production. Am J Physiol Lung Cell Mol Physiol 280:L400–L408
Moreland JG, Fuhrman RM, Wohlford-Lenane CL, Quinn TJ, Benda E, Pruessner JA, Schwartz DA (2001) TNF-alpha and IL-1 beta are not essential to the inflammatory response in LPS-induced airway disease. Am J Physiol Lung Cell Mol Physiol 280:L173–L180
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85
Howell K, Preston RJ, McLoughlin P (2003) Chronic hypoxia causes angiogenesis in addition to remodelling in the adult rat pulmonary circulation. J Physiol 547:133–145
Hopkins N, Cadogan E, Giles S, McLoughlin P (2001) Chronic airway infection leads to angiogenesis in the pulmonary circulation. J Appl Physiol 91:919–928
Bowler RP, Arcaroli J, Crapo JD, Ross A, Slot JW, Abraham E (2001) Extracellular superoxide dismutase attenuates lung injury after hemorrhage. Am J Respir Crit Care Med 164:290–294
Lehmann TG, Wheeler MD, Schoonhoven R, Bunzendahl H, Samulski RJ, Thurman RG (2000) Delivery of Cu/Zn-superoxide dismutase genes with a viral vector minimizes liver injury and improves survival after liver transplantation in the rat. Transplantation 69:1051–1057
O’Toole D, Hassett P, Contreras M, Higgins BD, McKeown ST, McAuley DF, O’Brien T, Laffey JG (2009) Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-kappaB dependent mechanism. Thorax 64:976–982
Quinlan GJ, Evans TW, Gutteridge JM (1994) Oxidative damage to plasma proteins in adult respiratory distress syndrome. Free Radical Res 20:289–298
Acknowledgments
This study was funded by the European Research Council (DOT, JGL), the Association of Anaesthetists of Great Britain and Ireland (JGL, BH) and the Health Research Board, Ireland (JD, BH, JGL). Dr O’Toole is a Research Fellow with the European Research Council (Grant No. ERC-2007-StG 207777) and Dr Devaney is a postdoctoral fellow with the Health Research Board, Ireland (Grant No. RP/2008/193).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Hassett, P., Curley, G.F., Contreras, M. et al. Overexpression of pulmonary extracellular superoxide dismutase attenuates endotoxin-induced acute lung injury. Intensive Care Med 37, 1680–1687 (2011). https://doi.org/10.1007/s00134-011-2309-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-011-2309-y