
Abstract
Type 1 diabetes is considered an autoimmune disease characterised by specific T cell-mediated destruction of the insulin-producing beta cells. Yet, except for insulin, no beta cell-specific antigens have been discovered. This may imply that the autoantigens in type 1 diabetes exist in modified forms capable of specifically triggering beta cell destruction. In other immune-mediated diseases, autoantigens targeted by the immune system have undergone post-translational modification (PTM), thereby creating tissue-specific neo-epitopes. In a similar manner, PTM of beta cell proteins might create beta cell-specific neo-epitopes. We suggest that the current paradigm of type 1 diabetes as a classical autoimmune disease should be reconsidered since the immune response may not be directed against native beta cell proteins. A modified model for the pathogenetic events taking place in islets leading to the T cell attack against beta cells is presented. In this model, PTM plays a prominent role in triggering beta cell destruction. We discuss literature of relevance and perform genetic and human islet gene expression analyses. Both direct and circumstantial support for the involvement of PTM in type 1 diabetes exists in the published literature. Furthermore, we report that cytokines change the expression levels of several genes encoding proteins involved in PTM processes in human islets, and that there are type 1 diabetes-associated polymorphisms in a number of these. In conclusion, data from the literature and presented experimental data support the notion that PTM of beta cell proteins may be involved in triggering beta cell destruction in type 1 diabetes. If the beta cell antigens recognised by the immune system foremost come from modified proteins rather than native ones, the concept of type 1 diabetes as a classical autoimmune disease is open for debate.
Similar content being viewed by others


Introduction
Since the early 1970s, type 1 diabetes has been considered to be the result of an immune reaction against the beta cells [1–3]. The disease is multifactorial, with both environmental and genetic factors contributing to type 1 diabetes risk. There is broad consensus that the pathogenesis of type 1 diabetes comprises two distinct phases: (1) the ‘initiation phase’, which is non-antigen-driven and non-lymphocyte-dependent. In this phase, initial beta cell damage is thought to be induced by environmental factors, e.g. a virus and/or cytokines secreted by activated monocytes, macrophages and dendritic cells resident in islets and/or possibly by beta cells themselves; and (2) the ‘amplification phase’ or ‘execution phase’, which is characterised by antigen-driven, lymphocyte-dependent progressive and selective destruction of the beta cells, causing clinical diabetes [4–6]. GAD 65 kDa (GAD65), insulin, protein tyrosine phosphatase (IA-2), zinc transporter 8 (ZnT8) and several additional proteins have been identified as autoantigens based on antibody or T cell reactivity in sera from type 1 diabetes patients [7, 8]. However, the paradigm of type 1 diabetes as a classical autoimmune disorder contains a paradox: none of the established autoantigens [9–12], with the possible exception of insulin, are exclusively expressed in the beta cells, thus failing to explain the selective immune-mediated destruction of the beta cells—a fact that has been largely neglected by researchers in the field.
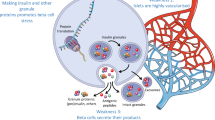
We suggest a simple pathogenetic model in which epitopes recognised by the immune system are not derived from native beta cell proteins but occur on proteins that have been post-translationally modified, creating immunogenic neo-epitopes in beta cells during the initiation phase (Fig. 1a). Such neo-epitopes have not been expressed in thymus during lymphocyte selection or in the normal, i.e. non-damaged, beta cell for induction of peripheral tolerance and are thus seen as ‘foreign’ by the immune system. Therefore, an immune attack is mounted to eliminate the target tissue (Fig. 1b). Implicit in this model is that type 1 diabetes is not caused by failure of normal clearance of autoreactive T cells. That neo-epitopes might play a role in the development of type 1 diabetes has also recently been considered by other researchers [13–15]. However, here we place the generation of neo-epitopes arising from modified beta cell proteins into a pathogenetic context (Fig. 1). We further propose that part of the genetic risk of type 1 diabetes is caused by variation in genes encoding proteins involved in post-translational modification (PTM) processes that could increase PTM of beta cell proteins in at-risk individuals.

Model for the pathogenesis of type 1 diabetes. (a) The initiation phase. Some degree of beta cell damage is induced in the initiation phase. This may be caused by environmental factors as indicated in genetically pre-disposed individuals directly in the beta cells or via activation of intra-islet DCs and/or macrophages (MΦs). The initial damage leads to upregulation of inducible nitric oxide synthase (iNOS) and production of ROS in the beta cell. This, in combination with alterations in the expression of genes encoding enzymes involved in PTM processes, leads to changes in PTMs of beta cell proteins during the process of beta cell death. The dying beta cells are phagocytosed by DCs and MΦs. Peptides arising from modified proteins in the phagocytosed beta cells are presented on MHC class II molecules on the DCs and MΦs. Alternatively (illustrated), the modified proteins or peptides hereof (neo-epitopes) are released by the beta cells and taken up by the DCs/MΦs. The process of antigen presentation stimulates the DCs/MΦs to produce and secrete IL-1β and nitric oxide (NO), causing more damage to the beta cells. The activated DCs/MΦs then travel to the pancreatic lymph node for MHC class II-antigen presentation to CD4+ T cells. (b) The amplification and executioner phase. In the lymph node, the DC/MΦ will present the neo-epitopes to CD4+ T cells resulting in activation of these including secretion of IFNγ and IL-2. Note that the presentation of several beta cell peptides by DCs/MΦs causes polyclonal activation of CD4+ T cells. Activated antigen-specific CD4+ T cells stimulate clonal expansion of B cells, leading to the production of antibodies. Upon islet infiltration, the CD4+ T cells activate cytotoxic CD8+ T cells bound to matching peptides presented on MHCI on the beta cells. Finally, the CD8+ T cells destroy the beta cells by several effector mechanisms, including Fas, perforin and granzyme B. The T cell-mediated beta cell killing creates a deleterious self-amplifying circle because the increase in beta cell death favours generation of additional beta cell-specific neo-epitopes that further escalate T cell activation and beta cell destruction. The result is clinical diabetes. BCR, B cell receptor
To support the concept that PTMs trigger the immune-mediated beta cell destruction in type 1 diabetes, we present relevant literature, measure the expression of genes encoding proteins involved in PTM processes in human islets and investigate these genes for type 1 diabetes-associated single nucleotide polymorphisms (SNPs). Of note, the purpose of this paper is not to give a comprehensive overview of the literature, but to present key findings in animal models and humans supporting the concept that PTM of beta cell antigens plays an important role in type 1 diabetes. We hope this will stimulate debate and lead to studies that will eventually solve the puzzle as to why the immune system destroys beta cells in type 1 diabetes.
Evidence from the literature
Post-translational modifications and autoimmunity
PTM of proteins is vital for cellular homeostasis, function and signalling pathways. PTM alters specific parts of a protein or peptide by the covalent attachment or removal of atoms, chemical groups or larger biomolecules such as sugars. PTM of proteins affects their properties, including folding, conformation and activity, thereby affecting their interaction with other proteins and their cellular function. While most PTMs are essential for proper cellular function, some PTMs can create neo-epitopes that can activate an immune response. This has been demonstrated in diseases such as rheumatoid arthritis, multiple sclerosis and celiac disease, where modified proteins play a major role for disease development [16–21]. PTM of proteins can be the result of both enzyme-catalysed and spontaneous reactions. For example, while acetylation, phosphorylation, glycosylation, palmitoylation, citrullination (also called deimination) and sumoylation are enzyme-catalysed modifications, oxidation, nitrosylation and glycation are typically spontaneous processes.
Under normal conditions, T cells that recognise self-antigens with high affinity are deleted in the thymus by negative selection [22]. However, lower affinity T cells may escape negative selection and migrate to the periphery. Depending on whether these naive T cells encounter their cognate antigen in the periphery, and the concentration at which the antigen is present, they may be deleted or become anergic [23]. Because many modified proteins are unique to the peripheral tissues, tolerance to some self-antigens exclusively depends on peripheral mechanisms [24]. Failure to eliminate self-reactive T cells results in an immune response. Autoimmune diseases are therefore classically considered to be the consequence of self-reactive T cells that escape either thymic or peripheral deletion. However, it is plausible that under certain conditions (e.g. viral infection and/or inflammation), alterations in PTMs are induced in the affected cells, thereby creating neo-epitopes from self-proteins that have not previously been exposed to the immune system either in the periphery or in the thymus.
Modified self-peptides or proteins may cause an immune response by various mechanisms. Modified peptides presented by MHC class II molecules on antigen-presenting cells (APCs) can modulate T cell receptor (TCR) binding to the MHC/antigen complex, leading to more efficient TCR binding, T cell signalling and proliferation. For example, citrullination (deimination of arginine) of a vimentin-related peptide enhances the binding affinity of the peptide for the MHC molecules and leads to increased T cell activation [25]. Similar to citrullination, glycosylation of peptides presented by MHC molecules has been shown to be important for T cell recognition and activation [26]. PTM may also influence the processing of an antigen, resulting in alternative cleavage of the peptide and/or lead to increased levels of a particular peptide epitope. Such an increase in epitope density could lead to the activation of low-avidity T cells in the periphery [27].
Post-translational modifications in type 1 diabetes
How and under which conditions could PTMs of beta cell proteins that could break immune tolerance in type 1 diabetes be generated? Islets contain resident APCs, including macrophages and dendritic cells (DCs) [28, 29]. APCs play a critical role in type 1 diabetes by secreting beta cell cytotoxic cytokines and by presenting beta cell antigens to the immune system. The essential role of APCs has been demonstrated in studies on spontaneous (BioBreeding [BB] rats and NOD mice) and induced (multiple low dose streptozotocin [MLD-STZ]) animal models of type 1 diabetes [30–32]. Activation of APCs leads to the production and release of cytokines and reactive oxygen species (ROS), creating an inflammatory and oxidative milieu that may produce many changes in PTM. Consistently, exposure of islets to cytokines causes several proteins to be modified as determined by mass spectrometry [33, 34]. Similar observations were made in islets from BB rats at different time points during diabetes development [34, 35].
Almost 20 years ago, we suggested for the first time the concept that beta cell antigens in type 1 diabetes may not be released in their native conformation, but in ROS-modified, and therefore more antigenic, forms [6]. In line with this, GAD65 has now been demonstrated to be modified to a slightly higher molecular weight form under oxidative conditions in rat islets. Importantly, anti-GAD65 autoantibodies from type 1 diabetes patients bind to modified GAD65 with higher affinity than they do to native GAD65 [36]. In a recent study, GAD65 was irradiated with UV light in the presence of hydrogen peroxide to induce oxidative modification of GAD65 [37]. This produced changes in structural, conformational and thermal denaturation profiles of GAD65. IgG from sera obtained from type 1 diabetic patients exhibited a significantly higher recognition of oxidatively modified GAD65 over native GAD65. In this regard, it is important to note that beta cells express lower levels of oxygen radical scavenger enzymes, including manganese superoxide dismutase and catalase, than other cell types [38]. This, together with high expression of inducible nitric oxide synthetase (iNOS) when challenged with cytokines [39, 40], is likely to make the beta cells particularly prone to oxidative damage, and hence oxidative modification of GAD65 and other proteins. Studies of beta cell maturation have shown that an acquired trait of the beta cell phenotype is increased sensitivity to the cytotoxic effects of cytokines [41, 42]. These unique features of beta cells may explain why other islet cell types and other secretory active cells are not destroyed by the immune system in type 1 diabetes.
In addition to modification of GAD65, a PTM of insulin—a disulfide bond between adjacent cysteine residues in the A-chain of human insulin—has been demonstrated. T cell clones isolated from type 1 diabetic patients showed increased recognition of the modified insulin epitope. Interestingly, the modified insulin peptide did not alter binding to MHC (HLA-DR4) [43].
Recently, a study demonstrated that patients with type 1 diabetes have antibodies to collagen type II and that antibody binding is significantly higher to oxidatively modified collagen type II over native collagen. Patients carrying the HLA-DRB1*04 allele showed the highest risk for autoimmunity against modified collagen, while carrying a DRB1*0301 allele was protective. Autoimmunity to native collagen type II was not associated with any DRB1 alleles [44]. This study therefore highlights that, in addition to the importance of formation of PTMs for creating neo-epitopes, specific HLA genotypes can be important for the presentation of these neo-epitopes to the immune system. It should be stressed, however, that without generation of cognate neo-epitopes, such HLA genotype effects may be insufficient to trigger disease.
Taken together these data indicate that, while modifications of proteins have been shown in type 1 diabetes contexts the exact pathogenetic importance and causative role of these is still unclear. However, it is noteworthy that two of the established major autoantigens in type 1 diabetes, GAD65 and insulin, can exist in modified forms that cause stronger immune responses.
Evidence for PTMs in animal models
A transgenic non-diabetes-prone C57/BL/6J mouse expressing CD154 under control of the rat insulin promoter (RIP) has been developed [45]. Beta cell expression of this transgene causes activation of DCs in the islets, leading to massive lymphocyte infiltration, insulitis and diabetes. Interestingly, adoptive transfer of spleen cells from transgenic diabetic mice to wild-type recipients does not cause insulitis or diabetes. Furthermore, wild-type islets transplanted into diabetic transgenic mice restore blood glucose levels to normal and the transplants are not destroyed. One possible interpretation of these data is that the immune system in this model does not recognise native proteins on undamaged islets, i.e. islets not exposed to activated DCs in situ. If the transgenic expression of CD154 on beta cells takes place in immuno-deficient mice, beta cell function is reduced but overt diabetes does not develop owing to lack of T cell-mediated beta cell destruction [45]. Thus, in these mice, an initiation phase occurs, but an execution phase does not. This model indicates that intra-islet activation of APCs induce PTMs of beta cell proteins creating neo-epitopes which trigger a T cell attack, i.e. the executioner phase.
A single high dose of STZ completely destroys the beta cells and causes diabetes in rodents without insulitis. However, administration of MLD-STZ induces insulitis followed by development of diabetes. Studies have shown that disease is not easily transferable by adoptive transfer of splenocytes from MLD-STZ-induced diabetic mice to recipients [46]. Only if the recipient mice have been sensitised by pretreatment with subimmunogenic doses of STZ can disease be adoptively transferred [47–49]. Furthermore, syngeneic islets transplanted under the renal capsule prior to MLD-STZ treatment show insulitis, but this does not happen if the islets are transplanted after STZ treatment. Also, if the donor islets were exposed in vitro to STZ before transplantation into MLD-STZ-treated mice, then insulitis developed in the syngeneic grafts [50]. We suggest that, in the MLD-STZ model, the repetitive beta cell insults by STZ generate PTM and beta cell-specific neo-epitopes that trigger an immune response.
It is of note that an initiation phase of sufficient magnitude is capable of producing insulitis and diabetes in models that are not diabetes-prone and do not have diabetes-associated MHC molecules, i.e. the RIP-CD154 and MLD-STZ models. In line with this it has been reported that there has been an increase in autoantibody-positive children not carrying type 1 diabetes-associated HLA molecules [51, 52].
Insulin plays a major role in driving the T cell response against the beta cells in NOD mice. Studies in both NOD mice and humans have shown that an important epitope from insulin recognised by diabetogenic T cells is derived from the B-chain of insulin (B:9–23) [53]. It remains unknown, however, whether the B:9–23 epitope is subjected to PTM. As mentioned, the most notable proof-of-principle that PTM of insulin can create immunogenic epitopes has been provided in man [43]. The important role of insulin as an antigen in both NOD mice and man is not a surprise. Firstly, insulin may be the only beta cell-specific antigen and, secondly, it is conceivable that the high concentration of insulin in the beta cells makes it particularly prone to modification under stress- and inflammatory conditions. Thus, PTM of insulin is likely to create causative neo-epitopes and may also favour epitope spreading.
Using diabetogenic T cell clones from NOD mice, recent work has shown that a naturally occurring proteolytic cleavage product (WE14) of chromogranin A, which is contained within the insulin granules, is converted into a highly antigenic T cell epitope by in vitro treatment with transglutaminase [54, 55]. This observation further demonstrates that PTM of antigens can significantly affect their antigenicity towards a stronger immune response. Very recently, human T cell clones that recognise PTM peptides derived from GAD65 and IA-2 were isolated from subjects with type 1 diabetes using HLA class II tetramers. These T cell clones preferentially recognised the citrullinated or transglutaminated form of their cognate peptide (personal communication, Edward James, Benaroya Research Institute, Seattle, WA, USA). This observation demonstrates that PTM can increase the antigenicity of self-epitopes in humans.
In summary, the studies discussed above provide circumstantial evidence for generation of PTMs leading to the creation of neo-epitopes during the pathogenetic initiation phase in animal models.
Apoptotic beta cells as sources of neo-epitopes
If beta cell damage creates beta cell-specific neo-epitopes in the pathogenetic initiation phase, it should be possible to prevent diabetes through immunisation with the modified antigens if this takes place early enough and before overt diabetes has developed. It has been demonstrated that induction of limited beta cell apoptosis by a single low-dose injection of STZ protected NOD mice from spontaneous diabetes [56]. Similarly, transfusion of in vitro-induced apoptotic (ultraviolet B irradiated) NIT-1 mouse beta cells into NOD mice prevented diabetes and correlated with suppression of anti-beta cell antibodies and induction of regulatory T cells [57]. Transfusion of non-apoptotic beta cells into NOD mice had no effect on diabetes. The results of these studies suggest that immune tolerance to beta cells in NOD mice can be attained by exposing the immune system to small amounts of modified beta cell antigens arising from apoptotic beta cells. This cannot be achieved by exposing the immune system to native proteins from non-apoptotic cells. Intriguingly, the tolerogenic effect of apoptotic cell transfusion can be achieved by transfusion of cells other than beta cells [58], suggesting that some of the neo-epitopes created by apoptotic cell death are common to all cell types. This, of course, argues that beta cell-specific neo-epitopes are responsible for the loss of immune tolerance in type 1 diabetes, but it is plausible that more uniform neo-epitopes arising from different cells, if presented to the immune system in disease-prone animal models, can provide sufficient tolerogenic effects to halt beta cell destruction. However, it will be important to determine whether immunisation with apoptotic non-beta cells can also lead to beta cell tolerance in models that are not prone to diabetes. Importantly, recent findings suggest that physiological beta cell death occurring in young NOD mice during organogenesis might be sufficient to initiate the immune response [59]. Collectively, the studies discussed above provide evidence that events are taking place in the beta cells during the initiation phase that make them a target for the adaptive immune system. These events are likely to be PTMs.
Analysis of genes involved in PTM processes
We hypothesised that part of the genetic component of type 1 diabetes is due to variation in genes regulating PTM processes. We first identified all known genes encoding proteins involved in different PTMs of potential relevance for type 1 diabetes: acetylation, glycosylation, amidation, amination, citrullination, sumoylation, methylation, nitrosylation/nitration and hydroxylation. These PTMs were chosen because they are either enzyme-catalysed and have previously been associated with other immune-mediated diseases or because they are produced by ROS, which are generated in islets during inflammatory stress. We identified a total of 291 genes encoding proteins involved in these PTM categories (electronic supplementary material [ESM] Table 1, ESM Methods).
We then examined whether cytokines (IL-1β + IFNγ + TNFα) change the mRNA expression of the identified genes in four individual human islet preparations. It was found that the expression of 39 genes was regulated by cytokines (p < 0.05, uncorrected value; ESM Table 2).
To examine for genetic variation in the cytokine-regulated genes, we took advantage of the genome-wide association study and meta-analysis performed by the Type 1 Diabetes Genetics Consortium [60]. The analysis revealed that 17 out of the 39 regulated genes contained nominally associated SNPs (ESM Table 2). For all 17 genes, the SNPs were located in either introns or up- or downstream of the gene (within 5 kb of the gene), suggesting that the polymorphisms in these genes might primarily affect the expression levels of the genes. We conclude that several genes involved in PTM processes are regulated by cytokines in human islets and also contain SNPs that are nominally associated with type 1 diabetes, providing support that such genes contribute to PTMs in islets that could create neo-epitopes.
Discussion
Of the established autoantigens in type 1 diabetes, insulin might be the only one that is exclusively expressed in beta cells [9–12]. The current paradigm of type 1 diabetes as a bona fide autoimmune disease therefore contains a paradox since the beta cell-specific destruction in type 1 diabetes cannot be explained by an immune response against the currently known antigens. We suggest that PTMs are induced during the initiation phase of pathogenesis and that these PTMs give rise to altered peptide structures, creating neo-epitopes not previously exposed to the immune system. There is both direct and circumstantial support for this concept in the published literature. Our analyses suggest that part of the non-HLA genetic susceptibility to type 1 diabetes may be explained by variation in the genes encoding enzymes involved in PTM processes specifically in islets. The mRNA expression of 39 of 291 identified genes encoding PTM enzymes of interest in human islets was changed following exposure to cytokines. Seventeen of these genes also harboured nominally associated SNPs, suggesting that they are of particular importance. It will be of importance to establish if these genes are also expressed and regulated at the protein level in islets and beta cells. Further, it will be highly relevant to determine whether the enzymatic activities of the PTM enzymes are altered in stressed (cytokine exposed) islets.
In support of the concept that cytokine-regulated genes may be involved in PTM processes that can affect type 1 diabetes, proteomic studies of islets exposed to cytokines in vitro and isolated islets from BB rats have shown that many PTM enzymes change expression levels and several proteins undergo PTM [33–35]. Consistent with an anticipated important pathogenetic role for PTM in the BB model, treatment of BB rats with a chemical inducer (CGP3466B) of the ‘PTM enzyme’ protein l-isoaspartate (d-aspartate) O-methyltransferase (PIMT) which recognises isomerised aspartate residues in proteins and converts them back to l-aspartate [61], delays disease onset and severity of disease and correlates with an increased number of beta cells in the islets [62]. These findings show that reversing a PTM by CGP3466B-induced upregulation of PIMT beneficially influences diabetes development in BB rats.
The observation that the transglutaminase (TGM)-2 gene contains a type 1 diabetes-associated polymorphism and is transcriptionally upregulated in human islets by cytokines is of interest. In celiac disease, TGM2 plays a prime role in breaking the immune tolerance to gluten via deamidation of glutamine residues in gluten peptides. Furthermore, this disease is characterised by autoantibodies to TGM2 [19]. The coexistence of type 1 diabetes and celiac disease [63] might thus, in part, be related to TGM2. In this context, it is interesting that the naturally occurring proteolytic cleavage product of chromogranin A, the WE14 peptide, becomes a highly antigenic T cell epitope following in vitro treatment with transglutaminase [54, 55].
Several diseases traditionally considered as autoimmune, e.g. rheumatoid arthritis, celiac disease and multiple sclerosis, are known to involve PTM of target proteins. The literature cited here and the experimental data presented support the hypothesis that PTM of beta cell proteins brought about by activation of members and mechanisms of the innate immune system during the initiation phase produce neo-epitopes on beta cell proteins. These neo-epitopes are presented on APCs and the beta cells on MHC class II and I molecules, respectively, and are foreign to the immune system. Therefore, the adaptive immune system will recognise them and mount a response to eliminate them. PTM of beta cell proteins are therefore causative rather than just a consequence of beta cell destruction. It is reasonable to assume that multiple antigens contribute to the activation of the polyclonal T cell attack against the beta cells.
Considering the complexity and polygenic nature of type 1 diabetes, it is plausible that the induced PTMs do not take place uniformly in the beta cells either within an individual islet or in different islets. Furthermore, there might be a great deal of interindividual variation. Thus, the same PTMs and proteins may not be equally important in triggering and exacerbating disease in different individuals.
If the PTM of beta cell proteins triggers beta cell destruction, the intriguing question is whether type 1 diabetes can be defined as truly ‘autoimmune’ when the T cells react against modified proteins that do not normally occur rather than native ones. Hence, it could be argued that there is nothing wrong with the immune system in type 1 diabetes in that it does what it is supposed to, i.e. destroy cells that contain modified and hence ‘foreign’ proteins. However, it is beyond doubt that part of the genetic contribution to type 1 diabetes is made by genes that have primary functions in immune cells. Variation in genes that modulate the effects of regulatory T cells might also be important. Such effects, however, are not causal, but are critical for the amplitude of the mounted immune response once initiated and are therefore important in determining how quickly clinical diabetes manifests itself.
To understand the pathogenesis of type 1 diabetes, the postulate of broken immunological tolerance as an underlying mechanism should be reconsidered. The hypothesis presented here is simple, but clearly in need of further experimental support. If correct, it leaves the concept of type 1 diabetes as a bona fide autoimmune disease open for debate. It is our aspiration that this For Debate article, together with the accompanying counter-debate by Lernmark [64] will stimulate researchers to re-think current dogmas and initiate experiments to confirm or refute a critical role of PTM in type 1 diabetes.
Abbreviations
- APC:
-
Antigen-presenting cell
- BB:
-
BioBreeding
- DC:
-
Dendritic cell
- GAD65:
-
GAD 65 kDa
- MLD:
-
Multiple low dose
- PTM:
-
Post-translational modification
- RIP:
-
Rat insulin promoter
- ROS:
-
Reactive oxygen species
- SNP:
-
Single nucleotide polymorphism
- STZ:
-
Streptozotocin
- TCR:
-
T cell receptor
- ZnT8:
-
Zinc transporter 8
References
Nerup J, Andersen OO, Bendixen G, Egeberg J, Poulsen JE (1971) Anti-pancreatic cellular hypersensitivity in diabetes mellitus. Diabetes 20:424–427
Nerup J, Platz P, Andersen OO et al (1974) HL-A antigens and diabetes mellitus. Lancet 2:864–866
Bottazzo GF, Florin-Christensen A, Doniach D (1974) Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 2:1279–1283
Eizirik DL, Colli ML, Ortis F (2009) The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol 5:219–226
Mathis D, Vence L, Benoist C (2001) β-Cell death during progression to diabetes. Nature 414:792–798
Nerup J, Mandrup-Poulsen T, Helqvist S et al (1994) On the pathogenesis of IDDM. Diabetologia 37(Suppl 2):S82–S89
Winter WE, Schatz DA (2011) Autoimmune markers in diabetes. Clin Chem 57:168–175
Mallone R, Brezar V, Boitard C (2011) T cell recognition of autoantigens in human type 1 diabetes: clinical perspectives. Clin Dev Immunol 2011:513210
Deniro M, Al-Mohanna FA (2012) Zinc transporter 8 (ZnT8) expression is reduced by ischemic insults: a potential therapeutic target to prevent ischemic retinopathy. PLoS One 7:e50360
Kim J, Richter W, Aanstoot HJ et al (1993) Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes 42:1799–1808
Murgia C, Devirgiliis C, Mancini E, Donadel G, Zalewski P, Perozzi G (2009) Diabetes-linked zinc transporter ZnT8 is a homodimeric protein expressed by distinct rodent endocrine cell types in the pancreas and other glands. Nutr Metab Cardiovasc Dis 19:431–439
Shimizu S, Saito N, Kubosaki A et al (2001) Developmental expression and localization of IA-2 mRNA in mouse neuroendocrine tissues. Biochem Biophys Res Commun 288:165–171
Atkinson MA, Bluestone JA, Eisenbarth GS et al (2011) How does type 1 diabetes develop? The notion of homicide or β-cell suicide revisited. Diabetes 60:1370–1379
Dunne JL, Overbergh L, Purcell AW, Mathieu C (2012) Posttranslational modifications of proteins in type 1 diabetes: the next step in finding the cure? Diabetes 61:1907–1914
Marrack P, Kappler JW (2012) Do MHCII-presented neoantigens drive type 1 diabetes and other autoimmune diseases? Cold Spring Harb Perspect Med 2:a007765
Kurien BT, Scofield RH (2008) Autoimmunity and oxidatively modified autoantigens. Autoimmun Rev 7:567–573
Wegner N, Lundberg K, Kinloch A et al (2010) Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev 233:34–54
Stoeckle C, Tolosa E (2010) Antigen processing and presentation in multiple sclerosis. Results Probl Cell Differ 51:149–172
Sollid LM, Jabri B (2011) Celiac disease and transglutaminase 2: a model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders. Curr Opin Immunol 23:732–738
Anderton SM (2004) Post-translational modifications of self antigens: implications for autoimmunity. Curr Opin Immunol 16:753–758
Doyle HA, Mamula MJ (2012) Autoantigenesis: the evolution of protein modifications in autoimmune disease. Curr Opin Immunol 24:112–118
Kyewski B, Klein L (2006) A central role for central tolerance. Annu Rev Immunol 24:571–606
Allen S, Turner SJ, Bourges D, Gleeson PA, van Driel IR (2011) Shaping the T cell repertoire in the periphery. Immunol Cell Biol 89:60–69
Walker LS, Abbas AK (2002) The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol 2:11–19
Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E (2003) Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol 171:538–541
Carbone FR, Gleeson PA (1997) Carbohydrates and antigen recognition by T cells. Glycobiology 7:725–730
Henrickson SE, Mempel TR, Mazo IB et al (2008) T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat Immunol 9:282–291
Calderon B, Unanue ER (2012) Antigen presentation events in autoimmune diabetes. Curr Opin Immunol 24:119–128
Coffey LC, Berman DM, Willman MA, Kenyon NS (2009) Immune cell populations in nonhuman primate islets. Cell Transplant 18:1213–1222
Charlton B, Bacelj A, Mandel TE (1988) Administration of silica particles or anti-Lyt2 antibody prevents beta-cell destruction in NOD mice given cyclophosphamide. Diabetes 37:930–935
Oschilewski M, Schwab E, Kiesel U et al (1986) Administration of silica or monoclonal antibody to Thy-1 prevents low-dose streptozotocin-induced diabetes in mice. Immunol Lett 12:289–294
Oschilewski U, Kiesel U, Kolb H (1985) Administration of silica prevents diabetes in BB-rats. Diabetes 34:197–199
D’Hertog W, Overbergh L, Lage K et al (2007) Proteomics analysis of cytokine-induced dysfunction and death in insulin-producing INS-1E cells: new insights into the pathways involved. Mol Cell Proteomics 6:2180–2199
Sparre T, Christensen UB, Mose LP et al (2002) IL-1β induced protein changes in diabetes prone BB rat islets of Langerhans identified by proteome analysis. Diabetologia 45:1550–1561
Sparre T, Christensen UB, Gotfredsen CF et al (2004) Changes in expression of IL-1β influenced proteins in transplanted islets during development of diabetes in diabetes-prone BB rats. Diabetologia 47:892–908
Trigwell SM, Radford PM, Page SR et al (2001) Islet glutamic acid decarboxylase modified by reactive oxygen species is recognized by antibodies from patients with type 1 diabetes mellitus. Clin Exp Immunol 126:242–249
Khan MW, Sherwani S, Khan WA, Ali R (2009) Characterization of hydroxyl radical modified GAD65: a potential autoantigen in type 1 diabetes. Autoimmunity 42:150–158
Acharya JD, Ghaskadbi SS (2010) Islets and their antioxidant defense. Islets 2:225–235
Corbett JA, McDaniel ML (1995) Intraislet release of interleukin 1 inhibits beta cell function by inducing beta cell expression of inducible nitric oxide synthase. J Exp Med 181:559–568
Strandell E, Buschard K, Saldeen J, Welsh N (1995) Interleukin-1β induces the expression of HSP70, heme oxygenase and Mn-SOD in FACS-purified rat islet β-cells, but not in α-cells. Immunol Lett 48:145–148
Nielsen K, Karlsen AE, Deckert M et al (1999) Beta-cell maturation leads to in vitro sensitivity to cytotoxins. Diabetes 48:2324–2332
Nielsen K, Kruhoffer M, Orntoft T et al (2004) Gene expression profiles during beta cell maturation and after IL-1β exposure reveal important roles of Pdx-1 and Nkx6.1 for IL-1β sensitivity. Diabetologia 47:2185–2199
Mannering SI, Harrison LC, Williamson NA et al (2005) The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med 202:1191–1197
Strollo R, Rizzo P, Spoletini M et al (2013) HLA-dependent autoantibodies against post-translationally modified collagen type II in type 1 diabetes mellitus. Diabetologia 56:563–572
Haase C, Skak K, Michelsen BK, Markholst H (2004) Local activation of dendritic cells leads to insulitis and development of insulin-dependent diabetes in transgenic mice expressing CD154 on the pancreatic beta-cells. Diabetes 53:2588–2595
Kiesel U, Kolb H, Freytag G (1981) Strain dependency of the transfer of experimental immune insulitis in mice. Clin Exp Immunol 43:430–433
Kim YT, Steinberg C (1984) Immunologic studies on the induction of diabetes in experimental animals. Cellular basis for the induction of diabetes by streptozotocin. Diabetes 33:771–777
Klinkhammer C, Popowa P, Gleichmann H (1988) Specific immunity to streptozocin. Cellular requirements for induction of lymphoproliferation. Diabetes 37:74–80
Zou XL, Zhao ZY, Wang YY, Su ZQ, Xiang M (2008) Diabetogenic T cells induce autoimmune diabetes in BALB/c mice. Chin Med Sci J 23:88–94
Weide LG, Lacy PE (1991) Low-dose streptozocin-induced autoimmune diabetes in islet transplantation model. Diabetes 40:1157–1162
Gillespie KM, Bain SC, Barnett AH et al (2004) The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet 364:1699–1700
Hermann R, Knip M, Veijola R et al (2003) Temporal changes in the frequencies of HLA genotypes in patients with type 1 diabetes—indication of an increased environmental pressure? Diabetologia 46:420–425
Sosinowski T, Eisenbarth GS (2013) Type 1 diabetes: primary antigen/peptide/register/trimolecular complex. Immunol Res 55:270–276
Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K (2012) Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes 61:3229–3246
Delong T, Baker RL, He J, Haskins K (2013) Novel autoantigens for diabetogenic CD4 T cells in autoimmune diabetes. Immunol Res 55:167–172
Hugues S, Mougneau E, Ferlin W et al (2002) Tolerance to islet antigens and prevention from diabetes induced by limited apoptosis of pancreatic β cells. Immunity 16:169–181
Xia CQ, Peng R, Qiu Y, Annamalai M, Gordon D, Clare-Salzler MJ (2007) Transfusion of apoptotic beta-cells induces immune tolerance to beta-cell antigens and prevents type 1 diabetes in NOD mice. Diabetes 56:2116–2123
Xia CQ, Qiu Y, Peng RH, Lo-Dauer J, Clare-Salzler MJ (2008) Infusion of UVB-treated splenic stromal cells induces suppression of beta cell antigen-specific T cell responses in NOD mice. J Autoimmun 30:283–292
Diana J, Simoni Y, Furio L et al (2013) Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med 19:65–73
Barrett JC, Clayton DG, Concannon P et al (2009) Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 41:703–707
Reissner KJ, Aswad DW (2003) Deamidation and isoaspartate formation in proteins: unwanted alterations or surreptitious signals? Cell Mol Life Sci 60:1281–1295
Wagner AM, Cloos P, Bergholdt R et al (2007) Post-translational protein modifications in type 1 diabetes: a role for the repair enzyme protein-l-isoaspartate (d-aspartate) O-methyltransferase? Diabetologia 50:676–681
Sud S, Marcon M, Assor E, Palmert MR, Daneman D, Mahmud FH (2010) Celiac disease and pediatric type 1 diabetes: diagnostic and treatment dilemmas. Int J Pediatr Endocrinol 2010:161285
Lernmark Å (2013) Is there evidence for post-translational modification of beta cell autoantigens in the aetiology and pathogenesis of type 1 diabetes? Diabetologia doi:10.1007/s00125-013-3041-7
Funding
This work was supported by funding from the Danish Research Council, European Foundation for the Study of Diabetes (joint EFSD/JDRF/Novo Nordisk grant), Novo Nordisk Foundation, Beckett Foundation and the Sehested-Hansen Foundation.
Duality of interest
JS, AJO, CAB, FP, CHB-B, JN, and F. Pociot declare no duality of interests. C.H. is employed by Novo Nordisk A/S and holds shares in Novo Nordisk A/S, which manufactures and markets drugs to treat diabetes.
Contribution statement
JS, JN and F. Pociot were responsible for the conception of the manuscript. All authors were responsible for the design of the manuscript, drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 14 kb)
ESM Table 1
(XLS 67 kb)
ESM Table 2
(PDF 20 kb)
Rights and permissions
About this article
Cite this article
Størling, J., Overgaard, A.J., Brorsson, C.A. et al. Do post-translational beta cell protein modifications trigger type 1 diabetes?. Diabetologia 56, 2347–2354 (2013). https://doi.org/10.1007/s00125-013-3045-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-3045-3