
Abstract
Aims/hypothesis
To understand the complex metabolic changes that occur long before the diagnosis of type 2 diabetes, we investigated differences in metabolomic profiles in plasma between prediabetic and normoglycaemic individuals for subtypes of prediabetes defined by fasting glucose, 2 h glucose and HbA1c measures.
Methods
Untargeted metabolomics data were obtained from 155 plasma samples from 127 Mexican American individuals from Starr County, TX, USA. None had type 2 diabetes at the time of sample collection and 69 had prediabetes by at least one criterion. We tested statistical associations of amino acids and other metabolites with each subtype of prediabetes.
Results
We identified distinctive differences in amino acid profiles between prediabetic and normoglycaemic individuals, with further differences in amino acid levels among subtypes of prediabetes. When testing all named metabolites, several fatty acids were also significantly associated with 2 h glucose levels. Multivariate discriminative analyses show that untargeted metabolomic data have considerable potential for identifying metabolic differences among subtypes of prediabetes.
Conclusions/interpretation
People with each subtype of prediabetes have a distinctive metabolomic signature, beyond the well-known differences in branched-chain amino acids.
Data availability
Metabolomics data are available through the NCBI database of Genotypes and Phenotypes (dbGaP, accession number phs001166; www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001166.v1.p1).
Similar content being viewed by others


Introduction
The diagnosis of type 2 diabetes is often made long after underlying metabolic changes have occurred. Individuals with prediabetes already experience a host of changes and are likely to develop overt diabetes [1]. Prediabetes is currently defined by three different but relevant tests: fasting glucose of 5.6–7.0 mmol/l, 2 h post-load glucose of 7.8–11 mmol/l from an OGTT and HbA1c of 39–46 mmol/mol (5.7–6.5%) [2]. Although all three tests predict diabetes, they differ somewhat in their biological underpinnings. For example, fasting and 2 h glucose differ in muscle insulin resistance, hepatic insulin sensitivity and first- and second-phase insulin responses [3]. Furthermore, only 12.3% of those with prediabetes in Starr County, TX, USA are so identified on all three criteria [1].
It has long been known that branched-chain amino acids (BCAAs) play an important role in insulin regulation and glucose metabolism [4, 5] and are associated with type 2 diabetes [6, 7] and/or prediabetes [8, 9]. A 2016 systematic review identified more than 20 studies testing cross-sectional associations between amino acids and (pre)diabetes and 33 studies testing prospective associations between metabolites and (pre)diabetes progression [10]. Meta-analyses of these studies showed that BCAAs (valine, leucine and isoleucine) and aromatic acids (tyrosine and phenylalanine) are positively associated with relative risks for type 2 diabetes, while glycine and glutamine are negatively associated. Most of the studies included in the review were targeted metabolomic studies, but two studies employed untargeted approaches [11, 12] and identified multiple metabolites associated with type 2 diabetes in addition to BCAAs. Interestingly, an untargeted metabolomics study in Mexican Americans from the San Antonio Family Heart Study suggested that future diabetes risk can be predicted from plasma lipidomic profiles [13]. These results show that metabolomic data provide important information about diabetes and its progression. It is also evident that most knowledge is still largely based on targeted metabolomics, with a need for and considerable potential in evaluating the untargeted metabolome in the context of (pre)diabetes development and progression.
Methods
To begin to investigate the metabolic underpinnings associated with progression to and subtypes of prediabetes, we conducted untargeted metabolomic and targeted lipidomic assays on 155 samples from 127 women without type 2 diabetes from Starr County, TX, USA. Participants for the present study were selected from the representative population of Mexican Americans from Starr County described in Hanis et al. [1]. Briefly, in a survey conducted from March 2002 to January 2006, we performed OGTTs on 1345 individuals aged 20–77 years. We were able to follow up a total of 768 of the 1345 individuals between December 2010 and January 2014, with a mean of 8.6 years between examinations. From these participants, we investigated all 127 unrelated women who did not have diabetes in either the original or the follow-up examination and who had exome sequencing data and Staphylococcus aureus carriage status available. A total of 28 of the 127 women were profiled using plasma samples collected approximately 10 years apart. These 28 individuals were selected based on their BMI profiles: the 14 who showed the greatest increases in BMI, and 14 individuals who showed no changes in BMI over the 10 years. Overall, 69 of the 127 women had prediabetes defined by at least one category: 38 by fasting glucose, 41 by 2 h glucose and 31 by HbA1c levels. Only seven individuals had prediabetes according to all three definitions. All study participants gave informed consent and the study was approved by the institutional review board at the University of Texas Health Science Center at Houston.
The Michigan Regional Comprehensive Metabolomics Resource Core (Ann Arbor, MI, USA) performed untargeted metabolomic and lipidomic assays. EDTA plasma aliquots had been stored at −80°C since their collection date. Individuals were aged 35–62 years at the time of sample collection. Lipids were extracted using a modified Bligh–Dyer method [14]. The lipid extract was then subjected to LC-MS lipidomic profiling using an Agilent (Santa Clara, CA, USA) 6520 qTOF operating in positive and negative ion modes using data-dependent MS/MS with dynamic mass exclusion. Lipids were identified using the LipidBlast package [15], resulting in 707 identified lipid species. To perform untargeted metabolomic profiling, a 1290 Infinity Binary LC system from Agilent was used for LC with a Waters Acquity (Milford, MA, USA) UPLC HSS T3 column. MS was performed using an Agilent 6530 qTOF system. Raw processing was conducted with Agilent MassHunter Qual (v. B.08.00) and Profinder (v. B.08.00) software, followed by data analysis with the Agilent Mass Profiler Pro package (v. B.14.9.1). A total of 7661 raw features with at least one measurement were obtained.
To reduce peak intensity drift, daily sample processing was accompanied by analysis of six pooled plasma samples along with process blanks. To perform normalisation, compounds present in the pooled samples were used to estimate ranks within each batch through inverse probability weighting. We further used the properly transformed and densely selected ranks to match observations of each batch to a targeted distribution through a local smoothing technique that is based on the concept behind quantile normalisation [16, 17]. As we observed some metabolites with measurements far exceeding others, we removed outliers to avoid results driven by only a few observations. Rather than setting an arbitrary threshold for deviation, we used Grubbs’ test to systematically set the threshold according to the number of available measures [18]. We also used the mean absolute difference statistic instead of SDs [19]. Outlier removal processing was applied for every metabolite with at least 30 measurements.
To test for statistical associations of amino acid levels with prediabetes, we standardised each amino acid level and applied linear regression tests, including age and BMI as covariates, by dichotomising individuals as normoglycaemic or having prediabetes using the three different definitions of prediabetes. For associations between all known metabolites and fasting glucose, 2 h glucose and HbA1c levels, we treated these three phenotypes as quantitative traits and performed linear regression between each and the metabolites while adjusting for age and BMI. Only metabolites with fewer than 50% missing rates were tested for associations. To assess the association of known metabolites with future changes, we looked at the 28 individuals with metabolomic profiles at two points in time. We used changes in fasting glucose, 2 h glucose and BMI levels between the measurements as dependent variables in the linear regression. We used BMI and age at baseline as covariates, together with each quantitative trait’s baseline measures.
With so many measures, even very low rates of missing data result in few samples with entirely complete data. Missingness is less problematic when analysing individual metabolites, but it restricts many multidimensional methods such as principal component analysis for multivariate analysis. We employed the ‘impute’ package from R Bioconductor (v. 3.8, www.bioconductor.org), which implements K-nearest-neighbour imputation [20], to avoid the missing data problem for our multivariate analyses. We imputed every metabolite with less than 30% missing rates, resulting in 3560 metabolites including 151 named metabolites. We used the MASS package (v. 7.3–50, https://cran.r-project.org/web/packages/MASS) for linear discriminant analyses (LDAs) and the glmnet package (v. 2.0–18, https://cran.r-project.org/web/packages/glmnet) for LASSO regression analysis. The cv.glmnet function was used to select the best parameter for least absolute shrinkage and selection operator (LASSO) regression employing leave-one-out (LOO) cross-validation. We chose to use the lambda.min value because it yields the most optimised model. Because our prediabetes definitions overlap, we chose the multiresponse Gaussian family provided by glmnet, which allows assigning multiple response variables in each instance. This method is useful when there are multiple correlated response variables, as the correlation structure is maintained such that the set of selected variables is shared across all target variables. To test classification performances, we performed another set of LOO cross-validation experiments by using LASSO and support vector machine (SVM) in conjunction, where in each LOO experiment we used 126 training samples to choose metabolites by LASSO (with tenfold cross-validation within 126 samples), and then used those metabolites to train an SVM classifier to classify each subtype of diabetes vs the rest. The LOO sample was used to measure classification accuracies and Matthews correlation coefficients (MCCs) of predictions. The MCC provides better insights on classification performances than accuracies when class distributions are imbalanced. For baseline comparisons, we also calculated SVM predictions using traditional risk factors (i.e. BMI, age, HDL-cholesterol, triacylglycerol, systolic BP and diastolic BP) as features. SVM was trained using the radial basis function kernel with C = (number of features), gamma = 1/(number of features). We used the e1071 package (v. 1.7–2, https://cran.r-project.org/web/packages/e1071/) for SVM analyses. We used R version 3.5.1 (R Foundation for Statistical Computing, Vienna, Austria) for all statistical analyses.
Results
Amino acids and prediabetes subtypes
We investigated differences in mean amino acid levels between individuals with prediabetes and those with normoglycaemia for 12 amino acids (Fig. 1) using the metabolomic profiles of 127 individuals. The results showed distinctive amino acid signatures for normoglycaemic and prediabetic individuals. We also determined the statistical significance of differences in amino acid distributions between normoglycaemic and prediabetic individuals (Table 1). Isoleucine, tryptophan and lysine levels were differentially distributed among individuals with overall prediabetes (by any criteria) and individuals with normoglycaemia at p < 0.05. Among the subcategories of prediabetes, valine, isoleucine, tryptophan, lysine and arginine were associated at p < 0.05 for prediabetes defined by fasting glucose, while isoleucine, proline and arginine were associated with prediabetes status defined by 2 h glucose levels. With a more stringent Bonferroni-adjusted significance threshold (p < 4.2 × 10−3, adjusted for 12 amino acids), isoleucine and lysine showed a significant association with overall prediabetes. In the subcategories, isoleucine and lysine were significantly associated with prediabetes status defined by fasting glucose only. Isoleucine was associated with both fasting glucose- and 2 h glucose-defined prediabetes, and lysine was associated with fasting glucose- and HbA1c-defined prediabetes. Individuals with prediabetes defined by fasting glucose had higher levels of valine and isoleucine, but these BCAA measures were not higher in individuals with prediabetes by HbA1c only. On the contrary, mean arginine levels were higher in individuals with HbA1c-defined prediabetes and were also higher in fasting glucose-defined prediabetes than in normoglycaemic individuals, but were lower in individuals with 2 h glucose-defined prediabetes than in normoglycaemic individuals. Tryptophan and lysine levels were consistently higher in individuals with all definitions of prediabetes than in individuals with normoglycaemia (Fig. 1).

Mean normalised amino acid levels in prediabetes and normoglycaemia by (a) any of the three criteria for prediabetes, (b) fasting blood glucose, (c) 2 h glucose and (d) HbA1c levels. Arg, arginine; Gln, glutamine; Glu, glutamate; Ile, isoleucine; Leu, leucine; Lys, lysine; Phe, phenylalanine; Pro, proline; Thr, threonine; Trp, tryptophan; Tyr, tyrosine; Val, valine. Data are expressed as means ± SEM
These results suggested that the three glycaemic measures defining prediabetes differ in their characteristics; hence, we calculated correlation among these three measures. We observed the strongest correlation between fasting glucose and HbA1c at 0.4459 (p < 1.49 × 10−7), followed by 0.3511 (p < 7.32 × 10−5) between fasting and 2 h glucose, and 0.1904 (p < 0.0357) between 2 h glucose and HbA1c.
Known compounds associated with glucose and HbA1c levels
The analyses were extended across 181 known metabolites with missing rates of less than 50% using a slightly different approach. Instead of classifying individuals as prediabetic or normoglycaemic, we tested for associations by using fasting glucose, 2 h glucose and HbA1c as quantitative traits in linear regression (see electronic supplementary material [ESM] Table 1). Not surprisingly, we found that the metabolite glucose was most significantly associated with fasting plasma glucose levels (p = 4.1 × 10−9). Glucose was also positively associated with HbA1c levels, albeit non-significantly (p = 5.2 × 10−3), but was not included in the top association results for 2 h glucose. We also found a statistically significant association between lysine and fasting glucose levels (p = 1.5 × 10−4) and non-significant associations between tryptophan, isoleucine and valine and fasting glucose levels. For 2 h glucose, we found significant associations with palmitate (4.6 × 10−6), palmitoleic acid (5.0 × 10−6), oleic acid (p = 2.5 × 10−6) and cis-7,10,13,16-docosatetraenoic acid (adrenic acid, p = 4.6 × 10−5). No known compound was identified to be significantly associated with HbA1c levels after Bonferroni correction (p < 3.1 × 10−4), but several amino acids including tryptophan, arginine, lysine and tyrosine were in the top association results. We also observed associations of HbA1c levels with kynurenine and xanthine (p < 0.05), which are metabolites involved in the tryptophan–kynurenine pathway.
Compounds associated with temporal changes
To understand which metabolites are indicative of future changes in diabetes-related traits, we analysed metabolites measured in samples from an earlier time point by testing for associations with changes in BMI, fasting glucose and 2 h glucose levels in 28 individuals with measures from two time points. We could not test HbA1c because it was not collected in the initial examinations. We performed linear regressions for changes in each phenotype adjusted by baseline age, BMI and baseline phenotype (ESM Table 2). Although we did not find any metabolites with p values lower than the Bonferroni level (p < 3.1 × 10−4) due to the small sample size, we observed several non-significant (p < 0.05) association signals. Baseline oleic acid and glucose levels were both negatively associated with future BMI changes. Lignoceric acid (p= 2.1 × 10−3) and glycocholate (p = 2.6 × 10−3) showed positive associations with fasting glucose changes. Glutamine showed a non-significant association (p = 1.4 × 10−3) with future changes in 2 h glucose levels.
Multidimensional analysis with unknown metabolites
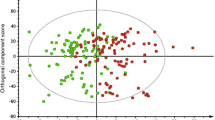
Although testing individual metabolites permits robust statistical interpretation, the richness of metabolomics provides more interesting perspectives with collective information from multiple metabolites. Because it is not possible to assess the statistical significance of high-dimensional multivariate analyses due to the exponentially increasing hypothesis space, we focused on qualitative aspects of discriminant analysis with variable selection strategies. To understand the overall information content of amino acid levels collectively, we performed LDA using all 12 measured amino acids (including redundant measures) to separate individuals with prediabetes from the rest by the three aforementioned criteria. Because LDA provides one-dimensional projections that best separate the given groups, we performed three separate LDAs with the three different definitions of prediabetes (Fig. 2a–c). We created the three-dimensional coordinates on these plots by using LDA with prediabetes status defined by fasting glucose levels, 2 h glucose and HbA1c levels. Figures 2a and b are shown with two of these three linear discriminants, by using the dimensions from fasting and 2 h glucose, while Fig. 2c is shown on the dimensions from fasting glucose and HbA1c. As expected, fasting glucose-defined prediabetes was best separated along the dimension created by LDA on fasting glucose status. Next, we extended our search space into all 151 known metabolites with missing rates of less than 30% (Fig. 2d–f), and then to all 3560 measured metabolites with missing rates of less than 30%, including the unknown ones (Fig. 2g–i). For these analyses, we applied LASSO [21] regression to select the subset of metabolites that yielded the best separation for all three subtypes of prediabetes while regularising the number of selected metabolites. LASSO regression selected 63 out of 151 known metabolites and 118 out of 3560 metabolites. We applied LDA onto these selected sets of metabolites to find the one-dimensional projections to visualise the separation. The 63 metabolites selected by LASSO regression provided better separation for all three prediabetes definitions (Fig. 2d–f) than using just amino acid levels, but the improvements were more noticeable in the 2 h glucose results than in the others. Using all available metabolites resulted in near-perfect separation for all three prediabetes definitions (Fig. 2g–i). To measure how well we could classify subtypes of diabetes based on the metabolomics data, we performed LOO classification experiments using both LASSO and SVM. The accuracy of fasting glucose-, 2 h glucose- and HbA1c-defined prediabetes was 0.87, 0.92 and 0.86, respectively, when we used features selected from all metabolites, which was greatly improved from the baseline accuracies of 0.55, 0.67 and 0.70 that were derived by using the traditional risk factors of age, BMI, HDL-cholesterol, triacylglycerol, systolic BP and diastolic BP. To take account of class label imbalances we also measured MCCs, which improved to 0.67, 0.82 and 0.59 from the baseline values of −0.27, 0.20 and 0.070 for fasting glucose-, 2 h glucose- and HbA1c-defined prediabetes predictions, respectively (ESM Table 3).

LDAs between normoglycaemic and prediabetic individuals defined by three criteria. Linear discriminant (LD)1 was projected by using fasting glucose (FG)-based prediabetes status as class labels, and LD2 and LD3 by using 2 h glucose (2hG) and HbA1c, respectively. (a–c) LDA was performed on 12 amino acid measures with missing rates of less than 30%, coloured by prediabetes status by (a) fasting glucose, (b) 2 h glucose and (c) HbA1c. (d–f) LDA was performed on 63 metabolites selected by LASSO regression on 151 known metabolites with missing rates of less than 30%, coloured by prediabetes status by (d) fasting glucose, (e) 2 h glucose and (f) HbA1c. (g–i) LDA was performed on 118 metabolites selected by LASSO regression on 3560 metabolites, including unknown metabolites, coloured by prediabetes status by (g) fasting glucose, (h) 2 h glucose and (i) HbA1c
It has been reported that tryptophan to kynurenine metabolism is upregulated in individuals with type 2 diabetes and it is also closely related to stress and inflammation [22, 23]. We further investigated the pairwise relationship between tryptophan and kynurenine levels and discovered that kynurenine levels are positively correlated with tryptophan levels (Fig. 3). More interestingly, we found that the correlation between tryptophan and kynurenine was higher in prediabetic individuals than in those with normoglycaemia (Fig. 3).

Correlation between tryptophan and kynurenine levels in normoglycaemic and prediabetic groups. *p < 0.05, **p < 0.001. Corr, correlation
Discussion
These results show that there are distinct differences in amino acid levels between prediabetic and normoglycaemic individuals. More interestingly, we observed differences in amino acid levels between subtypes of prediabetes. For example, the mean arginine levels of individuals with prediabetes defined by fasting glucose (i.e. impaired fasting glucose) were much higher than those of normoglycaemic individuals, while arginine levels for individuals with prediabetes defined by 2 h glucose (i.e. impaired glucose tolerance) was noticeably lower than those of normoglycaemic individuals. The mean arginine levels of individuals with prediabetes by HbA1c were also higher than normal, suggesting that arginine levels may interact differently with the metabolic drivers of fasting and post-load glucose levels. Mean isoleucine levels were consistently higher in individuals with prediabetes in all three subtypes of prediabetes, consistent with existing reports on the relationship between BCAAs and type 2 diabetes [8], but the difference was non-significant for HbA1c-defined prediabetes. The mean proline level was (p < 0.05) lower in prediabetic vs normoglycaemic individuals only for 2 h glucose-defined prediabetes, while the mean threonine level was lower (p < 0.05) only in HbA1c-defined prediabetes.
Our plasma samples from the second timepoint (127 samples) had been stored for 2–6 years and samples from the first timepoint (28 samples) had been stored frozen 10–16 years before metabolomic profiling. It has been reported that metabolites stay stable at −80°C for up to seven years, but longer storage of up to 16 years would affect the analyses [24]. We acknowledge that our results might have been affected by the lengthy storage of the samples.
It has been previously reported that metabolites involved in the tryptophan metabolism pathway show different signatures between normoglycaemic individuals and those with type 2 diabetes [22]. We also observed that mean tryptophan levels were higher in individuals with prediabetes (Fig. 1, Table 1). Our association analysis with all known metabolites (ESM Table 2) showed that tryptophan was positively associated with fasting glucose and HbA1c levels. More interestingly, we also found that the HbA1c level was positively associated with kynurenine (p = 4.2 × 10−3) and xanthine (p = 0.011), which are two of the main metabolites involved in the tryptophan metabolic pathway. Stronger correlations between kynurenine and tryptophan in individuals with prediabetes vs those with normoglycaemia (Fig. 3) suggest that the tryptophan–kynurenine metabolism is stronger in individuals with prediabetes.
Our discriminant analyses show that metabolomic profiling can separate the different subtypes of prediabetes. As few as 12 amino acid measures provided reasonable separation between individuals with fasting glucose-defined prediabetes and normoglycaemic control participants (Fig. 2a–c), while the separation was less clear in other subtypes. Obviously, more features provide better separation (Fig. 2d–i), but this might also result in overfitting in the absence of an exponentially larger sample size with the increased dimensionality. We tried to minimise the overfitting problem by employing regularised multivariate regression (LASSO). LOO classification results with LASSO and SVM clearly showed improvements obtained by additional metabolites in predicting subtypes of prediabetes.
We acknowledge that the statistical power of our results is limited by the moderate sample size and lengthy frozen storage of samples. Although we focused more on descriptive and exploratory analyses than on novel discovery in this study, our results are consistent with those of previous studies [4,5,6,7,8,9,10] and suggest a need for and great potential in evaluating untargeted metabolomics data to reveal the biological underpinnings of the early progression of (pre)diabetes. They also point to metabolically complex differences among individuals with prediabetes defined by different criteria. It is not known to what extent the same is true for the different definitions of overt diabetes.
In conclusion, we observed distinctive amino acid signatures between normoglycaemic and prediabetic individuals including BCAAs, as previously reported, with distinctive signatures between different subtypes of prediabetes. We identified various metabolites that are sensitive predictors of prediabetes status and its subtypes using statistical association tests. Our results show that metabolomics data provide a high-dimensional snapshot of metabolic changes in the early developmental stages of type 2 diabetes.
Data availability
The pilot metabolomics samples used in this study are part of the Starr County GWAS and Exome Sequencing Study, and the metabolomics data are available through the NCBI database of Genotypes and Phenotypes (dbGaP, accession number phs001166).
Abbreviations
- BCAA:
-
Branched-chain amino acid
- LASSO:
-
Least absolute shrinkage and selection operator
- LDA:
-
Linear discriminant analysis
- LOO:
-
Leave-one-out
- MCC:
-
Matthews correlation coefficient
- SVM:
-
Support vector machine
References
Hanis CL, Redline S, Cade BE et al (2016) Beyond type 2 diabetes, obesity and hypertension: an axis including sleep apnea, left ventricular hypertrophy, endothelial dysfunction, and aortic stiffness among Mexican Americans in Starr County, Texas. Cardiovasc Diabetol 15:86
American Diabetes Association (2014) Diagnosis and classification of diabetes mellitus. Diabetes Care 37(Suppl 1):S81–S90
Winner D, Norton L, Kanat M et al (2014) Strong association between insulin-mediated glucose uptake and the 2-hour, not the fasting plasma glucose concentration, in the normal glucose tolerance range. J Clin Endocrinol Metab 99:3444–3449
Luzi L, Castellino P, DeFronzo RA (1996) Insulin and hyperaminoacidemia regulate by a different mechanism leucine turnover and oxidation in obesity. Am J Phys 270:E273–E281
Luzi L, Petrides AS, De Fronzo RA (1993) Different sensitivity of glucose and amino acid metabolism to insulin in NIDDM. Diabetes 42:1868–1877
Tillin T, Hughes AD, Wang Q et al (2015) Diabetes risk and amino acid profiles: cross-sectional and prospective analyses of ethnicity, amino acids and diabetes in a south Asian and European cohort from the SABRE (Southall and Brent REvisited) study. Diabetologia 58:968–979
Floegel A, Stefan N, Yu Z et al (2013) Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 62:639–648
Wurtz P, Soininen P, Kangas AJ et al (2013) Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care 36:648–655
Ferrannini E, Natali A, Camastra S et al (2013) Early metabolic markers of the development of dysglycemia and type 2 diabetes and their physiological significance. Diabetes 62:1730–1737
Guasch-Ferre M, Hruby A, Toledo E et al (2016) Metabolomics in prediabetes and diabetes: a systematic review and meta-analysis. Diabetes Care 39:833–846
Drogan D, Dunn WB, Lin W et al (2015) Untargeted metabolic profiling identifies altered serum metabolites of type 2 diabetes mellitus in a prospective, nested case control study. Clin Chem 61:487–497
Zhao J, Zhu Y, Hyun N et al (2015) Novel metabolic markers for the risk of diabetes development in American Indians. Diabetes Care 38:220–227
Mamtani M, Kulkarni H, Wong G et al (2016) Lipidomic risk score independently and cost-effectively predicts risk of future type 2 diabetes: results from diverse cohorts. Lipids Health Dis 15:67
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917
Kind T, Liu KH, Lee DY, DeFelice B, Meissen JK, Fiehn O (2013) LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat Methods 10:755–758
Bolstad BM, Irizarry RA, Astrand M, Speed TP (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193
Liu LY, Wang N, Lupton JR, Turner ND, Chapkin RS, Davidson LA (2005) A two-stage normalization method for partially degraded mRNA microarray data. Bioinformatics 21:4000–4006
Grubbs FE (1969) Procedures for detecting outlying observations in samples. Technometrics 11:1–21
Leys C, Ley C, Klein O, Bernard P, Licata L (2013) Detecting outliers: do not use standard deviation around the mean, use absolute deviation around the median. J Exp Soc Psychol 49:764–766
Hastie T, Tibshirani R, Sherlock G, Eisen M, Brown P, Botstein D (1999) Imputing missing data for gene expression arrays. Technical report. Division of Biostatistics, Stanford University, Stanford
Zou H, Hastie T (2005) Regularization and variable selection via the elastic net. J R Statist Soc B 67:301–320
Oxenkrug GF (2015) Increased plasma levels of xanthurenic and kynurenic acids in type 2 diabetes. Mol Neurobiol 52:805–810
Oxenkrug G (2013) Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenine dinucleotide metabolic pathways. Mol Neurobiol 48:294–301
Wagner-Golbs A, Neuber S, Kamlage B, et al. (2019) Effects of long-term storage at −80°C on the human plasma metabolome. Metabolites 9(5)
Acknowledgements
We thank G. I. Bell (Department of Human Genetics, University of Chicago) for helpful advice and discussions. Some of the data in this study were presented as an abstract at the ADA 77th Scientific Sessions in 2017.
Funding
This study was supported by the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Disease (awards DK089503, DK097153 and DK118631).
Author information
Authors and Affiliations
Contributions
All authors significantly contributed to the manuscript and approved the final version for publication. CLH and CFB contributed to the study design, data acquisition, analysis and manuscript revision. CE and DA contributed to data acquisition, interpretation of data and manuscript revision. GJ contributed to data analysis, interpretation of data, manuscript drafting and revision. GJ is the guarantor of this work.
Corresponding author
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM
(PDF 126 kb)
Rights and permissions
About this article

Cite this article
Jun, G., Aguilar, D., Evans, C. et al. Metabolomic profiles associated with subtypes of prediabetes among Mexican Americans in Starr County, Texas, USA. Diabetologia 63, 287–295 (2020). https://doi.org/10.1007/s00125-019-05031-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-05031-4