
Abstract
Aims/hypothesis
Accumulation of adipose tissue macrophages is considered pivotal in the development of obesity-associated inflammation and insulin resistance. In addition, recent studies suggest an involvement of the intestine as the primary organ in inducing hyperglycaemia and insulin resistance. We have reported that the C-C motif chemokine receptor (CCR) CCR9 is associated with intestinal immunity and has a pathogenic role in various liver diseases. However, its contribution to type 2 diabetes is unknown. In the current study, we aimed to clarify the involvement of CCR9 in the pathology of type 2 diabetes and the potential underlying mechanisms.
Methods
To elucidate how CCR9 affects the development of metabolic phenotypes, we examined the impact of CCR9 deficiency on the pathogenesis of type 2 diabetes using male C57BL/6J (wild-type [WT]) and CCR9-deficient (CCR9 knockout [KO]) mice fed a 60% high-fat diet (HFD) for 12 weeks.
Results
WT and Ccr9KO mice fed an HFD exhibited a comparable weight gain; however, glucose tolerance and insulin resistance were significantly improved in Ccr9KO mice. Moreover, visceral adipose tissue (VAT) and the liver of Ccr9KO mice presented with less inflammation and increased expression of glucose metabolism-related genes than WT mice. Ccr9 and Ccl25 expression were specifically higher in the small intestine but was not altered by HFD feeding and type 2 diabetes development. Accumulation of IFN-γ-producing CD4+ T lymphocytes and increased intestinal permeability in the small intestine was observed in WT mice following HFD feeding, but these changes were suppressed in HFD-fed Ccr9KO mice. Adoptive transfer of gut-tropic CCR9-expressing T lymphocytes partially reversed the favourable glucose tolerance found in Ccr9KO mice via exacerbated inflammation in the small intestine and VAT.
Conclusions/interpretation
CCR9 plays a central role in the pathogenesis of type 2 diabetes by inducing an inflammatory shift in the small intestine. Our findings support CCR9 as a new therapeutic target for type 2 diabetes via the gut–VAT–liver axis.
Graphical abstract

Similar content being viewed by others



Introduction
Obesity-induced insulin resistance is thought to be a precursor phenotype of type 2 diabetes, which can accelerate the progression to non-alcoholic fatty liver disease (NAFLD). One of the key mechanisms of insulin resistance is low-grade inflammation in adipose tissue, with abnormal adipokine production [1, 2]. Adipose tissue macrophages (ATM) infiltrate the adipose tissue in a chemokine-dependent manner, specifically in response to the chemokines C-C motif ligand (CCL)2 and CCL5, and secrete inflammatory cytokines that inhibit insulin signalling in humans and rodents [3,4,5,6]. Therefore, the chemokine system plays an important role in both adipose tissue inflammation and insulin resistance.
Dysfunction in adipose tissues is not the only contributor to insulin resistance. Pathological changes in the intestine occur prior to the development of obesity, and leakage of gut-derived components from the disrupted intestinal barrier causes inflammation in insulin-sensitive organs, including visceral adipose tissue (VAT) and the liver, via portal blood flow [7,8,9,10]. Thus, it is possible that an inflammatory shift in the intestinal tract induced by a high-fat diet (HFD) can cause systemic endotoxaemia and subsequent chronic adipose tissue inflammation. One well-studied mechanism underlying the intestinal inflammation that is associated with the consumption of an HFD involves the gut microbiota. Germ-free mice fed an HFD do not develop type 2 diabetes, whereas germ-free mice transplanted with faecal microbiota from obese mice develop type 2 diabetes [11], suggesting that an inflammatory shift in the intestinal tract, caused by altered microbial flora, plays a central role in obesity and insulin resistance.
Inflammatory shift in the intestinal tract is also controlled by the chemokine system, as seen in adipose tissues [12]. However, it is unclear whether dysregulation of the chemokine system in the intestinal tract directly leads to systemic inflammation in downstream metabolic organs. Furthermore, a conventional intestinal inflammatory shift has been extensively studied in the colon (where the gut microbiota is more abundant), but not in the small intestine, which is indispensable for nutrient absorption. Therefore, the contribution of the small intestine to the pathogenesis of obesity and type 2 diabetes is yet to be elucidated.
The C-C motif chemokine receptor (CCR) CCR9 and its ligand CCL25 play a physiological role in early T cell differentiation in the thymus and in the recruitment of immune cells to the small intestine [13, 14]. In addition, we have reported the critical role of CCR9 in the pathogenesis of acute and chronic liver diseases via its direct effect on hepatic macrophages [15, 16]. However, the contribution of CCR9 to the pathogenesis of systemic diseases has not been well studied. In the current study, we aimed to clarify the role of this chemokine in metabolic organs during the progression of obesity-induced type 2 diabetes.
Methods
For detailed methods, please refer to the electronic supplementary material (ESM).
Mice
C57BL/6J (wild-type [WT]) CD45.2 mice (CLEA Japan, Japan; www.clea-japan.com/en/), WT CD45.1 mice and Ccr9−/− CD45.2 mice [15] were maintained under specific pathogen-free conditions in the Animal Care Facility of Keio University School of Medicine (Tokyo, Japan). Germ-free mice (C57BL/6 background strain) were kept in the germ-free Facility of Keio University School of Medicine. Experiments were performed with age- and sex-matched mice at 6–8 weeks of age. All experiments were approved by the animal ethics committee of Keio University, Tokyo, Japan and performed according to the guidelines. In this study, randomisation and blinding were not carried out.
Induction of obesity and insulin resistance in mice
Male WT and Ccr9 knockout (KO) mice were fed an HFD containing 60 kJ % fat (D-12492; Research Diets, New Brunswick, NJ, USA), or a normal diet (ND; CE-2; CLEA, Japan) as the control, for 12 weeks. Mice underwent tolerance tests and metabolic measurements 1 week before they were killed for further blood and tissue sample collection.
Gnotobiotic study design
Faecal samples were collected from WT or Ccr9KO mice after 12 weeks of HFD feeding. Samples were suspended in an equal amount (wt/vol.) of PBS containing 40% (vol./vol.) glycerol, snap-frozen and stored at −80°C until use. The frozen stocks were thawed, and each sample was suspended in a sixfold volume of PBS and passed through a cell strainer. Male germ-free mice were orally inoculated with 200 μl of the suspensions, followed by HFD feeding. Ten weeks after the inoculation, GTTs and ITTs were performed.
Adoptive transfer experiment of small intestinal lamina propria mononuclear cells
Small intestinal lamina propria mononuclear cells (LPMCs) isolated from CD45.1 WT mice were magnetically purified using CD45 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and resuspended in PBS. Isolated CD45+ leukocytes (1 × 107 cells/500 μl) or the same volume of PBS were intravenously administered to HFD-fed male CD45.2 Ccr9KO mice once a week for 8 weeks. Mice underwent tolerance tests and metabolic measurements 1 week before being killed for further blood and tissue sample collection.
GTT and ITT
Both OGTT and ITT were performed after 5 h of fasting. For OGTT, 20% or 40% (wt/vol.) glucose was administered; for ITT, insulin was administered intraperitoneally (2 U/Kg). A drop of peripheral blood was collected from the tail vein to measure blood glucose levels using a glucometer at 0, 15, 30, 60 and 120 min after glucose or insulin load.
Measurement of serum insulin concentration
Fasting and glucose-loaded serum insulin levels were measured by ELISA.
Serum and intrahepatic biochemical parameter analysis
Determination of serum alanine aminotransferase (ALT), total cholesterol (TCHO) and triacylglycerol was carried out using the DRI-CHEM 2500i Analyzer (FujiFilm, Tokyo, Japan). Lipopolysaccharide (LPS) levels were measured in blood by a ToxinSensor Chromogenic LAL Endotoxin Assay Kit (GenScript, NJ, USA). Soluble CD14 (sCD14) and LPS binding protein (LBP) concentrations were measured in the serum by ELISA. TCHO and triacylglycerol levels in the liver were measured using the Cholesterol/Cholesteryl Ester Quantitation Kit (BioVision, Mountain View, CA, USA) and a triglyceride kit (Wako, Fujifilm Wako Pure Chemical, Tokyo, Japan), respectively.
Histology and fluorescent immunohistochemistry
Formalin-fixed paraffin-embedded tissue was used for haematoxylin and eosin staining (liver and pancreas) and immunohistochemical analysis of zonula occludens-1 (ZO-1; small intestine). For fluorescent immunohistochemistry, non-fixed whole adipose tissue was sliced and stained using the anti-CD11b or anti-F4/80 antibody and analysed using confocal microscopy.
Real-time quantitative PCR (qPCR) and reverse transcription
Isolated liver tissue, visceral adipose tissue, subcutaneous adipose tissue, brown adipose tissue, or muscle, small intestine and colon tissue were immersed into RNAlater Stabilization Solution (Invitrogen, Carlsbad, CA, USA) and stored at −80°C before RNA isolation. Synthesised cDNA was subjected to quantification by TaqMan qPCR assays (Invitrogen). The following genes were analysed: Ccr9, Ccl25, Ccr2, Ccr5, Ccl2, Ccl3, Ccl4, Adgre1, Tnfa, Adipoq, Il1b, Nfkb, Tjp1, Ocln, Cldn7, Ppara, Slc2a4, Srebf1, Acaca, Fas, Scd1 and Pparg. Actb was used as the internal control.
Isolation of tissue immune cells
Isolation of intestinal LPMCs and liver mononuclear cells was conducted as described previously [17]. Isolation of visceral adipose tissue stromal vascular fraction was conducted as described previously, with slight modification [5]. Briefly, epididymal fat pads were minced and digested with Hanks’ balanced salt solution (HBSS; Nacalai Tesque, Japan) containing 1.5% FBS, 1.25 mg/ml collagenase D (Roche Diagnostics, Germany), 0.85 mg/ml collagenase V (Sigma-Aldrich, MO, USA), 1 mg/ml dispase (Gibco, Carlsbad, CA, USA) and 15 μg/ml DNase (Sigma-Aldrich) for 20 min at 37°C. The cell suspension was filtered, washed and collected as the stromal vascular fraction. All collected immune cells were resuspended in RPMI-1640 containing 10% (vol./vol.) FBS and 1% (vol./vol.) penicillin/streptomycin and subjected to flow cytometry.
Re-stimulation of isolated mononuclear cells
Isolated mononuclear cells were seeded on 24-well tissue-culture plates and stimulated with 50 ng/mg phorbol12-myristate13-acetate (PMA; Sigma-Aldrich) and 1 μg/ml ionomycin (Sigma-Aldrich) or 1 μg/mL LPS (Sigma-Aldrich) for 5 h. Following this, cells underwent staining for flow cytometry.
Flow cytometry analysis
For cell surface and intracellular staining, the cells were washed and stained, as previously described [17]. Details of each antibody used and clone-related information are summarised in ESM Table 1. The stained cells were analysed using a FACS Canto II flow cytometer (Becton Dickinson, NJ, USA) and the data were analysed using FlowJo software (v 10.4.2; Tree Star, OR, USA). A representative gating strategy for flow cytometry is shown in ESM Fig. 1.
DNA extraction from faecal samples
Fresh small intestine stool samples were collected from mice, snap-frozen in liquid nitrogen and stored at −80°C until processing. Bacterial DNA was isolated by the enzymatic lysis method, as mentioned previously [18].
Microbial analysis
The hypervariable V3–V4 regions of the 16S rRNA gene were amplified using Ex Taq Hot Start (TaKaRa) and subsequently purified using AMPure XP (Beckman Coulter, CA, USA). Approximately equal amounts of each amplified DNA were then sequenced using the Miseq Reagent Kit V3 (600 cycle) and the Miseq sequencer (Illumina, CA, USA). Sequences were analysed using the QIIME2 software package version 2019.10 (https://qiime2.org). Principal coordinate analysis (PCoA) was expressed by the qiime2R (v 0.99.13) and ggforce (v 0.3.1) packages in R software (v 3.6.1). Taxa barplots were generated using the R package qiime2R (v 0.99.13), phyloseq (v 1.30.0) and MicrobeR (v 0.3.2) [18].
Western blotting
After 5 h fasting, mice were intraperitoneally administered with insulin (0.5 U/kg) or vehicle (saline; 154 mmol/l NaCl) to assess the insulin sensitivity. After 15 min, mice were killed and freshly isolated VAT and liver tissues were lysed with RIPA buffer containing phosphatase inhibitors. Lysates were subjected to SDS-PAGE using the NuPAGE kit according to the kit manual, transferred to PVDF membranes and immunoblotted with anti-p-Akt and anti-Akt antibodies. Relative expression of p-Akt and Akt was determined using densitometry by ImageQuant TL software (v 8.1.0.0; GE Healthcare, Little Chalfont, UK).
Determination of adipocyte size by computer image analysis
Adipocyte size was manually calculated for up to 20 cells in four independent sites of the same slide using NDP2.view software (v 2.8; Hamamatsu Photonics, Hamamatsu, Japan). In accordance with the manual, we calculated the size of each adipocyte manually using the size measurement tool.
Chemotaxis assay
Cell-migration assays were performed using isolated LPMCs from the small intestine of HFD-fed WT or KO mice using 5 μm pore 24-well Transwell plates. LPMCs were seeded in the upper chamber of the plates and serum-free medium with or without CCL25 (500 ng/ml) was added to the lower chamber. After 4 h, migrated CD4+ T cells in the lower chamber were immunostained and counted by flow cytometry.
In vitro T lymphocytes activation study
Isolated LPMCs from the small intestine of HFD-fed WT or Ccr9KO mice were seeded with a range of T cell stimulants (phytohemagglutinin, concanavalin A, CD3/CD28 Dynabeads [Invitrogen] and CCL25). After 4 h culture at 37°C, CD69+7-amino-actinomycin D (AAD)−CD3+CD4+ activated T lymphocytes were analysed by flow cytometry.
Statistical analysis
Data were analysed using GraphPad Prism software (v 8.4.2; GraphPad software, CA, USA). Differences between two groups were evaluated using an unpaired Student’s t test or Mann–Whitney U test. Comparisons of more than two groups was performed with one-way ANOVA followed by Tukey–Kramer’s multiple comparison test. Differences were considered statistically significant if p < 0.05. Data are expressed as means ± SEM. No data were excluded from statistical analysis throughout the study.
Results
Ccr9 deficiency ameliorates glucose tolerance and insulin resistance in HFD-induced obesity
To explore the contribution of CCR9 to the pathogenesis of type 2 diabetes, we first examined glucose tolerance and insulin sensitivity in HFD-fed WT and Ccr9KO mice. During the 12 weeks of HFD feeding, both mouse strains exhibited severe obesity without any differences in body weight and food consumption throughout type 2 diabetes progression between the two groups (Fig. 1a,b). The Ccr9KO mice fed an HFD demonstrated significantly improved glucose tolerance compared with HFD-fed WT mice (Fig. 1c). Furthermore, ITT results indicated that the HFD-fed Ccr9KO mice were more insulin-sensitive than HFD-fed WT mice (Fig. 1d). This improvement started as early as 6 weeks after commencement of HFD feeding (ESM Fig. 2a, b) and was sustained for up to 35 weeks (ESM Fig. 2c). Hyperinsulinaemia was also moderate in the Ccr9KO mice, and insulin secretion in response to glucose exposure was significantly suppressed in HFD-fed Ccr9KO mice as compared with HFD-fed WT mice (Fig. 1e). Consistently, histological analyses of pancreas tissue revealed improved histology with Ccr9 deletion in that the size of islets of HFD-fed Ccr9KO mice was smaller in tissue from WT mice (ESM Fig. 2d). These results collectively suggested that Ccr9 deficiency improved glucose tolerance and insulin resistance without affecting body weight.

Ccr9 deficiency ameliorates glucose tolerance and insulin resistance in HFD-induced obesity. WT and Ccr9KO mice were fed either an ND or HFD for 12 weeks and glucose tolerance and insulin resistance were examined. (a) Body weight of ND- or HFD-fed WT and Ccr9KO mice from 0 to 12 weeks after HFD onset (WT/ND and WT/HFD, n=4; KO/ND and KO/HFD, n=5). Data are representative of five independent experiments. (b) Food consumption for 3 days at 12 weeks of the experiment (n=3). Data are representative of three independent experiments. (c) Serum glucose concentration during an OGTT (WT/ND and WT/HFD, n=4; KO/ND and KO/HFD, n=5). Data are representative of five independent experiments. (d) Serum glucose levels during an ITT (WT/ND and WT/HFD, n=4; KO/ND and KO/HFD, n=5). Data are representative of two independent experiments. (e) Serum insulin concentration pre- and 15 min post-glucose load after 5 h of fasting (n=4–5). Data are mean±SEM. *p<0.05, †p<0.05, ††p<0.01, HFD-fed Ccr9KO vs HFD-fed WT mice
Hepatic inflammation is reduced in Ccr9KO mice, with a reduction in ectopic fat accumulation in the liver that is independent of Ccr9 expression
Next, we examined the contribution of CCR9 to HFD-feeding-induced inflammation in the liver, which could lead to the development of type 2 diabetes. Serum ALT levels and TCHO levels, but not triacylglycerols, were significantly decreased in HFD-fed Ccr9KO mice vs HFD-fed WT mice (Fig. 2a,b, ESM Fig. 3a). The number of lipid droplets and inflammation in the liver were milder in the Ccr9KO mice fed an HFD vs HFD-fed WT mice (Fig. 2c,d). Both intrahepatic triacylglycerols and TCHO levels were notably suppressed in HFD-fed Ccr9KO mice vs WT mice, but these findings were only significant for intrahepatic TCHO (Fig. 2e,f). Genes associated with fatty acid oxidation and insulin sensitivity (Ppara and Pparg) were upregulated, while genes associated with the synthesis of fatty acids (Srebf1, Acaca, Fas, and Scd1) were downregulated in the HFD-fed Ccr9KO mice vs WT mice (Fig. 2g–l). Consistently, phosphorylated Akt levels were upregulated in the liver of HFD-fed Ccr9KO mice compared with HFD-fed WT mice and the difference was prominent under insulin-stimulated conditions, while levels were comparable between ND-fed WT mice and ND-fed Ccr9KO mice (Fig. 2m, ESM Fig. 3b). These results collectively suggest that Ccr9 deficiency suppressed HFD-induced ectopic fat accumulation and liver inflammation, whilst also ameliorating hepatic metabolic function.

Hepatic inflammation is reduced in Ccr9KO mice and ectopic fat deposits are decreased in the liver, independent of Ccr9 expression. WT and Ccr9KO mice were fed either an ND or HFD for 12 weeks and the phenotype in the liver was analysed. (a) Serum ALT levels (n=4); representative data of three independent experiments. (b) Serum TCHO concentration (n=4–5); representative data of three independent experiments. (c) Representative photomicrographs of haematoxylin and eosin stained sections of the liver. Scale bars, 100 μm. (d) NAFLD activity score (n=4–5). (e, f) Intrahepatic triacylglycerol (e) and TCHO (f) concentrations per unit weight (n=4–5). (g–l) Expression of Ppara (g), Pparg (h), Srebf1 (i), Acaca (j), Fas (k) and Scd1 (l) in the liver (n=5). (m) Representative western blot of p-Akt, Akt and β-Actin expression in the liver with and without insulin sensitisation, and quantitative graph of p-Akt/Akt ratio (n=6). (n, p, r) Frequency of CD4+ T cells (n), CD8+ T cells (p) and invariant NKT (iNKT) cells (r) in the liver (n=4–5). (o, q, s) Representative histograms of CCR9 expression in CD4+ T cells (o), CD8+ T cells (q) and iNKT cells (s). (t) Frequency of CD11b+ macrophages. (u) Representative histogram of CCR9 expression in CD11b+ macrophages. (v) Representative surface CCR9 and intracellular TNF-α staining in CD11b+-gated whole mononuclear cells in the liver from ND- or HFD-fed WT mice. (w) Frequency of TNF-α+ cells in CD11b+-gated liver mononuclear cells (n=4–5). Mφ, macrophages; TG, triacylglycerol. Data are mean±SEM. *p<0.05, **p<0.01
The analysis of liver immunocompetent cells by flow cytometry revealed that the frequency of CD4+ T cells and CD8+ T cells were increased in the liver of HFD-fed WT mice compared with ND-fed WT mice; this increase was ameliorated in the Ccr9KO mice (Fig. 2n,p). A decrease in the number of CD1d 4mer+ natural killer T (NKT) cells, a representative state of immune activation [19], in the liver of HFD-fed vs ND-fed WT mice was reversed in the HFD-fed Ccr9KO mice (Fig. 2r). Although the frequency of CD11b+ macrophages was comparable between HFD-fed WT and Ccr9KO mice (Fig. 2t), TNF-α production by CD11b+ macrophages after in vitro LPS stimulation was significantly reduced in the Ccr9KO mice compared with that in the HFD-fed WT mice (Fig. 2v,w). Interestingly, CCR9 expression was not upregulated in any of the hepatic immune cells in HFD-fed WT mice as compared with ND-fed WT mice (Fig. 2o,q,s,u), indicating that the improvement in hepatic inflammation and metabolic function in the Ccr9KO mice was not brought about by the direct involvement of CCR9 in the liver.
Ccr9 deficiency results in attenuated inflammation of VAT with improved glucose metabolism
We next assessed whether Ccr9 deficiency affected the inflammatory state of VAT that is located upstream of the liver, along the portal blood flow. As expected, the frequency of CD11b+F4/80+ ATMs, well-known pathological players in adipose tissue inflammation and glucose tolerance in VAT, was significantly lower in the VAT of HFD-fed Ccr9KO mice than in the HFD-fed WT mice (Fig. 3a). Furthermore, crown-like structures (CLSs), assessed by immunofluorescent staining with anti-F4/80 and anti-CD11b antibodies, were markedly reduced in the VAT of HFD-fed Ccr9KO mice vs WT mice (Fig. 3c). Accordingly, expression of genes relating to inflammatory transcription, Adgre1 and Tnfa, was significantly lower in HFD-fed Ccr9KO mice than in the WT mice (Fig. 3d,e). In contrast, Adipoq expression was higher in HFD-fed Ccr9KO mice, although this finding was not significant (Fig. 3f). These results indicate that the Ccr9KO mice developed severe obesity upon HFD feeding with less inflammation and ATM infiltration in adipose tissues as compared with WT mice. Consequently, glucose metabolism-related genes, such as Slc2a4 and Pparg were upregulated in the VAT of HFD-fed Ccr9KO mice compared with WT mice (Fig. 3g,h). Upregulation of Akt phosphorylation in the VAT of HFD-fed Ccr9KO mice in response to insulin sensitisation further supported a recovered metabolic function (Fig. 3i). We also confirmed that the size of adipocytes in the VAT was significantly smaller in HFD-fed Ccr9KO mice than in HFD-fed WT mice (Fig. 3j–n), indicating a hyperplastic characteristic of these cells. Of note, HFD-fed Ccr9KO mice had decreased liver weight, whereas these mice also had increased VAT and subcutaneous adipose tissue (SAT) weight compared with HFD-fed WT mice (ESM Fig. 4a), leading to a comparable body weight. As observed in the liver, upregulation of CCR9 was not detected in ATM or other immune cells in the VAT of HFD-fed WT mice compared with ND-fed WT mice (Fig. 3b, ESM Fig. 4b), supporting the notion that the improvement in glucose metabolism in VAT in HFD-fed Ccr9KO mice does not directly involve CCR9.

Ccr9 deficiency results in an attenuated inflammation in VAT with improved glucose metabolism. WT and Ccr9KO mice were fed either an ND or HFD for 12 weeks and the phenotype in the adipose tissue was analysed. (a) Representative CD11b and F4/80 staining on CD45+-gated immune cells in the stromal vascular fraction (SVF) of VAT from ND- or HFD-fed WT and Ccr9KO mice. The frequency of CD11b+ F4/80+ ATM in the CD45+-gated cells of the SVF of VAT is also shown in the bar chart (n=3–4). (b) Representative histogram of CCR9 expression in CD11b+F4/80+ ATM. (c) Representative photomicrographs of immunofluorescent staining with anti-CD11b or anti-F4/80 antibodies for visualising crown-like structures (CLS) in VAT. Scale bars, 100 μm. (d–h) Expression of Adgre1 (d), Tnfa (e), Adipoq (f), Slc2a4 (g) and Pparg (h) in VAT (n=4–5). (i) Representative western blot of p-Akt, Akt, and β-Actin expression, with and without insulin sensitisation, in VAT. A quantitative graph of p-Akt/Akt ratio is also shown (n=6). (j) Representative photomicrographs of haematoxylin and eosin stained sections of VAT. Scale bars, 10 μm. (k–n) Profile of the distribution of adipocytes cell size in ND-fed WT (k), ND-fed KO (l), HFD-fed WT (m) and HFD-fed KO (n) mice (n=4). Data are mean±SEM. *p<0.05, ** p<0.01
Ccr9 and Ccl25 expression is specifically upregulated in the small intestine, regardless of the occurrence of type 2 diabetes
Next, we analysed the expression of Ccr9 and Ccl25 in various tissues to explore the responsible organs involved in the pathogenesis of type 2 diabetes. For this analysis, the liver, muscle, small intestine, colon, brown adipose tissue (BAT), and white adipose tissue (WAT), including VAT and SAT, were isolated from ND- and HFD-fed WT mice. As previously reported [20, 21], Ccr9 and Ccl25 expression was elevated in the small intestine in a group comparison with the other organs, both under the ND and HFD feeding condition (Fig. 4a,b). Notably, high Ccr9 and Ccl25 expression in the small intestine was not affected by HFD feeding (Fig. 4a,b), whereas expression of Ccr2, Ccr5, and their ligands were significantly upregulated in the WAT by HFD feeding (ESM Fig. 5), consistent with previous reports [3, 5]. These findings suggested that the physiological functions of CCR9/CCL25 in the small intestine might be critically involved in the development of type 2 diabetes.

Ccr9 and Ccl25 expression is specifically upregulated in the small intestine, regardless of type 2 diabetes status. WT mice were fed either an ND or HFD for 12 weeks and expression of Ccr9 and Ccl25 was analysed in the liver, WAT, BAT, muscle, small intestine (SI) and colon. (a) Ccr9 and (b) Ccl25 expression in the indicated tissues of ND-fed and HFD-fed WT mice. Expression levels are relative to expression in the liver of ND-fed WT mice. Data are mean±SEM (n=4). **p<0.01, one-way ANOVA
HFD-induced inflammatory shift in the small intestine is suppressed in Ccr9KO mice via abrogated recruitment of IFN-γ-producing T lymphocytes
We next evaluated the intestines of the HFD-fed WT and Ccr9KO mice to verify our hypothesis that the small intestine plays a fundamental role in type 2 diabetes progression. Intestinal inflammation and epithelial barrier dysfunction cause LPS leakage and initiate VAT inflammation in the early phases of type 2 diabetes [12, 22]. First, we confirmed that the status of intestinal permeability, assessed by serum LPS, LBP and sCD14 concentration, was increased in HFD-fed WT mice compared with ND-fed WT mice at 12 weeks, while it was significantly reduced in HFD-fed Ccr9KO mice compared with HFD-fed WT mice (Fig. 5a–c). In addition, gene expression of epithelial junction markers related to gut permeability was significantly higher in the small intestine of Ccr9KO mice vs WT mice after 4 weeks of HFD feeding (Fig. 5d). Consistently, a decreased and disturbed expression of ZO-1 in the small intestine was observed in HFD-fed WT mice vs ND-fed mice, but was less prominent in the HFD-fed Ccr9KO mice (Fig. 5e). As a consequence, Lbp expression in the VAT was significantly downregulated in HFD-fed Ccr9KO mice compared with HFD-fed WT mice (ESM Fig. 6a). Of note, epithelial junction markers in the colon were not affected by Ccr9 deficiency (ESM Fig. 6b). Furthermore, proinflammatory gene transcripts in HFD-fed Ccr9KO mice were significantly suppressed in the small intestine but not in the colon, as compared with HFD-fed WT mice (Fig. 5f, ESM Fig. 6c). These results indicate that CCR9 mainly affects the permeability in the small intestine but not in the colon, which is consistent with the distribution of Ccr9 and Ccl25 expression.

HFD-induced inflammatory shift in the small intestine is suppressed in Ccr9KO mice via abrogated recruitment of IFN-γ-producing T lymphocytes. WT and Ccr9KO mice were fed either an ND or HFD for 12 weeks and the phenotype in the small intestine was analysed. (a–c) Concentration of serum LPS (a), LBP (b) and sCD14 (c) (n=4–5). (d) Expression of Tjp1, Ocln and Cldn7 in the small intestine of HFD-fed KO mice relative to HFD-fed WT mice at each time point (n=4–5). (e) Representative immunohistochemical images of ZO-1 staining in the small intestine. Scale bars, 10 μm. (f) Expression of Adgre1, Il1b, Tnfa and Nfkb in the small intestine of HFD-fed KO mice relative to HFD-fed WT mice at each time point (n=4–5). (g) Representative surface CD4 and intracellular IFN-γ staining in CD3+-gated small intestine LPMC from ND- or HFD-fed WT and Ccr9KO mice. (h, j) Frequency of IFN-γ+CD4+ Th1 cells in CD3+-gated T lymphocytes (h) and IFN-γ+γδTCR+ γδT cells in γδTCR+-gated γδT lymphocytes (j) in the small intestine. (i, k) Representative histogram of CCR9 expression in IFN-γ+CD4+ Th1 cells (i) and IFN-γ+γδTCR+ γδT cells (k). SI, small intestine; TCR, T cell receptor. Data are mean ± SEM. *p<0.05, **p<0.01
In vitro Transwell migration assays demonstrated that CD4+ T cells derived from HFD-fed WT mice, but not Ccr9KO mice, exhibited a migratory potential toward CCL25, while CCL25 did not activate these cells in vitro (ESM Fig. 6d, e). In addition, the frequency of IFN-γ-producing CD4+ T and γδT cells was significantly higher in HFD-fed WT mice than in the Ccr9KO mice (Fig. 5g,h,j). Of note, Ccr9 expression appeared to be upregulated in these cell subsets of the small intestine in WT mice compared with Ccr9KO mice (Fig. 5i,k), suggesting that CCR9 regulated the inflammatory shift in the small intestine by directly affecting the homing ability of IFN-γ-producing T lymphocytes. Consistently, these findings were not observed in the colon of the HFD-fed Ccr9KO mice vs HFD-fed WT mice (ESM Fig. 6f). Furthermore, the frequency of inflammatory macrophages and dendritic cells (DCs) that produce IFN-γ, IL-1β, and TNF-α was also reduced in the small intestine of the HFD-fed Ccr9KO mice vs WT mice (ESM Fig. 6g, h). Together with previous findings [23,24,25], these results suggest that CCR9 deficiency contributes to the improvement of small intestinal permeability by suppressing the HFD-induced inflammatory condition, which may lead to protection against metabolic disorder progression.
Microbiota in the small intestine did not directly affect the improvement in HFD-induced insulin resistance that accompanies Ccr9 deficiency
To explore the role of gut microbiota in the control of intestinal inflammation by CCR9, we analysed the composition of gut microbiota in the small intestine of the four groups of mice. As previously reported, HFD feeding induced a marked change in the microbiota in both WT and Ccr9KO mice (Fig. 6a–c). Notably, there was little difference in the composition of microbiota between WT and Ccr9KO mice under ND feeding, while there was a prominent difference under HFD feeding (Fig. 6a). These results suggest that the microbial change was not brought by genetic differences, but rather by HFD-induced inflammation in the small intestine, which was milder in Ccr9KO mice. To validate this hypothesis, we inoculated germ-free mice with faecal samples either from HFD-fed WT mice or HFD-fed Ccr9KO mice, followed by HFD feeding for 10 weeks (Fig. 6d). There was no difference in the body weight, OGTT and insulin levels between the mice (Fig. 6e–g). These results collectively suggest that microbiota exerts minimal impact on the attenuated insulin resistance observed with Ccr9 deficiency.

Microbiota in the small intestine do not directly induce the improvement in HFD-induced insulin resistance observed with Ccr9 deficiency. (a) Principal-coordinate (PC) analysis based on the weighted UniFrac analysis of bacterial community structures of the indicated groups (WT/ND, KO/ND and KO/HFD, n=5; WT/HFD, n=4). Dissimilarities between two groups were evaluated by permutational multivariate ANOVA. Tukey–Kramer multiple comparison test was used. The p values between the groups were as follows: ND-fed WT vs ND-fed KO, p=0.377; HFD-fed WT vs HFD-fed KO, p=0.006; ND-fed WT vs HFD-fed WT, p=0.010; ND-fed KO vs HFD-fed KO, p=0.011. (b) The number of operational taxonomic units (OTUs). (WT/ND, KO/ND and KO/HFD, n=5; WT/HFD, n=4). (c) Family-level taxonomic distribution in each mouse. Values represent the relative abundance (%). Key: 1, Remainder; 2, kingdom (k__)Bacteria; phylum (p__)Firmicutes; class (c__)Clostridia; order (o__)Clostridiales; family (f__)Clostridiaceae; 3, k__Bacteria; p__Firmicutes; c__Clostridia; o__Clostridiales; f__unclassified; 4, k__Bacteria; c__Verrucomicrobia; c__Verrucomicrobiae; o__Verrucomicrobiales; f__Verrucomicrobiaceae; 5, k__Bacteria; p__Actinobacteria; c__Coriobacteriia; o__Coriobacteriales; f__Coriobacteriaceae; 6, k__Bacteria; p__Firmicutes; c__Clostridia; o__Clostridiales; f__Lachnospiraceae; 7, k__Bacteria; p__Actinobacteria; c__Actinobacteria; o__Bifidobacteriales; f__Bifidobacteriaceae; 8, k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__S24−7; 9, k__Bacteria; p__Firmicutes; c__Erysipelotrichi; o__Erysipelotrichales; f__Erysipelotrichaceae; 10, k__Bacteria; p__Firmicutes; c__Bacilli; o__Lactobacillales; f__Streptococcaceae; 11, k__Bacteria; p__Firmicutes; c__Bacilli; o__Lactobacillales; f__Lactobacillaceae. (d) Design of gnotobiotic studies: germ-free (GF) mice were either transplanted with faecal samples derived from HFD-fed WT mice (n=5) or HFD-fed Ccr9KO mice (n=5), followed by HFD feeding for 10 weeks. SI, small intestine. (e, f) Body weight (e; n=5) and serum glucose concentration during OGTT (f; n=5) in gnotobiotic mice. (g) Serum insulin concentration pre- and 15 min post-glucose load after 5 h of fasting in gnotobiotic mice. Data are mean ± SEM. *p<0.05, **p<0.01
Small intestine homing property of the transferred small intestinal LPMCs is critical for glucose intolerance
To elucidate whether small intestine-derived immune cells directly contribute to HFD-induced hyperglycaemia, LPMCs were isolated from the small intestine of WT (CD45.1) mice and intravenously transferred into the HFD-fed Ccr9KO (CD45.2) mice once a week for 8 weeks. We confirmed that the transferred CD45.1+ cells were specifically localised in the small intestine of the Ccr9KO mice (Fig. 7a,b), which is consistent with previous reports [26, 27]. We also confirmed that the majority of the migrated CD45.1+ cells were CCR9+CD4+ T cells with a potential for IFN-γ production in the lamina propria of the small intestine (Fig. 7c–f). Seven days after the last cell transfer, we performed OGTT to determine the extent of glucose tolerance. Transfer of LPMCs resulted in worsened glucose tolerance in the HFD-fed Ccr9KO mice without any impact on body weight (Fig. 7g,h). In particular, LPMC-transferred mice showed disturbed tight junctions in the small intestine compared with the control mice (Fig. 7i). Furthermore, the number of inflammatory macrophages and Lbp gene expression in the VAT increased in the LPMC-transferred HFD-fed Ccr9KO mice vs control mice (Fig. 7j,k). These results corroborate the pathogenic role of CCR9-dependent homing of IFN-γ-producing CD4+ T cells in small intestinal inflammation and subsequent glucose intolerance in VAT.

Accumulation of LPMCs in the small intestine plays a causative role in the development of type 2 diabetes. LPMCs isolated from the small intestine of CD45.1 WT mice were intravenously transplanted into HFD-fed CD45.2 Ccr9KO mice once a week for 8 weeks. (a, b) Representative histogram (a) and frequency (b) of localised CD45.1+ donor cells in the small intestine and indicated tissues of HFD-fed recipient Ccr9KO mice (n=4–5). (c) Representative CD45.1 and CD45.2 staining of indicated immune-cell subsets in 7AAD−CD45+-gated LPMC in the small intestine of recipient Ccr9KO mice. cDC, conventional dendritic cells; pDC, plasmacytoid dendritic cells. (d) Frequency of localised CD45.1+ donor cells in the small intestine of HFD-fed Ccr9KO mice (n=4–5). For (b) and (d), one-way ANOVA was used for the statistical analysis. (e, f) Representative histograms and the frequency of CCR9+ (e) and IFN-γ+ cells (f) in CD45.1+- or CD45.2+-gated CD4+ T cell populations in HFD-fed recipient Ccr9KO mice (n=4). (g) Body weight of mice adoptively transferred with either PBS or CD45.1 LPMCs at the time of the OGTT experiment (n=5). (h) Serum glucose concentration during OGTT (n=5). Statistical notation indicates a difference between mice adoptively transferred with PBS vs mice adoptively transferred with CD45.1 LPMCs at the same time point. (i) Representative immunohistochemical images of ZO-1 staining in the small intestine in mice adoptively transferred with either PBS or CD45.1 LPMCs. Scale bars, 10 μm. (j) Frequency of infiltrated CD11b+F4/80+ macrophages in the VAT of mice adoptively transferred with either PBS or CD45.1 LPMCs (n=4). (k) Lbp gene expressions in the VAT of mice adoptively transferred with either PBS or CD45.1 LPMCs (n=5). Mφ, macrophages, SI, small intestine. Data are mean±SEM. *p<0.05, **p<0.01
Discussion
Although the role of immune cell infiltration and subsequent inflammation in adipose tissues in the pathogenesis of insulin resistance has been well studied [1, 2], whether and how immune cells in the intestinal tract contribute to disease development is still a controversial topic. Moreover, most studies have focused on colonic immunity that is influenced by altered gut microbiota [8, 12, 24, 28, 29], but not on the small intestine, a central organ that is involved in nutrient absorption. In this study, we demonstrated that the CCR9/CCL25 axis directly regulates inflammatory shift in the small intestine as a master organ in the pathogenesis of type 2 diabetes, and CCR9 deficiency results in improved glucose metabolism by modulating the inflammatory state of downstream insulin-sensitive organs.
Ccr9 deficiency leads to loss of T lymphocyte homing ability in the small intestine but not in the colon [14, 26]. Consistently, the expression of Ccr9 in the immune cell population was only affected in the small intestine in this study; therefore, we hypothesised that the small intestine may serve as a central pathogenic organ in the development of type 2 diabetes. The change in the inflammatory state of the small intestine by HFD feeding has been reported to occur in the following order: (1) alteration of intestinal flora [30]; (2) disruption of epithelial barrier [31]; and (3) inflammatory shift after 6 weeks of HFD feeding [32, 33]. These profiles are, in part, consistent with our results, and we further demonstrated that the improvement in glucose tolerance lasted for over 30 weeks after initiating HFD feeding in the Ccr9KO mice. These results suggest a sustained immunological change and persistent changes in inflammation in the small intestine through HFD feeding [12, 22, 32]. We found an increase in the number of IFN-γ-producing CD4+ T cell (type 1 helper T cells [Th1]) and γδT cells in the small intestine during HFD feeding, which is consistent with other reports [8, 22]. In contrast, another study has reported that Th17 cells in the small intestine contribute to insulin resistance [34]. Differences in environmental factors, such as microbiota, housing conditions, vendors or diet of mice may explain the discrepancy in findings. Regarding the causative role of gut microbiota in the small intestine, our results from gnotobiotic mice suggest that microbiota have a minimal impact on the attenuated insulin resistance observed with Ccr9 deficiency.
In HFD-fed Ccr9KO mice, migration and inflammation of ATM in the VAT were attenuated, with improved adipocyte morphology and metabolic function compared with WT mice. Since the expression of Ccr9 and Ccl25 in WAT was not enhanced by HFD feeding, the role of CCR9 in type 2 diabetes progression might be different from that of other disease-associated chemokine receptors, such as CCR2 and CCR5, the expression of which was upregulated in infiltrated ATM (ESM Fig. 5) [3,4,5]. Instead, we speculated that suppressed inflammation in adipose tissues during HFD feeding might be secondarily induced by a quiescent inflammatory state in the small intestine of the Ccr9KO mice. Regarding the mechanism that links the intestinal inflammation and ATM accumulation, we confirmed that Ccr9KO mice transferred with LPMCs demonstrated disrupted tight junctions in the small intestine, which enabled bacterial translocation. Indeed, the number of CD11b+ macrophages and Lbp expression in the VAT increased in these mice. There results collectively reinforce the notion that Th1 cells in the small intestine directly cause inflammatory shift in a CCR9-dependent manner, which eventually leads to insulin resistance.
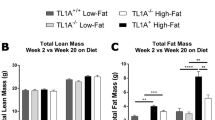
Our group has reported a pathogenic role for the CCR9/CCL25 axis in acute liver injury and liver fibrosis, with the direct involvement of liver macrophages [15, 16]. Thus, we initially assumed that Ccr9 deficiency affected immune cells in the inflamed liver and suppressed NAFLD progression. On the other hand, pathogen-associated molecular patterns from leaky gut induce hepatic inflammation and contribute to the development of type 2 diabetes or NAFLD [7,8,9,10]. We assumed that the improved phenotype in the livers of the Ccr9KO mice was a secondary effect of the improvement in inflammation in small intestine for the following reasons: (1) Ccr9 expression in immune cells was not enhanced by HFD feeding in the liver; (2) the number of infiltrating hepatic macrophages in HFD-fed Ccr9KO mice was comparable to that in HFD-fed WT mice; and (3) Ccr9 deficiency also improved hepatic metabolic function. Since there was a variation in the distribution of fat mass in HFD-fed Ccr9KO mice, CCR9 may regulate HFD-induced fat deposition in the liver through its activity in the small intestine. Further study is warranted to clarify this.
The chemokine ligand/receptor axis has attracted worldwide attention as another therapeutic target in patients with NAFLD/non-alcoholic steatohepatitis (NASH) and type 2 diabetes [3,4,5]. Moreover, an anti-β7 integrin monoclonal antibody is currently approved for the treatment of inflammatory bowel disease, and administration of 5-aminosalicyclic acid, a suppressive agent for bowel inflammation, has been reported to improve metabolic variables [8]. This evidence, along with our findings, suggest that regulation of immune-cell accumulation and subsequent inflammation in the small intestine could directly benefit glucose metabolism in humans. Collectively, these results provide further insight into chemokine signalling pathways as potential therapeutic targets in type 2 diabetes.
Data availability
All data generated and analysed during this study are included in this published article.
Abbreviations
- ALT:
-
Alanine aminotransferase
- ATM:
-
Adipose tissue macrophage
- BAT:
-
Brown adipose tissue
- CCL:
-
C-C motif ligand
- CCR:
-
C-C motif chemokine receptor
- HFD:
-
High-fat diet
- KO:
-
Knockout
- LBP:
-
Lipopolysaccharide binding protein
- LPMC:
-
Lamina propria mononuclear cell
- LPS:
-
Lipopolysaccharide
- NAFLD:
-
Non-alcoholic fatty liver disease
- ND:
-
Normal diet
- SAT:
-
Subcutaneous adipose tissue
- sCD14:
-
Soluble CD14
- TCHO:
-
Total cholesterol
- Th:
-
Helper T cell
- VAT:
-
Visceral adipose tissue
- WAT:
-
White adipose tissue
- WT:
-
Wild-type
- ZO-1:
-
Zonula occludens-1
References
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259(5091):87–91. https://doi.org/10.1126/science.7678183
Neels JG, Olefsky JM (2006) Inflamed fat: what starts the fire? J Clin Invest 116(1):33–35. https://doi.org/10.1172/JCI27280
Weisberg SP, Hunter D, Huber R et al (2006) CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116(1):115–124. https://doi.org/10.1172/JCI24335
Kanda H, Tateya S, Tamori Y et al (2006) MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116(6):1494–1505. https://doi.org/10.1172/JCI26498
Kitade H, Sawamoto K, Nagashimada M et al (2012) CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes 61(7):1680–1690. https://doi.org/10.2337/db11-1506
Xue W, Fan Z, Li L, Lu J, Zhai Y, Zhao J (2019) The chemokine system and its role in obesity. J Cell Physiol 234(4):3336–3346. https://doi.org/10.1002/jcp.27293
Konrad D, Wueest S (2014) The gut-adipose-liver axis in the metabolic syndrome. Physiology 29(5):304–313. https://doi.org/10.1152/physiol.00014.2014
Luck H, Tsai S, Chung J et al (2015) Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab 21(4):527–542. https://doi.org/10.1016/j.cmet.2015.03.001
Monteiro-Sepulveda M, Touch S, Mendes-Sa C et al (2015) Jejunal T Cell Inflammation in Human Obesity Correlates with Decreased Enterocyte Insulin Signaling. Cell Metab 22(1):113–124. https://doi.org/10.1016/j.cmet.2015.05.020
Garidou L, Pomie C, Klopp P et al (2015) The Gut Microbiota Regulates Intestinal CD4 T Cells Expressing RORgammat and Controls Metabolic Disease. Cell Metab 22(1):100–112. https://doi.org/10.1016/j.cmet.2015.06.001
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444(7122):1027–1031. https://doi.org/10.1038/nature05414
Kawano Y, Nakae J, Watanabe N et al (2016) Colonic Pro-inflammatory Macrophages Cause Insulin Resistance in an Intestinal Ccl2/Ccr2-Dependent Manner. Cell Metab 24(2):295–310. https://doi.org/10.1016/j.cmet.2016.07.009
Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY (2004) Retinoic acid imprints gut-homing specificity on T cells. Immunity 21(4):527–538. https://doi.org/10.1016/j.immuni.2004.08.011
Kunkel EJ, Campbell JJ, Haraldsen G et al (2000) Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: Epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. J Exp Med 192(5):761–768. https://doi.org/10.1084/jem.192.5.761
Nakamoto N, Ebinuma H, Kanai T et al (2012) CCR9+ macrophages are required for acute liver inflammation in mouse models of hepatitis. Gastroenterology 142(2):366–376. https://doi.org/10.1053/j.gastro.2011.10.039
Chu PS, Nakamoto N, Ebinuma H et al (2013) C-C motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology 58(1):337–350. https://doi.org/10.1002/hep.26351
Amiya T, Nakamoto N, Chu PS et al (2016) Bone marrow-derived macrophages distinct from tissue-resident macrophages play a pivotal role in Concanavalin A-induced murine liver injury via CCR9 axis. Sci Rep 6:35146. https://doi.org/10.1038/srep35146
Kim SW, Suda W, Kim S et al (2013) Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res 20(3):241–253. https://doi.org/10.1093/dnares/dst006
Wilson MT, Johansson C, Olivares-Villagomez D et al (2003) The response of natural killer T cells to glycolipid antigens is characterized by surface receptor down-modulation and expansion. Proc Natl Acad Sci U S A 100(19):10913–10918. https://doi.org/10.1073/pnas.1833166100
Mizuno S, Kanai T, Mikami Y et al (2012) CCR9+ plasmacytoid dendritic cells in the small intestine suppress development of intestinal inflammation in mice. Immunol Lett 146(1–2):64–69. https://doi.org/10.1016/j.imlet.2012.05.001
Amersi FF, Terando AM, Goto Y et al (2008) Activation of CCR9/CCL25 in cutaneous melanoma mediates preferential metastasis to the small intestine. Clin Cancer Res 14(3):638–645. https://doi.org/10.1158/1078-0432.CCR-07-2025
Winer DA, Luck H, Tsai S, Winer S (2016) The Intestinal Immune System in Obesity and Insulin Resistance. Cell Metab 23(3):413–426. https://doi.org/10.1016/j.cmet.2016.01.003
Cani PD, Amar J, Iglesias MA et al (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56(7):1761–1772. https://doi.org/10.2337/db06-1491
Cani PD, Bibiloni R, Knauf C et al (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57(6):1470–1481. https://doi.org/10.2337/db07-1403
Amar J, Chabo C, Waget A et al (2011) Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med 3(9):559–572. https://doi.org/10.1002/emmm.201100159
Stenstad H, Ericsson A, Johansson-Lindbom B et al (2006) Gut-associated lymphoid tissue-primed CD4+ T cells display CCR9-dependent and -independent homing to the small intestine. Blood 107(9):3447–3454. https://doi.org/10.1182/blood-2005-07-2860
Wendland M, Czeloth N, Mach N et al (2007) CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc Natl Acad Sci U S A 104(15):6347–6352. https://doi.org/10.1073/pnas.0609180104
Martinez-Medina M, Denizot J, Dreux N et al (2014) Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 63(1):116–124. https://doi.org/10.1136/gutjnl-2012-304119
Kim KA, Gu W, Lee IA, Joh EH, Kim DH (2012) High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One 7(10):e47713. https://doi.org/10.1371/journal.pone.0047713
Hamilton MK, Boudry G, Lemay DG, Raybould HE (2015) Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am J Physiol Gastrointest Liver Physiol 308(10):G840–G851. https://doi.org/10.1152/ajpgi.00029.2015
Tomas J, Mulet C, Saffarian A et al (2016) High-fat diet modifies the PPAR-gamma pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc Natl Acad Sci U S A 113(40):E5934–E5943. https://doi.org/10.1073/pnas.1612559113
Ding S, Chi MM, Scull BP et al (2010) High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One 5(8):e12191. https://doi.org/10.1371/journal.pone.0012191
Araujo JR, Tomas J, Brenner C, Sansonetti PJ (2017) Impact of high-fat diet on the intestinal microbiota and small intestinal physiology before and after the onset of obesity. Biochimie 141:97–106. https://doi.org/10.1016/j.biochi.2017.05.019
Hong CP, Park A, Yang BG et al (2017) Gut-Specific Delivery of T-Helper 17 Cells Reduces Obesity and Insulin Resistance in Mice. Gastroenterology 152(8):1998–2010. https://doi.org/10.1053/j.gastro.2017.02.016
Acknowledgements
We thank T. Teratani, T. Suzuki, R. Aoki, S. Chiba, and Y. Kawano (Keio University School of Medicine, Tokyo, Japan) for providing technical assistance. We would like to thank Editage (www.editage.jp) for English language editing.
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Funding
This study was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid (C) 16K09374 and Keio University Medical Fund.
Author information
Authors and Affiliations
Contributions
TA helped design the study, performed experiments, analysed the data and wrote the paper; NN conceived and designed the study, analysed the data and wrote the paper; NT, P-SC, YK, AY, SS and RM performed experiments and assisted in the preparation of the manuscript; KM performed microbiological analysis and critically reviewed the manuscript; JI and HI helped design the study, analysed the data and critically reviewed the manuscript; and TK helped conceive and supervise the study, contributed to analysis and interpretation of data and critically reviewed the manuscript. All the authors have approved the final version of this manuscript. NN and TK are the guarantors of this work.
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM
(PDF 2238 kb)
Rights and permissions
About this article

Cite this article
Amiya, T., Nakamoto, N., Irie, J. et al. C-C motif chemokine receptor 9 regulates obesity-induced insulin resistance via inflammation of the small intestine in mice. Diabetologia 64, 603–617 (2021). https://doi.org/10.1007/s00125-020-05349-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-020-05349-4