
Abstract
Aims/hypothesis
Glucagon-like peptide 1 (GLP-1) receptor (GLP-1R) agonists and dipeptidyl peptidase-4 (DPP-4) inhibitors attenuate postprandial lipaemia through mechanisms that remain unclear. As dyslipidaemia is a contributing risk factor for cardiovascular disease in type 2 diabetes, we examined the mechanisms linking pharmacological and physiological regulation of GLP-1 action to control of postprandial lipid metabolism.
Methods
Postprandial lipid synthesis and secretion were assessed in normal and fructose-fed hamsters and in wild-type mice that were treated with or without sitagliptin. Apolipoprotein B-48 (ApoB-48) synthesis and secretion were also examined in primary enterocyte cultures. The importance of exogenous vs endogenous GLP-1R signalling for regulation of intestinal lipoprotein synthesis and secretion was assessed in mice and hamsters treated with the GLP-1R agonist exendin-4, the GLP-1R antagonist exendin(9-39) and in Glp1r +/+ vs Glp1r −/− mice.
Results
Sitagliptin decreased fasting plasma triacylglycerol, predominantly in the VLDL fraction, as well as postprandial triacylglycerol-rich lipoprotein (TRL)-triacylglycerol, TRL-cholesterol and TRL-ApoB-48 in hamsters and mice. GLP-1R activation with exendin-4 alone also decreased plasma and TRL-ApoB-48 in hamsters and mice, and reduced secretion of ApoB-48 in hamster enterocyte cultures. Conversely, blockade of endogenous GLP-1R signalling by the antagonist exendin(9-39) or genetic elimination of GLP-1R signalling in Glp1r −/− mice enhanced TRL-ApoB-48 secretion in vivo. Co-administration of exendin(9-39) also abolished the hypolipidaemic effect of sitagliptin.
Conclusions/interpretation
Potentiation of endogenous incretin action via DPP-4 inhibition or pharmacological augmentation of GLP-1R signalling reduces intestinal secretion of triacylglycerol, cholesterol and ApoB-48. Moreover, endogenous GLP-1R signalling is essential for the control of intestinal lipoprotein biosynthesis and secretion.
Similar content being viewed by others

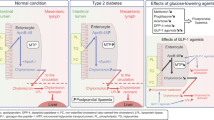

Introduction
The growing incidence of type 2 diabetes is a major problem [1] and may be associated with a variety of lipid abnormalities that pose cardiovascular disease risk factors, including hypertriacylglycerolaemia, increased levels of small dense LDL and low levels of HDL [2]. At the top of this cascade are intestinally derived chylomicrons containing triacylglycerol-rich apolipoprotein B-48 (ApoB-48), which are secreted following fat ingestion and peripherally catabolised to generate remnant particles [3]. Insulin resistance is positively correlated with remnant-like particle cholesterol [4] and remnant-like particle triacylglycerol is greatly increased in some diabetic populations [5]. Thus, it is of interest to identify therapeutic strategies that can limit postprandial triacylglycerol-rich lipoprotein (TRL) secretion.
The ingestion of nutrients, including fat, provokes the secretion of gut-derived hormones, including two incretins: gastric inhibitory peptide (GIP) from duodenal K cells [6] and glucagon-like peptide-1 (GLP-1) from ileal enteroendocrine L cells [7]. These peptides stimulate insulin secretion in a glucose-dependent manner and preserve pancreatic beta cell function and mass in preclinical studies, as reviewed [8]. A number of extra-pancreatic effects have also been demonstrated following exogenous administration of these two hormones. GLP-1 slows gastric emptying and induces anorectic effects [9], while GIP has anabolic effects on adipose tissue [10] and regulates adipokine secretion [11]. However, GLP-1 and GIP are rapidly inactivated by dipeptidyl peptidase-4 (DPP-4)-mediated cleavage, thus limiting their duration of action. In type 2 diabetes, the incretin response is blunted, due largely to reduced incretin action [12]. Therefore, there is considerable interest in enhancing incretin action for the treatment of type 2 diabetes mellitus.
DPP-4 inhibitors protect endogenous GLP-1 and GIP from N-terminal degradation thereby prolonging their bioactivity. DPP-4 inhibition lowers fasting and postprandial glycaemia in patients with type 2 diabetes mellitus [13, 14]; hence there is considerable interest in understanding the effects of incretins and DPP-4 inhibitors on plasma lipid profiles [15]. Acute administration of GIP reduces circulating chylomicrons, probably by promoting triacylglycerol catabolism by adipose tissue [16, 17]; GLP-1 also attenuates postprandial triacylglycerol secretion [18, 19], although the exact underlying mechanisms are not clear.
In the present study, we used wild-type mice and a hamster model of postprandial dyslipidaemia [20] to study the role of incretin action in controlling intestinal lipid and lipoprotein metabolism. The fructose-fed hamster exhibits intestinal overproduction of TRL [21], along with aberrant insulin signalling in the absorptive enterocyte [22]. Mice and hamsters were treated with sitagliptin or exendin-4 to potentiate incretin action with or without exendin(9-39) to assess the importance of exogenous GLP-1 receptor (GLP-1R) activation vs endogenous basal GLP-1R signalling for regulation of intestinal lipoprotein metabolism.
Methods
Animals
Male Syrian golden hamsters (Mesocricetus auratus) weighing 130 to 150 g were purchased from Charles River (Montreal, QC, Canada) and housed individually with room lighting set for a 12 h light–dark cycle and free access provided to food and water. Animals were acclimatised for 1 week prior to being given either a standard chow diet or a fructose-enriched pelleted hamster diet containing 60% fructose and 20% casein (Dyets, Bethlehem, PA, USA) for 10 days to induce insulin resistance [23]. Hamsters were randomised to receive either sitagliptin phosphate monohydrate (Merck, Whitehouse Station, NJ, USA) (5 mg/kg) or water by oral gavage, each given daily. Chow-fed hamsters were given sitagliptin for 2 weeks while fructose-fed hamsters received sitagliptin for 3 weeks. Blood was collected in the morning for ambient (fed) measurements and after a 6 h fast by retro-orbital bleeding. The hamsters were then killed for the ex vivo protocol or underwent the in vivo protocol. All animal protocols were approved by the Animal Ethics Committee of the Hospital for Sick Children, University of Toronto. Wild-type C57BL/6 J mice (Jackson Laboratory, Bar Harbor, ME, USA), Glp1r −/− and age and sex-matched Glp1r +/+ littermate controls fed normal chow were studied at 10 to 12 weeks of age and maintained under a 12 h light–dark cycle. Treatments in mice used a single dose of sitagliptin (10 mg/kg body weight) or exendin-4 (24 nmol/kg) (administration, see figure legends).
Assessment of intestinal lipoprotein production by in vivo Triton-WR1339 infusion
Hamsters were anaesthetised with isoflurane administered through a vaporiser. A cannula was inserted into the right jugular vein, exteriorised at the back of the neck, filled with heparinised saline (40 IU/ml) and sealed. The hamsters were allowed to recover and after a 12 h fast, conscious hamsters were given a 200 μl olive oil load via oral gavage, followed 20 min later by vehicle (saline), exendin-4 (5 nmol/kg) (Bachem, Torrance, CA, USA), exendin(9-39) (50 nmol/kg) (Bachem) or both, administered via intraperitoneal injection, and by a separate bolus of Triton-WR1339 that was diluted 20% (wt/vol.) (0.5 g/kg) and injected into the jugular vein to inhibit lipoprotein catabolism and uptake [24, 25]. Blood (300 μl) was collected from the jugular cannula into lithium heparin-coated tubes (BD Biosciences, Franklin Lakes, NJ, USA) at baseline and at 30, 60 and 120 min. The oral gavage, peptide administration, Triton infusion and blood collection were all performed on conscious animals in the absence of anaesthetics. The jejunum was excised under isoflurane anaesthesia.
Mice fasted for 5 or 16 h were given 200 μl olive oil orally. At 20 min after gavage, a blood sample was collected via the tail vein (time 0). Acute sitagliptin administration was given orally to conscious mice 20 min prior to olive oil gavage. At 20 min after fat load, Triton WR-1339 (0.5 g/kg body weight of a 15% [wt/vol.] solution prepared in PBS) was injected via the tail vein with or without exendin-4 (24 nmol/kg). Blood samples were collected via tail bleed without anaesthetics for plasma triacylglycerol and cholesterol assay, and ApoB-48 immunoblotting. At 90 min after Triton WR-1339 injection, mice were killed and cardiac puncture was performed for plasma triacylglycerol and cholesterol assay, ApoB-48 western blot analysis and TRL purification as described below.
Isolation of triacylglycerol-rich lipoproteins
To isolate the TRL fraction of the plasma, blood samples were first centrifuged for 15 min at 4°C and 2,300 g to separate the plasma layer. Plasma (150 μl) was overlayed with 4 ml potassium bromide solution (density 1.006 g/ml) in a 5 ml ultracentrifuge tube and centrifuged for 70 min at 116,140 g and at 15°C using a rotor (SW 55 Ti; Beckman Coulter, Mississauga, ON, Canada). The TRL fraction (Svedberg flotation rate [Sf] > 400) was collected as the top 300 μl of the tube content.
Chemiluminescent immunoblotting
ApoB-48 immunoblotting was performed on TRL fractions and diluted plasma (1:200) by SDS-PAGE analysis as previously described [23]. Membranes were then incubated in electrochemiluminescence detection reagents (GE Healthcare, Piscataway, NJ, USA) and exposed to Hyperfilm (Denville Scientific, Metuchen, NJ, USA). Quantitative analysis was performed using an AlphaImager imaging densitometer (Alpha Innotech, San Leandro, CA, USA).
Ex vivo metabolic labelling of intact primary enterocytes
Primary enterocytes were isolated from hamster intestinal tissue as described [21]. Resuspended villi were incubated with 8 mmol/l glucose and 100 pmol/l exendin-4. Hamster enterocytes were preincubated in methionine-free MEM at 37°C for 45 min and pulsed with 1.11 to 1.85 MBq/ml of [35S]methionine for 30 min. The cells were then chased with unlabelled 40 mmol/l methionine-enriched DMEM.
Immunoprecipitation, SDS-PAGE and fluorography
ApoB-48 immunoprecipitation and SDS-PAGE were performed as described previously [20]. The ApoB-48 bands were excised and quantitative analysis was performed using a liquid scintillation counter. No major differences were noticed in the trichloroacetic acid protein precipitation counts between the control and experimental groups. Media counts were normalised to initial cellular trichloroacetic acid counts. Radioactivity incorporation into ApoB-48 was also visualised with the Storm 840 Phosphor Imager (Molecular Dynamics, Sunnyvale, CA, USA).
Plasma measurements
Plasma triacylglycerol and cholesterol were determined by an enzymatic-based colorimetric assay (Randox, Crumlin, UK).
Fast protein liquid chromatography of plasma lipoproteins
Hamster plasma (200 μl) was filtered through a 0.45 μmol/l micro-spin polysulphone filter (Alltech; Mandel Scientific, Lachine, QC, Canada) to remove macroparticles. Plasma was then subjected to gel filtration through a HR 10/300 GL Superose 6 column (Pharmacia, Uppsala, Sweden) with a solution of 10 mmol/l Tris, 150 mmol/l NaCl, 2 mmol/l CaCl2, 100 μmol/l DTPA and 0.02% NaN3 (wt/vol.), pH 7.4 pumped at a flow rate of 0.5 ml/min.
Statistical analysis
All results are presented as mean±SEM. Statistical comparisons were performed using Student’s t test when comparing two groups and two-way ANOVA with the Bonferroni post hoc test as indicated in the text and figure legends.
Results
A DPP-4 inhibitor attenuates dyslipidaemia in fructose-fed hamsters
As increased incretin action can attenuate postprandial plasma lipid accumulation [18, 19, 26], we examined the consequences of DPP-4 inhibition for accumulation of triacylglycerol and cholesterol in hamsters fed a high-fructose diet (60%), which promotes dyslipidaemia and mild insulin resistance [23]. A high-fructose diet for 10 days prior to start of sitagliptin treatment produced a significant increase in plasma triacylglycerol and cholesterol (p = 0.0002 and p = 0.006, respectively) (Fig. 1b, c), without changes in blood glucose or body weight (Table 1) and no differences in postprandial glucose excursions (data not shown). After 3 weeks of sitagliptin treatment, the fructose-induced increase of plasma triacylglycerol, but not that of plasma cholesterol was significantly attenuated (p < 0.05) (Fig. 1b, c).

Changes in plasma lipids following chronic administration of sitagliptin. a Blood samples were drawn from hamsters fed either regular chow or a high-fructose diet supplemented with vehicle or sitagliptin for 2 to 3 weeks. b Plasma triacylglycerol and cholesterol (c) levels were determined prior to and after each dosing period. White bars, chow vehicle; hatched bars, chow sitagliptin; black bars, fructose vehicle; crossed bars, fructose sitagliptin. d Lipoproteins were separated from plasma of hamsters fed high-fructose diet supplemented with vehicle or sitagliptin as shown. Separation was by FPLC fractionation into VLDL/chylomicron remnants, LDL and HDL. e Levels of lipoprotein triacylglycerol and cholesterol (f) were determined. Black bars, vehicle; crossed bars, sitagliptin. n = 9–16 for plasma lipid variables analysed by paired t test (b, c); n = 4 for FPLC variables analysed by Student’s t test (e, f); *p < 0.05 fructose-fed sitagliptin vs fructose-fed vehicle; ***p < 0.001 fructose-fed vehicle vs chow-fed vehicle or fructose-fed sitagliptin vs chow-fed sitagliptin
Fast protein liquid chromatography (FPLC) was performed to separate the various densities of lipid particles. In fructose-fed hamsters, levels of VLDL- and LDL-triacylglycerol and -cholesterol were significantly higher than in control animals (data not shown). Sitagliptin significantly reduced levels of VLDL-triacylglycerol by threefold (Fig. 1e). LDL- and HDL-triacylglycerol levels were slightly but not significantly decreased by sitagliptin treatment (Fig. 1e). Whereas total plasma cholesterol was not altered by sitagliptin, LDL-cholesterol was reduced and a significant decrease in VLDL- and HDL-cholesterol (Fig. 1f, d) was observed in sitagliptin-treated vs control animals (p < 0.05).
A DPP-4 inhibitor decreases intestinal production of TRL-triacylglycerol and TRL-cholesterol
To understand how sitagliptin modulates plasma lipoprotein levels, we assessed intestinal lipoprotein production and secretion after treatment of chow fed hamsters with sitagliptin for 2 weeks. Sitagliptin slightly reduced TRL cholesterol (Fig. 2b) and ApoB-48 (Fig. 2d) in TRL fraction after an acute fat load, while the accumulation of TRL-triacylglycerol was unaffected (Fig. 2c). In contrast, sitagliptin significantly reduced the amount of triacylglycerol in TRL fraction 90 min after the fat load (Fig. 2g) in fructose-fed hamsters and lowered TRL-cholesterol (Fig. 2f) and ApoB-48 (Fig. 2h) compared with vehicle-treated high-fructose fed animals. Slope calculations for TRL-cholesterol and -triacylglycerol secretion in fructose-fed hamsters (cholesterol: 7.25 ± 1.55 × 10−3 vs 1.11 ± 0.27 × 10−2 mmol l−1 min−1, sitagliptin vs control; triacylglycerol 0.0175 ± 0.0034 vs 0.0252 ± 0.0037 mmol l−1 min−1, sitagliptin vs control) suggested that sitagliptin retards intestinal postprandial cholesterol and triacylglycerol secretion.

Changes in TRL lipid mass following chronic sitagliptin administration. a Hamsters were fed regular chow (b–d) or (e) a high-fructose diet (f–h) and administered either vehicle or sitagliptin for 2 to 3 weeks. Hamsters were then fat-loaded, administered Triton WR-1339 and blood was drawn at 0, 30, 60, 90 and 120 min (a, e). Plasma was spun to isolate TRL fraction and levels of cholesterol, triacylglycerol and ApoB-48 were determined. Graphs show TRL levels of (b) cholesterol, (c) triacylglycerol and (d) ApoB-48 in chow-fed hamsters given vehicle (white circles) or sitagliptin (black circles). f Cholesterol, (g) triacylglycerol and (h) ApoB-48 content of TRL fractions from fructose-fed hamsters given vehicle (white squares) or sitagliptin (black squares). n = 4 for each group; *p < 0.05 as analysed by two-way ANOVA with the Bonferroni post hoc test
To determine whether the effect of sitagliptin on plasma levels of cholesterol and triacylglycerol was restricted to hamsters, chylomicron production was assessed in chow-fed mice. Sitagliptin significantly decreased cholesterol in plasma (Fig. 3b) and in the TRL-enriched fraction (Table 2) after an acute fat load; furthermore, plasma triacylglycerol (Fig. 3c) and TRL-triacylglycerol (Table 2) were significantly reduced 90 min after triton injection in sitagliptin-treated mice. The accumulation of ApoB-48 in plasma was also decreased by sitagliptin (Fig. 3d).

Changes in postprandial lipid secretion following a single acute administration of sitagliptin in mice. a Wild-type mice fasted for 5 h were given 10 mg/kg of sitagliptin (black circles) prepared in water by oral gavage or water only (white circles). At 20 min after sitagliptin treatment, mice were administered 200 μl olive oil and injected intravenously 20 min later with Triton WR1339 (0.5 g/kg ). Blood samples were collected prior to i.v. injection (time 0), as well as at 30, 60 and 90 min after injection for measurement of plasma (b) cholesterol, (c) triacylglycerol and (d) ApoB-48 as described. n = 4 per group; *p < 0.05, ***p < 0.001 as determined by two-way ANOVA with the Bonferroni post hoc test
Pharmacological activation of GLP-1R mimics the effects of sitagliptin on intestinal lipid absorption
As GLP-1 and GIP regulate plasma lipid levels and are DPP-4 substrates, we measured intestinal lipid absorption in mice after acute administration of the DPP-4-resistant peptides, D-Ala2-GIP and exendin-4. As shown in Fig. 4, D-Ala2-GIP significantly increased levels of plasma triacylglycerol and ApoB-48 after an acute fat load (Fig. 4b, c). GIP, therefore, could not have been responsible for the actions of sitagliptin on intestinal lipid absorption. In contrast, the GLP-1R agonist exendin-4 reduced plasma and TRL-triacylglycerol and ApoB-48 accumulation (Fig. 4e, f, Table 2), but no effect was noted on plasma and TRL-cholesterol (Fig. 4c, Table 2). Hence, activation of GLP-1R but not GIP receptor signalling mimics the actions of sitagliptin on intestinal lipid absorption.

Pharmacological activation of GLP-1R mimics the effect of sitagliptin on postprandial lipid excursion. Wild-type mice fasted for 5 h were gavaged with olive oil at −20 (a–f) or −60 min (g–i) and injected (at 0 min) with triton WR-1339 (0.5 g/kg body weight) and with (b, c) D-Ala2-GIP (24 nmol/kg, small black squares) or (d–f) exendin-4 (24 nmol/kg, large black squares). White symbols, control. Blood samples were collected prior to i.v. injection (time 0), as well as at 30, 60 and 90 min after injection. (b, e, h) Triacylglycerol, (d) cholesterol and (c, f, i) ApoB-48 secretion were measured. n = 3–6 per group; *p < 0.05 as determined by two-way ANOVA with the Bonferroni post hoc test
Insulin and glucagon are involved in the regulation of lipaemia and activation of GLP-1R signalling has been shown to stimulate insulin secretion and inhibit glucagon secretion. Accordingly, we measured plasma insulin and glucagon levels after an oral fat load, with or without sitagliptin or exendin-4 treatment. Both sitagliptin and exendin-4 significantly increased plasma insulin levels compared with controls (Electronic supplementary material [ESM] Fig. 1a). However, this transient difference in plasma insulin levels is unlikely to account for the effect of sitagliptin or exendin-4 on intestinal lipid absorption or for the difference in magnitude of the effect of those two treatments, as plasma insulin levels were no longer significantly different between the groups 20 min after the fat load (ESM Fig. 1a). More importantly, exogenous insulin administration had no effect on triacylglycerol accumulation in mice (ESM Fig. 1b). Furthermore, acute administration of sitagliptin or exendin-4 to fasted mice in association with a subsequent oral fat load had no significant effect on plasma glucagon levels (ESM Fig. 1c).
As exendin-4 is a rapid and potent inhibitor of gastric emptying, which in turn might contribute to decreased intestinal lipid absorption, we monitored intestinal lipid absorption when exendin-4 was administered 1 h after the oral fat load to facilitate entry of olive oil into the mouse small bowel. Under these conditions, exendin-4 still significantly decreased triacylglycerol and ApoB-48 accumulation in plasma (Fig. 4h, i) and TRL fraction (Table 2).
Endogenous GLP-1R signalling is required for control of postprandial lipaemia
We next investigated the role of GLP-1 in the regulation of intestinal lipid absorption in mice and hamsters after an acute fat load. Exendin-4 decreased triacylglycerol and cholesterol in VLDL/chylomicron remnant-sized lipoproteins in chow-fed hamsters not given a bolus of Triton WR-1339 (Fig. 5b) and significantly reduced levels of ApoB-48 in the TRL fraction 90 min after a fat load (Fig. 5c). The GLP-1R antagonist, exendin(9-39), blocked the exendin-4-mediated reduction of TRL-ApoB-48 and, administered alone, augmented levels of TRL-ApoB-48 120 min after fat load (Fig. 5c), indicating that endogenous basal GLP-1R signalling modulates postprandial lipaemia.

Role of GLP-1 in postprandial lipaemia and sitagliptin action. a–c Acute effect of the GLP-1R agonist exendin-4 and antagonist exendin(9-39) on ApoB-48 metabolism in chow-fed hamsters. (b) Representative FPLC profile of plasma from chow-fed hamsters injected intraperitoneally with exendin-4 without intravenous Triton WR-1339. (c) Hamsters were challenged with a fat load prior to Triton WR-1339 administration. TRL ApoB-48 mass was measured by immunoblotting. Data are normalised to baseline values for each treatment. n = 3 per group; **p < 0.01 for exendin(9-39) or exendin(9-39) + exendin-4 vs control; ***p < 0.001 exendin-4 vs control as determined by two-way ANOVA with the Bonferroni post hoc test. d–f Glp1r −/− mice (black squares) or littermate control mice (white diamonds) fasted for 16 h were given orally 200 μl of olive oil. At 20 min after gavage, mice were injected i.v. with Triton WR1339 (0.5 g/kg). Blood samples were collected prior to i.v. injection (time 0), as well as at 30, 60 and 90 min after injection. Graphs show triacylglycerol (e) and ApoB-48 (f) measurement as described. n = 6 per group; *p < 0.05 as determined by two-way ANOVA with the Bonferroni post hoc test. g–i Mice were fasted for 5 h and administered sitagliptin (10 mg/kg; black circles) or vehicle (white circles) by oral gavage. GLP-1R signalling was blocked by co-administering the GLP-1R antagonist exendin9-39 (black diamonds) or PBS (white squares) by intraperitoneal injection. After 20 min (−20 min), mice were challenged with an oral fat load and given Triton WR-1339 by intravenous injection at 0 min. (h) Cholesterol and (i) triacylglycerol secretion was measured as described. n = 5–6 per group; *p < 0.05, **p < 0.01 sitagliptin vs control or sitagliptin + exendin(9-39) as determined by two-way ANOVA with the Bonferroni post hoc test
Consistent with data obtained using the GLP1-R antagonist exendin(9-39) in hamsters, triacylglycerol accumulation in plasma (Fig. 5e) and TRL fraction (Table 2) was significantly enhanced in Glp1r −/− mice compared with Glp1r +/+ littermate controls, despite the fact that Glp1r −/− mice have a gastric emptying rate similar to Glp1r +/+ littermate controls [27]. There was also significantly greater TRL ApoB-48 mass in GLP-1R-deficient mice (Table 2). Therefore, the modulatory actions of basal GLP-1R signalling on intestinal lipid absorption are not due to regulation of gastric emptying and implicate the endogenous GLP-1R in the control of intestinal lipid absorption.
Intact GLP-1R signalling is required for the hypolipidaemic action of sitagliptin
To determine whether the actions of sitagliptin on postprandial lipid secretion required GLP-1R signalling, the GLP-1R antagonist exendin(9-39) was co-administered with sitagliptin 20 min before the fat load. Sitagliptin attenuated postprandial triacylglycerol excursions in mice (Fig. 5i), but did not significantly affect plasma cholesterol levels (Fig. 5h). Furthermore, the GLP-1R antagonist exendin(9-39) eliminated the hypolipidaemic actions of sitagliptin on plasma triacylglycerol levels (Fig. 5i).
Exendin-4 directly reduces enterocyte ApoB-48 secretion
We next evaluated the effect of exendin-4 treatment on enterocytes isolated from chow-fed hamsters. Pulse chase studies in primary enterocytes showed that cellular ApoB-48 was not changed in exendin-4-treated hamsters (Fig. 6a). In contrast, exendin-4 decreased the amount of secreted ApoB-48 from enterocytes of chow-fed hamsters (Fig. 6b) (p < 0.01 at 60 min, p < 0.001 at 90 min). Hence GLP-1R activation directly regulates enterocyte ApoB-48 secretion ex vivo.

Direct effects of exendin-4 on intestinal ApoB-48 metabolism ex vivo. Enterocytes from chow-fed hamsters were analysed. (a) Cellular and secreted (b) levels of ApoB-48 were determined in primary enterocytes metabolically labelled with [35S]methionine in medium containing 100 pmol/l exendin-4. Direct treatment with exendin-4 significantly decreased secretion in newly synthesised ApoB-48. White circles, control; black circles, exendin-4. n = 3 per group; **p < 0.01, ***p < 0.001 as determined by two-way ANOVA with the Bonferroni post hoc test
Discussion
Dipeptidyl peptidase-4 inhibitors exert their actions in part via augmentation of GLP-1 action, which leads to reduction of glucagon, increases in insulin and reduced glycaemia in human participants [28, 29]. Moreover, GLP-1 administration reduced postprandial circulating lipid levels in human participants [18], although the underlying mechanisms remain uncertain. In the current study, we found that sitagliptin decreased triacylglycerol accumulation, specifically in the VLDL fraction, in hamsters and mice. Sitagliptin treatment also decreased ApoB-48 accumulation in plasma, specifically in the intestinally produced TRL fraction. Treatment with DPP-4 inhibitors can significantly decrease postprandial triacylglycerol, cholesterol and ApoB-48 levels in humans [30, 31]; however, DPP-4 inhibition has minimal effects on fasting lipid levels [30]. Our data indicate that a prominent aspect of sitagliptin’s effects on plasma triacylglycerol levels is the decrease in levels of VLDL. This suggests that the major effect of augmenting GLP-1 action is the reduction in number and/or size of large TRL particles.
Our observations demonstrate that sitagliptin and exendin-4 exert qualitatively similar effects on postprandial lipid profiles, consistent with the notion that increased GLP-1 action is likely to be responsible for the reduced circulating lipid levels and reduced intestinally derived TRL observed following sitagliptin administration. Nevertheless, exendin-4 more potently reduced postprandial triacylglycerol synthesis and secretion compared with sitagliptin, consistent with the relatively greater GLP-1R activation achieved with exendin-4 relative to sitagliptin. Moreover, inhibition of DPP-4 activity is also associated with potentiation of GIP action, which might be expected to partially attenuate the hypolipidaemic effects mediated through the GLP-1R. Although increased levels of insulin may also reduce plasma triacylglycerol levels and ApoB-48 secretion from the intestine [32], administration of exogenous insulin did not affect intestinal lipid absorption (ESM Fig. 1b), ruling out insulin as a mediator of sitagliptin’s or exendin-4’s effects on postprandial lipaemia.
We also present data clarifying the putative role(s) of GIP or glucagon in the actions of sitagliptin on intestinal lipid secretion. Although glucagon has been reported to inhibit lipid secretion from hepatocytes [33], circulating levels of glucagon were not reduced following sitagliptin administration in our current experiments. Furthermore D-Ala2-GIP did not reduce, but actually promoted postprandial triacylglycerol and ApoB-48 secretion. These experiments, taken together with data using exendin(9-39) and Glp1r −/− mice implicate GLP-1 as the predominant mediator of sitagliptin action on intestinal ApoB-48 secretion.
Our data also demonstrate overlapping effects of sitagliptin and exendin-4 treatment on plasma and TRL lipoprotein levels. Sitagliptin decreased cholesterol, triacylglycerol and ApoB-48 levels in the TRL fraction of fructose-fed hamsters. Similarly, exendin-4 decreased ApoB-48 secretion in chow-fed hamsters and in freshly isolated primary enterocyte cultures. These observations suggest for the first time that GLP-1 directly regulates lipoprotein assembly and/or secretory machinery in the enterocyte. However, additional work is required to understand the molecular mechanism underlying the suppressive effect of GLP-1 on ApoB-48-TRL production.
An important aspect of our studies is the demonstration that reduction or elimination of GLP-1R signalling results in detectable changes in postprandial lipoprotein profiles. Specifically, administration of exendin(9-39) alone resulted in increased levels of TRL ApoB-48 in hamsters, whereas levels of ApoB-48 and triacylglycerol mass were increased in Glp1r −/− compared with Glp1r +/+ mice. Hence, these findings establish an essential role for basal levels of GLP-1R signalling in the control of intestinal lipoprotein synthesis/secretion in vivo.
In summary, our data support an important role for the GLP-1R signalling system in regulating intestinal lipid and lipoprotein metabolism. Augmentation of GLP-1R signalling lowers postprandial circulating levels of triacylglycerol, cholesterol-rich TRL and chylomicrons containing ApoB-48. The current study suggests that enhanced GLP-1 action, achieved via DPP-4 inhibition or use of GLP-1R agonists, may contribute to control of postprandial lipid excursion through control of intestinal lipoprotein synthesis and secretion.
Abbreviations
- ApoB-48:
-
Apolipoprotein B-48
- DPP4:
-
Dipeptidyl peptidase-4
- FPLC:
-
Fast protein liquid chromatography
- GIP:
-
Gastric inhibitory peptide
- GLP-1:
-
Glucagon-like peptide 1
- GLP-1R:
-
GLP-1 receptor
- TRL:
-
Triacylglycerol-rich lipoprotein
References
Zimmet P, Alberti KG, Shaw J (2001) Global and societal implications of the diabetes epidemic. Nature 414:782–787
Ginsberg HN (2000) Insulin resistance and cardiovascular disease. J Clin Invest 106:453–458
Mero N, Syvanne M, Taskinen MR (1998) Postprandial lipid metabolism in diabetes. Atherosclerosis 141(Suppl 1):S53–S55
Ohnishi H, Saitoh S, Takagi S et al (2002) Relationship between insulin-resistance and remnant-like particle cholesterol. Atherosclerosis 164:167–170
Schaefer EJ, McNamara JR, Shah PK et al (2002) Elevated remnant-like particle cholesterol and triglyceride levels in diabetic men and women in the Framingham Offspring Study. Diabetes Care 25:989–994
Lu WJ, Yang Q, Sun W et al (2008) Using the lymph fistula rat model to study the potentiation of GIP secretion by the ingestion of fat and glucose. Am J Physiol Gastrointest Liver Physiol 294:G1130–G1138
Iakoubov R, Izzo A, Yeung A, Whiteside CI, Brubaker PL (2007) Protein kinase Czeta is required for oleic acid-induced secretion of glucagon-like peptide-1 by intestinal endocrine L cells. Endocrinology 148:1089–1098
Drucker DJ (2006) The biology of incretin hormones. Cell Metab 3:153–165
Baggio LL, Huang Q, Brown TJ, Drucker DJ (2004) Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology 127:546–558
Miyawaki K, Yamada Y, Ban N et al (2002) Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 8:738–742
Hansotia T, Maida A, Flock G et al (2007) Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest 117:143–152
Meier JJ, Nauck MA (2006) Incretins and the development of type 2 diabetes. Curr Diab Rep 6:194–201
Aschner P, Kipnes MS, Lunceford JK et al (2006) Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 29:2632–2637
Raz I, Hanefeld M, Xu L et al (2006) Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 49:2564–2571
Amori RE, Lau J, Pittas AG (2007) Efficacy and safety of incretin therapy in type 2 diabetes: systematic review and meta-analysis. JAMA 298:194–206
Wasada T, McCorkle K, Harris V et al (1981) Effect of gastric inhibitory polypeptide on plasma levels of chylomicron triglycerides in dogs. J Clin Invest 68:1106–1107
Kim SJ, Nian C, McIntosh CH (2007) Activation of lipoprotein lipase by glucose-dependent insulinotropic polypeptide in adipocytes. A role for a protein kinase B, LKB1, and AMP-activated protein kinase cascade. J Biol Chem 282:8557–8567
Meier JJ, Gethmann A, Gotze O et al (2006) Glucagon-like peptide 1 abolishes the postprandial rise in triglyceride concentrations and lowers levels of non-esterified fatty acids in humans. Diabetologia 49:452–458
Qin X, Shen H, Liu M et al (2005) GLP-1 reduces intestinal lymph flow, triglyceride absorption, and apolipoprotein production in rats. Am J Physiol Gastrointest Liver Physiol 288:G943–G949
Taghibiglou C, Rashid-Kolvear F, Van Iderstine SC et al (2002) Hepatic very low density lipoprotein-ApoB overproduction is associated with attenuated hepatic insulin signaling and overexpression of protein-tyrosine phosphatase 1B in a fructose-fed hamster model of insulin resistance. J Biol Chem 277:793–803
Haidari M, Leung N, Mahbub F et al (2002) Fasting and postprandial overproduction of intestinally derived lipoproteins in an animal model of insulin resistance. Evidence that chronic fructose feeding in the hamster is accompanied by enhanced intestinal de novo lipogenesis and ApoB48-containing lipoprotein overproduction. J Biol Chem 277:31646–31655
Federico LM, Naples M, Taylor D, Adeli K (2006) Intestinal insulin resistance and aberrant production of apolipoprotein B48 lipoproteins in an animal model of insulin resistance and metabolic dyslipidemia: evidence for activation of protein tyrosine phosphatase-1B, extracellular signal-related kinase, and sterol regulatory element-binding protein-1c in the fructose-fed hamster intestine. Diabetes 55:1316–1326
Taghibiglou C, Carpentier A, Van Iderstine SC et al (2000) Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J Biol Chem 275:8416–8425
Taggart C, Gibney J, Owens D et al (1997) The role of dietary cholesterol in the regulation of postprandial apolipoprotein B48 levels in diabetes. Diabet Med 14:1051–1058
Zoltowska M, Ziv E, Delvin E et al (2003) Cellular aspects of intestinal lipoprotein assembly in Psammomys obesus: a model of insulin resistance and type 2 diabetes. Diabetes 52:2539–2545
Ebert R, Nauck M, Creutzfeldt W (1991) Effect of exogenous or endogenous gastric inhibitory polypeptide (GIP) on plasma triglyceride responses in rats. Horm Metab Res 23:517–521
Baggio LL, Huang Q, Brown TJ, Drucker DJ (2004) A recombinant human glucagon-like peptide (GLP)-1-albumin protein (albugon) mimics peptidergic activation of GLP-1 receptor-dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis. Diabetes 53:2492–2500
Bergman AJ, Stevens C, Zhou Y et al (2006) Pharmacokinetic and pharmacodynamic properties of multiple oral doses of sitagliptin, a dipeptidyl peptidase-IV inhibitor: a double-blind, randomized, placebo-controlled study in healthy male volunteers. Clin Ther 28:55–72
Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP (2007) Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 9:194–205
Matikainen N, Manttari S, Schweizer A et al (2006) Vildagliptin therapy reduces postprandial intestinal triglyceride-rich lipoprotein particles in patients with type 2 diabetes. Diabetologia 49:2049–2057
Boschmann M, Engeli S, Dobberstein K et al (2009) Dipeptidyl-peptidase-IV inhibition augments postprandial lipid mobilization and oxidation in type 2 diabetic patients. J Clin Endocrinol Metab 94:846–852
Levy E, Sinnett D, Thibault L et al (1996) Insulin modulation of newly synthesized apolipoproteins B-100 and B-48 in human fetal intestine: gene expression and mRNA editing are not involved. FEBS Lett 393:253–258
Longuet C, Sinclair EM, Maida A et al (2008) The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab 8:359–371
Acknowledgements
This work was supported in part by CIHR operating grants MOP-53093 (to K. Adeli) and MOP-82700 (to D. J. Drucker), and by the Canada Research Chairs Program (D. J. Drucker).
Duality of interest
D. J. Drucker has served as a consultant to Merck Inc. The remaining authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Hsieh and C. Longuet are joint first authors; D. J. Drucker and K. Adeli are joint senior co-authors.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 41 kb)
Rights and permissions
About this article
Cite this article
Hsieh, J., Longuet, C., Baker, C.L. et al. The glucagon-like peptide 1 receptor is essential for postprandial lipoprotein synthesis and secretion in hamsters and mice. Diabetologia 53, 552–561 (2010). https://doi.org/10.1007/s00125-009-1611-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1611-5