
Abstract
Aims/hypothesis
Maternal diabetes during pregnancy increases the risk of congenital malformations such as neural tube defects (NTDs). Although the mechanism of this effect is uncertain, it is known that levels of nitric oxide synthase (NOS) and nitric oxide are elevated in embryos of a mouse model of diabetes. We postulated that overproduction of nitric oxide causes diabetes-induced congenital malformations and that inhibition of inducible NOS (iNOS) might prevent diabetic embryopathy.
Methods
Mice were rendered hyperglycaemic by intraperitoneal injection of streptozotocin. The incidence of congenital malformations including NTDs was evaluated on gestational day 18.5. We assessed the involvement of iNOS in diabetes-induced malformation by administering ONO-1714, a specific inhibitor of iNOS, to pregnant mice with streptozotocin-induced diabetic mice and by screening mice with iNOS deficiency due to genetic knockout (iNos −/−).
Results
ONO-1714 markedly reduced the incidence of congenital anomalies, including NTDs, in fetuses of a mouse model of diabetes. It also prevented apoptosis in the head region of fetuses, indicating that iNOS is involved in diabetes-related congenital malformations. Indeed, no NTDs were observed in fetuses of diabetic iNos −/− mice and the incidence of other malformations was also markedly reduced.
Conclusions/interpretation
We conclude that increased iNOS activity during organogenesis plays a crucial role in the pathogenesis of diabetes-induced malformations and suggest that inhibitors of iNOS might help prevent malformations, especially NTDs, in diabetic pregnancy.
Similar content being viewed by others

Introduction
Maternal diabetes during pregnancy significantly increases the risk of congenital anomalies in offspring. The incidence of congenital malformation in type 1 diabetic pregnancy is estimated to be 4.2% to 9.4% compared with approximately 1% in the general population [1–3]. Although a planned pregnancy with optimal blood glucose control and folic acid supplements at the time of conception reduces the risk of congenital malformations, a recent prospective study showed that the incidence of congenital malformations is still elevated in women with type 1 diabetes [4]. The congenital malformations associated with diabetic pregnancy affect many major organs, including the central nervous, cardiovascular, gastrointestinal, urogenital and musculoskeletal systems [5, 6]. The pathogenesis of congenital anomalies is complex and still poorly understood, although it has been suggested that excessive reactive oxygen species (ROS) associated with hyperglycaemia are responsible for the increased risk of malformation [7–11].
Nitric oxide, a signalling molecule, is synthesised from L-arginine by three distinct forms of nitric oxide synthase (NOS): neuronal, endothelial and inducible NOS (nNOS, eNOS and iNOS respectively) [12]. nNOS and eNOS are constitutively expressed at low levels and are regulated by calcium-activated calmodulin. In contrast, iNOS is induced when macrophages and other cells are activated by inflammatory mediators and, unlike the constitutively expressed types, can be expressed at relatively high levels [13]. Nitric oxide has been shown to be involved in the differentiation, proliferation and apoptosis of neuronal cells [14–16]; the effect of nitric oxide is either physiological or cytotoxic, depending on its level. For example, a low level of nitric oxide has antioxidant and anti-apoptotic properties and plays an important role in normal development. Traister et al. reported that NOS is expressed and is active in the chick neuroepithelium during neural tube closure [17]. Other reports show that intra-amniotic injection of either a nitric oxide donor or a non-specific NOS inhibitor is dysmorphogenic in embryos of rats [18] and that nitric oxide levels affect the balance between cell proliferation and apoptosis in the neural tube [19]. These studies suggest that NOSs are essential for normal development of the neural tube. On the other hand, overproduction of nitric oxide, which is mainly induced by iNOS, has hazardous effects on development. Such overproduction has been shown to exert an apoptotic effect in a variety of cells and is implicated in the pathogenesis of many disorders, including diabetic complications [20, 21]. Nitric oxide reacts with superoxide to form the powerful oxidant, peroxynitrite (ONOO−), which is thought to play a role in nitric oxide-dependent cytotoxicity [22]. It has been reported that NOS activity and nitric oxide levels are elevated and that peroxynitrite is formed in the maldeveloped neural tube of embryos of diabetic dams [23]. Based on these findings, we hypothesised that maternal hyperglycaemia might induce iNOS expression in the neural tube of embryos and that the overproduction of nitric oxide is involved in diabetic embryopathy. We further postulated that inhibition of iNOS activity might prevent neural tube defects (NTDs). To test these hypotheses, we investigated the effect of pharmacological inhibition of iNOS on the incidence of diabetic embryopathy. In addition, we screened iNOS-deficient mice to determine whether loss of function of iNOS prevented NTDs and other embryopathies induced by maternal diabetes.
Methods
Animal protocol
ICR mice (9–10 weeks old, body weight 30 g; Chubu Science Materials, Nagoya, Japan) were housed in a standard animal facility under conditions of constant temperature (23°C) and a 12 h light/dark cycle. Mice had free access to standard chow and tap water. Female mice were rendered hyperglycaemic by an intraperitoneal injection of streptozotocin (240 mg/kg body weight: Sigma Chemical, St Louis, MO, USA). At 7 days after the streptozotocin injection, plasma glucose concentrations were measured in tail vein blood using a compact glucose analyser (MediSafe; Terumo, Tokyo, Japan). The diabetic state was defined as a plasma glucose concentration exceeding 19.5 mmol/l. The plasma glucose concentration was measured in the early morning. The success rate of diabetes induction was approximately 70% and the survival rate of the injected mice approximately 95%. The diabetic female mice were mated overnight with non-diabetic ICR males. Embryonic day (E) 0.5 was considered to be noon of the day the vaginal plug was found. All procedures described above were performed in accordance with institutional guidelines for animal care in Nagoya University, which conform to the National Institutes of Health animal care guidelines.
NOS inhibitor treatment
From E7 to E10, pregnant mice were given a daily intraperitoneal injection of the non-selective NOS inhibitor N ω-nitro-l-arginine methyl ester (l-NAME, Sigma) or ONO-1714 (gift from ONO Pharmaceutical, Osaka, Japan), a selective inhibitor of iNOS. l-NAME and ONO-1714 were dissolved in isotonic saline and injected at a dose of 10 and 0.3 mg/kg body weight, respectively. For the non-diabetic group and the diabetic group treated with vehicle, an equal volume of saline was injected.
iNOS knockout mice
C57BL/6-backcrossed iNos −/− mice (B6.129P2-Nos2 tm1Lau/J) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The genetic background of iNos −/− mice is C57BL/6J. The iNos −/− line of mice has been previously established [24, 25]; although they are deficient in iNOS expression, they are nevertheless fertile and no breeding problems have been reported [26]. The mice were bred under specific pathogen-free conditions at the Animal Facility of Nagoya University. Wild-type C57BL/6J mice (iNos +/+) were purchased from Chubu Science Materials (Nagoya, Japan). Diabetes was induced in wild-type (diabetic iNos +/+) and iNos −/− mice (diabetic iNos −/−) by an intraperitoneal injection of streptozotocin (240 mg/kg body weight). Diabetic female iNos −/− or iNos +/+ mice were mated with a non-diabetic male of the same strain.
Analysis of malformations
On E18.5, pregnant mice were anaesthetised with ether and blood samples for serum glucose determination were obtained. The females were then killed and the fetuses extracted from the uterus, weighed and their crown-to-rump lengths measured. Using a stereomicroscope, the fetuses were screened for external malformation as described previously [8]. The number of resorptions was determined and the rate of resorption expressed as a percentage of the total number of implantations per litter. The incidence of malformations was expressed as the percentage of the total number of viable fetuses per litter. After evaluation for external malformations, fetuses were killed by an overdose of ether inhalation. Then, approximately half the fetuses of each litter were placed in Bouin’s solution and subsequently examined for visceral malformation. The remaining fetuses were fixed in 95% (vol./vol.) ethanol, stained with Alizarin Red S and Alcian Blue and examined for skeletal malformations. The incidence of congenital visceral and skeletal malformations was expressed as percentages of the total number of live fetuses per litter analysed for visceral and skeletal malformations, respectively.
Quantitative RT-PCR
Total RNAs were extracted from embryos at E8.5 using a kit (RNeasy; Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. Real-time PCR was carried out using an ABI-PRISM 7000 (Applied Biosystems, Foster City, CA, USA) and the Power SYBR Green Master Mix (Applied Biosystems) as described previously [27]. The sequences of the primers used for real-time PCR can be viewed in the Electronic supplementary material (ESM) Table 1. The level of mRNA expression was normalised by that of β-actin mRNA.
Measurement of nitrite and nitrate
As an index of nitric oxide production, nitrite and nitrate levels in whole embryos were measured using a Griess reagent kit (colorimetric NO2/NO3 Assay Kit-C ll; Wako Pure Chemical Industries, Osaka, Japan) according to the manufacturer’s protocol. Two embryos were used together for each measurement of nitrates and nitrites. Values were corrected for protein content, which was determined using a kit (BCA Protein Reagent kit; Pierce, Rockford, IL, USA).
Histological analysis
A total of 36 sections (six each from six embryos at E9.5) from each group (non-diabetic, diabetic control or diabetes + ONO-1714) was used for whole-mount immunohistochemistry. Whole-mount immunohistochemistry was performed as described previously [28]. Briefly, embryos were fixed with 4% (wt/vol.) paraformaldehyde in PBS at 4°C, washed with PBS containing 0.1% Tween 20 (wt/vol.), dehydrated through a methanol series, bleached in 5% hydrogen peroxide (wt/vol.) and stored in methanol at −20°C until use. Embryos were rehydrated and blocked with PBS containing 1% BSA and 0.1% TritonX-100. Embryos were incubated with one of the following primary antibodies: monoclonal mouse anti-iNOS antibody (1:10,000 dilution; BD Biosciences, San Jose, CA, USA), monoclonal mouse anti-eNOS antibody (1:1,000 dilution; BD Biosciences) and monoclonal mouse anti-nNOS antibody (1:250 dilution; BD Biosciences). Binding of the primary antibodies was detected using horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies. All antibodies were diluted in PBS containing 1% BSA and 0.1% TritonX-100. A total of 36 sections (six each from six embryos at E9.5) from each group (non-diabetic, diabetic control or diabetes + ONO-1714) was used for whole-mount TUNEL staining. Whole-mount TUNEL staining was performed using a kit (In Situ Apoptosis Detection; Takara Biomedicals, Tokyo, Japan) according to the manufacturer’s instructions with minor modifications. Briefly, embryos were incubated for 8 min at 37°C with 15 µg/ml proteinase K. After being washed with PBS containing 0.1% Tween 20, embryos were fixed again in 4% paraformaldehyde for 5 min. After blocking endogenous peroxidase by 3% hydrogen peroxide in methanol, fragmented DNA was labelled with deoxyuridine 5′-triphosphate (dUTP) in the presence of terminal deoxynucleotidyl transferase (TdT) for 120 min at 37°C. To visualise signals, embryos were incubated with 0.3 mg/ml diaminobenzidine (Sigma). Whole-mount diaminobenzidine-stained embryos were examined using a VB-G25 microscope (Keyence, Osaka, Japan). After imaging, embryos were embedded in paraffin, cut into 5 µm sections and counterstained with Methyl Green. Some embryos were embedded in paraffin and the sections (10 µm) were collected on glass microscope slides. Immunostaining was performed as described previously [29]. The sections were incubated with monoclonal mouse anti-iNOS (1:5,000 dilution; BD Biosciences). FITC-labelled sections were examined using a Zeiss LSM 510 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Statistics
Unless otherwise stated, results are expressed as means ± SEM. Statistical analyses were performed using a one-way ANOVA followed by Fisher’s protected least significant difference test. A value of p < 0.05 was regarded as significant.
Results
Reproductive outcome and fetal size
Reproductive outcome and fetal sizes are summarised in Table 1. The mean numbers of implantations per litter were similar among the four groups. The resorption rate in the diabetic control group was significantly higher than that of the non-diabetic group. Following treatment with l-NAME or ONO-1714, the rates of resorption fell but not by a significant amount. Fetal body weights and crown-to-rump lengths were significantly decreased in the diabetic control group compared with the non-diabetic group. Treatment with either l-NAME or ONO-1714 significantly increased fetal sizes, but did not restore them to those of the non-diabetic group (Table 1).
iNOS inhibitors decrease congenital malformations in fetuses of diabetic mice
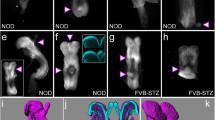
No congenital malformations were observed in the non-diabetic group (Fig. 1f, Table 2). In contrast, external, visceral and skeletal malformations were observed in the offspring of diabetic females (Fig. 1f, Table 2). The most common type of anomaly was NTDs, representative examples of which are shown in Fig. 1a. The other external malformations observed were agnathia, micrognathia, omphalocele, cleft face, anophthalmia, microphthalmia, cleft palate and limb hyperflexion. Most of these external malformations were complicated with NTDs. Visceral analysis revealed a high incidence in the diabetic control group of cardiovascular malformations (CVMs), including cardiac outflow tract defects such as truncus arteriosus (Fig. 1c) and transposition of the great vessels. The other less frequent visceral malformations were diaphragmatic hernia, kidney agenesis, enlarged kidney, abnormal lung lobation, ovarian aplasia and malpositioned ovary. Skeletal malformations such as caudal regression syndrome (Fig. 1e), infusion of ribs, supernumerary ribs, agnathia, micrognathia and centrum defects were also observed. Treatment with l-NAME or ONO-1714 during organogenesis significantly decreased the incidence of NTDs, CVMs and skeletal malformations (Fig. 1f). There were no significant differences in the effectiveness of the l-NAME and ONO-1714 treatments (Fig. 1f), suggesting that iNOS is the likely target of these compounds. Treatments with NOS inhibitors did not affect maternal blood glucose levels (Fig. 1g).

The effects of pharmacological inhibition of iNOS in diabetic and non-diabetic mice on the incidence of fetal congenital malformations and on maternal blood glucose concentrations. a–e Fetal morphologies at E18.5. a White arrows, myeloschisis associated with exencephaly in a fetus of the diabetic control group. b, c Hearts of fetuses from the non-diabetic (b) and diabetic control group (c). For clarity, the atria were removed. Ao, aorta; PT, pulmonary trunk. c Truncus arteriosus. White arrow, the single arterial vessel. d, e Alizarin Red S and Alcian Blue staining of fetuses from the non-diabetic (d) and diabetic control (e) group. e Caudal regression syndrome with retardation of ossification. Black arrow, absence of sacrum and a defect of the lumbar spine. f The incidence of NTDs, CVMs and skeletal malformations in the fetuses. No malformations were observed in the non-diabetic group. g Maternal blood glucose concentrations at E0.5 and E18.5. Values (f, g) are expressed as means ± SEM. *p < 0.05 vs non-diabetic group; † p < 0.05 vs diabetic control group. White bars, non-diabetic; black bars, diabetic control; horizontally striped bars, diabetic + l-NAME; hatched bars, diabetic + ONO-1714
Maternal diabetes increases iNOS levels, nitric oxide production and levels of TNF-α, IFN-γ and NADPH oxidase components in embryos
Since the disclosure of neural tube is thought to begin at E8.5 and be completed at E10.5 [30, 31], we chose the middle of the disclosure process, i.e. E9.5, to evaluate nitric oxide production and iNOS protein levels. Nitric oxide production in whole embryos of the diabetic control group at E9.5 was higher than that in the non-diabetic group (Fig. 2a). To determine whether NOS was involved in neural tube development, we examined the expression of iNOS, eNOS and nNOS by immunohistochemistry. At E9.5, iNOS staining was present in the hindbrain of whole-mount immunostained embryos of the diabetic control group (Fig. 2d), but not in the non-DM group (Fig. 2b). In transverse sections cut from whole-mount embryos stained with anti-iNOS antibodies, immunoreactivity was located around the neural tubes of the diabetic control mice (Fig. 2e), but no staining was present around those of non-diabetic mice (Fig. 2c). We also performed immunofluorescent staining of iNOS in paraffin-embedded sections from embryos at E9.5. Strong iNOS immunoreactivity was present in the neural tube of diabetic control mice (Fig. 2g), whereas the staining was not observed in the non-diabetic group (Fig. 2f). Thus, we confirmed that iNOS was produced and distributed in neural tubes of diabetic control mice. Immunostaining for eNOS or nNOS was faint in both non-diabetic and diabetic groups, and no differences were found in immunoreactivity between the two groups (data not shown).

Maternal diabetes increases iNOS expression, nitric oxide production and expression of IFN-γ and NADPH oxidase components in embryos. a Nitrite and nitrate (NOx) contents of non-diabetic (non-DM) and diabetic (DM) control group embryos at E9.5. n = 9. b–g iNOS immunostaining of E9.5 embryos. Whole-mount iNOS immunostaining of embryos from the non-DM (b) and the DM-control group (d). c, e Transverse sections of the same (b, d) embryos, respectively. Sections were counterstained with Methyl Green. Black arrow (d), iNOS immunoreactivity in the dorsal edge of the hindbrain region. e iNOS-positive cells are present in the neuroepithelium of the neural tube (black arrows). f, g Fluorescence immunostaining for iNOS in the neural tube of non-DM (f) and of DM control group (g). Scale bar, 50 µm. h–n mRNA levels of Tnf-α, Ifn-γ and NADPH oxidase components in E8.5 embryos. The amounts of mRNA were determined by quantitative RT-PCR. Values are normalised against β-actin RNA, n = 12–14. All values are expressed as means ± SEM. *p < 0.05 vs non-DM group
To evaluate mechanisms by which iNOS expression is increased in embryos in the diabetic control group, a quantitative RT-PCR analysis was carried out. Since levels of iNOS protein were increased at E9.5, we performed RT-PCR analysis one day before the increase in iNOS protein, i.e. at E8.5. We found that expression of mRNAs for Ifn-γ (also known as Ifng) and components of the NADPH oxidase complex, including p47 phox (also known as Ncf1) and p67 phox (also known as Ncf2), were increased at E8.5 in the diabetic control group compared with the non-diabetic group (Fig. 2h–n). On the other hand, expression of Tnf-α, gp91 phox, p22 phox and p40 phox was unchanged.
Inhibition of iNOS decreases apoptosis in embryos of diabetic mice
To determine whether inhibition of iNOS prevents apoptosis in embryos of diabetic mice, we used TUNEL staining of whole-mount embryos at E9.5. The numbers of apoptotic cells in the brain regions of diabetic control group embryos (Fig. 3b) were larger than in the non-diabetic group (Fig. 3a). Treatment with ONO-1714 reduced the rate of apoptotic cells in the diabetic control group embryos (Fig. 3c). In transverse sections cut from whole-mount TUNEL-stained embryos, we found that the increase in apoptotic cells was principally located in the neural tubes in diabetic control group embryos (Fig. 3e). Apoptotic cells were rarely seen in diabetic group embryos after maternal treatment with ONO-1714 treatment (Fig. 3f).

Reduction in apoptosis in embryos of diabetic mice treated with an iNOS inhibitor. Whole-mount TUNEL staining of E9.5 embryos of the non-diabetic (a), diabetic control (b) and diabetic + ONO-1714 (c) groups (n = 36). Apoptotic cells (arrow heads, stained brown) were increased in the diabetic control (b) compared with the non-diabetic group (a) in the head region. c The numbers of apoptotic cells fell in the head region following maternal ONO-1714 treatment. d–f Transverse sections of the embryos shown above (a–c), respectively. Arrows, apoptotic cells. Sections were counterstained with Methyl Green. Many apoptotic cells were present in the neuroepithelium of the neural tube (e), but not in other (d, f) images. Scale bars, 50 µm
Incidence of congenital malformations in fetuses of diabetic iNos+/+ and iNos−/− mice
Next, we used mice lacking iNOS expression (iNos −/−) to determine whether loss of iNOS function could prevent congenital anomalies induced by maternal diabetes. As iNos −/− mice have a normal life span and are fertile [26], the mutant strain was propagated by mating of iNos −/− homozygotes. In agreement with a previous report [26], iNos −/− fetuses were indistinguishable from wild-type fetuses in external appearance and also did not exhibit histological abnormalities. As described above for ICR mice, NTDs, CVMs and skeletal malformations were observed at high incidences in the iNos +/+ fetuses of diabetic mice (Fig. 4, Table 3). The frequencies of these malformations in fetuses of diabetic iNos −/− mice were markedly decreased compared with the diabetic iNos +/+ group (Fig. 4). It was of particular interest that no NTDs were observed in the fetuses of diabetic iNos −/− mice (Fig. 4, Table 3). There were no significant differences in blood glucose levels between the diabetic iNos +/+ and diabetic iNos −/− mice at E0.5 and E18.5 (E0.5 iNos +/+ 24.0 ± 2.2 mmol/l, iNos −/− 29.4 ± 2.2 mmol/l; E18.5 iNos +/+ 33.3 ± 3.2 mmol/l, iNos −/− 34.8 ± 4.2 mmol/l). These results clearly demonstrate that targeted disruption of the iNos gene diminished diabetes-induced congenital malformations and, in particular, prevented NTDs.

The incidence of congenital malformations in fetuses of diabetic iNos +/+ (white bars) and iNos −/− (black bars) mice, as shown, on E18.5. No NTDs were observed in the diabetic iNos −/− groups. Values are the means ± SEM. *p < 0.05 vs diabetic iNos +/+ group
Discussion
One of the novel findings in this study is that maternal diabetes induces iNOS expression in the neural tube of embryos and increases mRNA levels of the NADPH oxidase components and Ifn-γ in embryos during organogenesis. Previous reports have shown that NOS activity is present in the developing neural tube of the chick and is elevated in the embryos of diabetic mice [17, 32]. However, these studies did not attempt to identify the particular isotype of NOS that is increased in the neural tube. In this study, we demonstrated that the only NOS isotype to display an increase in immunoreactivity in the neural tubes of E9.5 embryos of diabetic mice was iNOS. Immunoreactivity for eNOS or nNOS was faint in non-diabetic and diabetic groups. Although it has been reported that E9.5 embryos display eNOS expression in the murine heart and limb skeletal myocytes [33, 34], these studies did not analyse eNOS expression in the neural tube. It is well known that the expression of iNos mRNA is regulated by nuclear factor kappa B (NFκB) activated by proinflammatory cytokines such as TNF-α and IFN-γ. ROS, generated mainly by NADPH oxidase, are also known to activate NFκB. The NADPH oxidase complex consists of the membrane-associated flavocytochrome b558 protein, which is composed of gp91phox and p22phox, and the cytosolic components p47phox, p67phox and p40phox [35]. It has been reported that the diacylglycerol–protein kinase C pathway is stimulated in the malformed embryos of diabetic mice [36] and that protein kinase C stimulates NADPH oxidase expression [37]. Thus, the data we present here suggest that an increase in NADPH oxidase expression and subsequent ROS production result in the elevation of iNOS expression in embryos of diabetic mice. In this study, we also found that Ifn-γ mRNA was increased. Since it has been reported that IFN-γ and TNF-α synergistically increase iNOS expression in macrophages [38], we postulate that the increased expression of IFN-γ enhanced iNOS expression in the embryos of diabetic mice by activation of NFκB. However, it is not yet clear whether iNos mRNA is regulated by NFκB in embryos, since this pathway may act differently in embryos to the way it acts in adults [39]. In addition, it has been shown that IFN-γ and NADPH oxidase components as well as TNF-α are not affected in diabetic embryopathy [40]. Therefore, further studies are required to clarify the mechanism of increased iNOS expression in the embryos of diabetic mice.
Increased iNOS expression results in overproduction of nitric oxide. As expected, therefore, we detected elevated nitric oxide levels in E9.5 embryos of diabetic mice compared with those in non-diabetic mice. Although nitric oxide is a weak free radical, its oxidant properties are enhanced when nitric oxide reacts with a superoxide to form peroxynitrite [22]. Jawerbaum et al. reported that intense nitrotyrosine immunostaining indicative of peroxynitrite was detected in the neural tube and neural folds of embryos of diabetic rats [23]. These findings suggest that increased expression of iNOS in diabetic embryos results in an excess of nitric oxide, which then leads to peroxynitrite accumulation. Peroxynitrite is known to cause DNA damage, protein lipid peroxidation and protein degradation [22, 41]. Thus, the present study suggests that the accumulation of peroxynitrite causes apoptosis during organogenesis, resulting in NTDs.
In order to further investigate the role of iNOS, we examined the effect of inhibition of iNOS on the incidence of NTDs. Treatment of pregnant diabetic females with ONO-1714, a specific iNOS inhibitor, decreased the fetal incidence of NTDs by 83%. Interestingly, loss of function of iNOS using iNos −/− mice completely prevented NTDs. We also found that ONO-1714 prevented apoptosis, particularly around the neural tubes. These findings demonstrate that iNOS plays a critical role in the pathogenesis of NTDs and suggest that iNOS-induced apoptosis is, at least in part, responsible for NTDs.
In this study, l-NAME and ONO-1714 were similarly effective in preventing diabetes-induced malformations. However, previous studies have shown that l-NAME is teratogenic in non-diabetic animals. Tiboni et al. showed that maternal treatment with l-NAME at doses of 40 mg/kg or higher produced axial skeletal defects in ICR mice [42]. Lee et al. reported that microinjection of N G-monomethyl-l-arginine (l-NMMA), a non-specific NOS inhibitor like l-NAME, into the amniotic fluids of cultured whole-rat conceptuses caused failure of anterior and posterior neural tube closure [18]. These findings suggest important roles for optimal levels of nitric oxide in normal embryonic development and that excess NOS inhibition may be dysmorphogenic. However, since no malformations have been shown in iNos −/− mice, the teratogenic effect of total inhibition of NOS is most likely to be due to the inhibition of eNOS and/or nNOS, but not due to iNOS inhibition. Another possibility why we did not find an increase in malformations in l-NAME-treated diabetic animals compared with diabetic controls is that the dose of l-NAME used in our study was much less than that used in the previous study by Lee et al [18].
Maternal diabetes is known to increase the incidence of CVMs in humans [43]. The data we present here show that approximately one third of all fetuses from diabetic mice developed CVMs and that pharmacological or genetic inhibition of iNOS decreased the incidence of CVMs by 76 and 82%, respectively. However, the mechanism by which the inhibition of iNOS prevented CVMs remains unclear. It is known that the migration of neural crest cells (NCCs) plays an essential role in cardiovascular development [44]. Failure of NCC migration results in CVMs, particularly those involving malformations of the great vessels and cardiac outflow tract defects, such as truncus arteriosus [44]. Bloch et al. reported that iNOS expression starts at an early stage (E8.5) of murine/rat heart development [33]. Several lines of evidence show that iNOS expression is induced by proinflammatory cytokines, such as TNF-α and IL-1β, and that the resulting increase in nitric oxide generation causes cardiac cell degeneration and apoptosis [45, 46]. Overall, it seems likely that enhanced iNOS expression and consequent cell degeneration and apoptosis in cardiomyocytes or NCCs may lead to defective migration of NCCs, resulting in CVMs in the offspring of diabetic mothers. However, loss-of-function of iNOS did not completely prevent CVMs, in contrast to its effect in NTDs. Therefore, in addition to the iNOS-mediated pathway, other metabolic and developmental processes are likely to be involved in the CVMs induced by a diabetic pregnancy.
Flodstrom et al. reported that iNOS inactivation makes animals more resistant to multiple low-dose streptozotocin-induced diabetes [47], whereas we administered a single injection of high-dose streptozotocin to induce diabetes. Multiple low-dose streptozotocin makes animals diabetic via an immune-mediated process in which iNOS is involved [47]. On the other hand, a single high-dose injection of streptozotocin makes animals diabetic by its direct cytotoxic effect on pancreatic beta cells [48]. This pathway may be independent of iNOS [48]. Indeed, in the present study, there were no significant differences in the plasma glucose levels between iNos −/− and iNos +/+ mice.
In view of the current epidemic of type 2 diabetes at younger ages [49], the number of diabetic pregnancies will undoubtedly increase. There is, therefore, an urgent need to increase our understanding of diabetic embryopathy at the molecular level in order to develop new strategies to prevent increased rates of children born with congenital malformations. In the present study, we demonstrated that iNOS plays an important role in the development of diabetic embryopathy. Thus, iNOS inhibitors are strong candidates for use in treatments to prevent congenital malformations, especially, NTDs, in the offspring of diabetic mothers.
Abbreviations
- CVM:
-
Cardiovascular malformation
- E:
-
Embryonic day
- eNOS:
-
Endothelial nitric oxide synthase
- iNOS:
-
Inducible nitric oxide synthase
- l-NAME:
-
N ω-Nitro-l-arginine methyl ester
- NCC:
-
Neural crest cells
- NFκB:
-
Nuclear factor kappa B
- nNOS:
-
Neuronal nitric oxide synthase
- NOS:
-
Nitric oxide synthase
- NTD:
-
Neural tube defect
- ROS:
-
Reactive oxygen species
References
Hawthorne G, Robson S, Ryall EA et al (1997) Prospective population based survey of outcome of pregnancy in diabetic women: results of the Northern Diabetic Pregnancy Audit, 1994. BMJ 3151:279–281
Casson IF, Clarke CA, Howard CV et al (1997) Outcomes of pregnancy in insulin dependent diabetic women: results of a five year population cohort study. BMJ 3151:275–278
Suhonen L, Hiilesmaa V, Teramo K (2000) Glycaemic control during early pregnancy and fetal malformations in women with type I diabetes mellitus. Diabetologia 43:79–82
Evers IM, de Valk HW, Visser GH (2004) Risk of complications of pregnancy in women with type 1 diabetes: nationwide prospective study in the Netherlands. BMJ 3281:915
Mills JL (1982) Malformations in infants of diabetic mothers. Teratology 251:385–394
Becerra JE, Khoury MJ, Cordero JF, Erickson JD (1990) Diabetes mellitus during pregnancy and the risks for specific birth defects: a population-based case-control study. Pediatrics 851:1–9
Hagay ZJ, Weiss Y, Zusman I et al (1995) Prevention of diabetes-associated embryopathy by overexpression of the free radical scavenger copper zinc superoxide dismutase in transgenic mouse embryos. Am J Obstet Gynecol 1731:1036–1041
Siman CM, Eriksson UJ (1997) Vitamin E decreases the occurrence of malformations in the offspring of diabetic rats. Diabetes 461:1054–1061
Sivan E, Lee YC, Wu YK, Reece EA (1997) Free radical scavenging enzymes in fetal dysmorphogenesis among offspring of diabetic rats. Teratology 561:343–349
Chang TI, Horal M, Jain SK et al (2003) Oxidant regulation of gene expression and neural tube development: Insights gained from diabetic pregnancy on molecular causes of neural tube defects. Diabetologia 461:538–545
Sakamaki H, Akazawa S, Ishibashi M et al (1999) Significance of glutathione-dependent antioxidant system in diabetes-induced embryonic malformations. Diabetes 481:1138–1144
Nathan C, Xie QW (1994) Nitric oxide synthases: roles, tolls, and controls. Cell 781:915–918
Aktan F (2004) iNOS-mediated nitric oxide production and its regulation. Life Sci 751:639–653
Peunova N, Enikolopov G (1995) Nitric oxide triggers a switch to growth arrest during differentiation of neuronal cells. Nature 3751:68–73
Moncada S, Erusalimsky JD (2002) Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat Rev Mol Cell Biol 31:214–220
Packer MA, Stasiv Y, Benraiss A et al (2003) Nitric oxide negatively regulates mammalian adult neurogenesis. Proc Natl Acad Sci USA 1001:9566–9571
Traister A, Abashidze S, Gold V et al (2002) Evidence that nitric oxide regulates cell-cycle progression in the developing chick neuroepithelium. Dev Dyn 2251:271–276
Lee QP, Juchau MR (1994) Dysmorphogenic effects of nitric oxide (NO) and NO-synthase inhibition: studies with intra-amniotic injections of sodium nitroprusside and NG-monomethyl-L-arginine. Teratology 491:452–464
Plachta N, Traister A, Weil M (2003) Nitric oxide is involved in establishing the balance between cell cycle progression and cell death in the developing neural tube. Exp Cell Res 2881:354–362
Zhao X, Haensel C, Araki E, Ross ME, Iadecola C (2000) Gene-dosing effect and persistence of reduction in ischemic brain injury in mice lacking inducible nitric oxide synthase. Brain Res 8721:215–218
Pacher P, Obrosova IG, Mabley JG, Szabo C (2005) Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem 121:267–275
Kim PK, Zamora R, Petrosko P, Billiar TR (2001) The regulatory role of nitric oxide in apoptosis. Int Immunopharmacol 11:1421–1441
Jawerbaum A, Higa R, White V et al (2005) Peroxynitrites and impaired modulation of nitric oxide concentrations in embryos from diabetic rats during early organogenesis. Reproduction 1301:695–703
Laubach VE, Shesely EG, Smithies O, Sherman PA (1995) Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci USA 921:10688–10692
Wei XQ, Charles IG, Smith A et al (1995) Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 3751:408–411
MacMicking JD, Nathan C, Hom G et al (1995) Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 811:641–650
Futaki S, Hayashi Y, Yamashita M et al (2003) Molecular basis of constitutive production of basement membrane components. Gene expression profiles of Engelbreth–Holm–Swarm tumor and F9 embryonal carcinoma cells. J Biol Chem 2781:50691–50701
Davis CA (1993) Whole-mount immunohistochemistry. Methods Enzymol 2251:502–516
Takefuji S, Murase T, Sugimura Y et al (2007) Role of microglia in the pathogenesis of osmotic-induced demyelination. Exp Neurol 2041:88–94
Harris MJ, Juriloff DM (1999) Mini-review: toward understanding mechanisms of genetic neural tube defects in mice. Teratology 601:292–305
Juriloff DM, Harris MJ (2000) Mouse models for neural tube closure defects. Hum Mol Genet 91:993–1000
Jawerbaum A, Sinner D, White V et al (2001) Modulation of PGE2 generation in the diabetic embryo: effect of nitric oxide and superoxide dismutase. Prostaglandins Leukot Essent Fatty Acids 641:127–133
Bloch W, Fleischmann BK, Lorke DE et al (1999) Nitric oxide synthase expression and role during cardiomyogenesis. Cardiovasc Res 431:675–684
Teichert AM, Scott JA, Robb GB et al (2008) Endothelial nitric oxide synthase gene expression during murine embryogenesis: commencement of expression in the embryo occurs with the establishment of a unidirectional circulatory system. Circ Res 1031:24–33
Babior BM (1999) NADPH oxidase: an update. Blood 931:1464–1476
Hiramatsu Y, Sekiguchi N, Hayashi M et al (2002) Diacylglycerol production and protein kinase C activity are increased in a mouse model of diabetic embryopathy. Diabetes 511:2804–2810
Inoguchi T, Sonta T, Tsubouchi H et al (2003) Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J Am Soc Nephrol 141:S227–S232
Chan ED, Winston BW, Uh ST et al (1999) Evaluation of the role of mitogen-activated protein kinases in the expression of inducible nitric oxide synthase by IFN-gamma and TNF-alpha in mouse macrophages. J Immunol 1621:415–422
Torchinsky A, Toder V (2004) To die or not to die: the function of the transcription factor NF-kappaB in embryos exposed to stress. Am J Reprod Immunol 511:138–143
Lian Q, Dheen ST, Liao D, Tay SS (2004) Enhanced inflammatory response in neural tubes of embryos derived from diabetic mice exposed to a teratogen. J Neurosci Res 751:554–564
Szabo C (2003) Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett 140–1411:105–112
Tiboni GM, Giampietro F, Di Giulio C (2003) The nitric oxide synthesis inhibitor N ω-nitro-l-arginine methyl ester (l-NAME) causes limb defects in mouse fetuses: protective effect of acute hyperoxia. Pediatr Res 541:69–76
Wren C, Birrell G, Hawthorne G (2003) Cardiovascular malformations in infants of diabetic mothers. Heart 891:1217–1220
Kirby ML, Waldo KL (1995) Neural crest and cardiovascular patterning. Circ Res 771:211–215
Arstall MA, Sawyer DB, Fukazawa R, Kelly RA (1999) Cytokine-mediated apoptosis in cardiac myocytes: the role of inducible nitric oxide synthase induction and peroxynitrite generation. Circ Res 851:829–840
Song W, Lu X, Feng Q (2000) Tumor necrosis factor-alpha induces apoptosis via inducible nitric oxide synthase in neonatal mouse cardiomyocytes. Cardiovasc Res 451:595–602
Flodstrom M, Tyrberg B, Eizirik DL, Sandler S (1999) Reduced sensitivity of inducible nitric oxide synthase-deficient mice to multiple low-dose streptozotocin-induced diabetes. Diabetes 481:706–713
Strandell E, Eizirik DL, Korsgren O, Sandler S (1988) Functional characteristics of cultured mouse pancreatic islets following exposure to different streptozotocin concentrations. Mol Cell Endocrinol 591:83–91
Pinhas-Hamiel O, Zeitler P (2005) The global spread of type 2 diabetes mellitus in children and adolescents. J Pediatr 1461:693–700
Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research from the Japanese Society for the Promotion of Science 16590875 (to T. Murase).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM Table 1
Sequences of the primers used for quantitative RT-PCR (PDF 41 kb)
Rights and permissions
About this article
Cite this article
Sugimura, Y., Murase, T., Oyama, K. et al. Prevention of neural tube defects by loss of function of inducible nitric oxide synthase in fetuses of a mouse model of streptozotocin-induced diabetes. Diabetologia 52, 962–971 (2009). https://doi.org/10.1007/s00125-009-1312-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1312-0