
Abstract
Aims/hypothesis
The effect of the benzopyran derivative T33, a novel non-thiazolidinedione agent, was studied on peroxisome proliferator-activated receptors (PPARs), insulin signalling and glucose uptake in adipocytes and skeletal muscle. We hypothesised that T33 could activate PPARγ and exert a beneficial effect on insulin action on glucose uptake and lipid metabolism.
Materials and methods
Using a cell-based reporter gene assay, T33 was identified as a PPARα/γ dual agonist, which activated human PPARγ and PPARα with EC50 values of 19 and 148 nmol/l, respectively. The effect of T33 on glucose metabolism was studied in cultured 3T3-L1 adipocytes and L6 myotubes. In vivo effects of T33 on skeletal muscle were determined in ob/ob mice treated with 8 mg/kg T33. The effect of T33 on metabolic abnormalities was observed in diet-induced obese mice.
Results
Exposure of 3T3-L1 adipocytes to T33 for 4 days increased basal and insulin-stimulated glucose uptake, with no effect noted in L6 myotubes. Treatment of ob/ob mice for 20 days with T33 normalised basal and insulin-stimulated glucose uptake and increased phosphorylation of Akt and p38 mitogen-activated protein kinase in skeletal muscle. In contrast, phosphorylation of AMP-activated protein kinase was unaltered. Moreover, T33 improved insulin sensitivity and lipid metabolism in diet-induced obese mice.
Conclusions/interpretation
T33 is non-thiazolidinedione PPARα/γ dual agonist which directly increases basal and insulin-stimulated glucose uptake in adipocytes and secondarily improves insulin action on insulin signalling and glucose metabolism in skeletal muscle from diabetic ob/ob mice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The global incidence of non-insulin-dependent type 2 diabetes mellitus is dramatically increasing and the greatest number of newly diagnosed patients is expected to be among young adults [1]. Type 2 diabetes is characterised by abnormal carbohydrate, lipid and protein metabolism, which is attributed to diminished production of insulin or defective insulin action (insulin resistance) [2]. Insulin resistance is a fundamental characteristic of type 2 diabetes, which is associated with hyperglycaemia, hyperinsulinaemia, dyslipidaemia, obesity and atherosclerotic vascular disease [3]. Ameliorating insulin resistance is one important strategy in the development of new pharmacological treatments for type 2 diabetes.
Under insulin-stimulated conditions, approximately 80% of glucose disposal occurs in skeletal muscle, whereas adipose tissue accounts for a comparatively smaller fraction of whole-body glucose uptake [4, 5]. Glucose transport is the first rate-limiting step in glucose metabolism and a major site of insulin resistance in type 2 diabetes [6]. Impaired insulin signalling is associated with defects in glucose uptake in skeletal muscle and adipose tissue in insulin-resistant humans [7]. Strategies to enhance insulin signalling and glucose uptake may be efficacious in improving the altered glucose homeostasis characteristic in type 2 diabetes.
The peroxisome proliferator-activated receptors (PPARs) are members of a nuclear transcription factor superfamily. Three PPAR subtypes, PPAR α, δ and γ have been discovered, and different PPAR subtypes play specialised roles in metabolic disease [8]. PPARγ is produced mainly in adipose tissue and plays a central role in adipogenesis and glucose homeostasis [9, 10]. PPARα is present at high concentrations in the liver and contributes to hepatic lipid metabolism. Activation of PPARγ modulates the expression of genes associated with carbohydrate, lipid and protein metabolism, which influence glucose uptake and insulin sensitivity [9]. Because of the central role of PPAR isoforms in metabolism, they have become attractive targets for drug discovery aimed at improving insulin sensitivity.
Thiazolidinediones (TZDs) are widely used oral glucose-lowering drugs which are high-affinity ligands for PPARγ and have hypoglycaemic and insulin-sensitising effects. TZDs improve glucose homeostasis by increasing insulin sensitivity in peripheral tissue [11–13]. The lipid-modulating activities of fibrates are presumably due to specific activation of PPARα. Activation of PPARα lowers triacylglycerol and elevates plasma HDL-cholesterol levels [14, 15]. Since type 2 diabetic patients often develop hyperglycaemia, dyslipidaemia and insulin resistance, novel approaches aiming to combine the insulin-sensitising effect of PPARγ agonists with the hypolipidaemic effects of PPARα agonists have been considered. Dual PPARα/γ agonists are expected to be more beneficial in ameliorating major metabolic disorders than either a PPARα- or PPARγ-selective agonist alone. Whether PPARγ agonists or PPARα/γ dual agonists have direct effects on skeletal muscle or adipocytes is unknown.
The benzopyran derivative T33, originally named T11, is a novel non-thiazolidinedione agent which stimulates differentiation of 3T3-L1 pre-adipocytes to adipocytes [16]. Since PPARγ is the major modulator controlling the pre-adipocyte switch to fully differentiated adipocytes, we hypothesised that T33 activates PPARγ. We determined the effect of T33 on PPARs, as well as the acute and chronic effect on glucose uptake in adipocytes and skeletal muscle using in vitro and in vivo approaches in ob/ob and dietary-induced insulin-resistant mice.
Materials and methods
Cell-based reporter gene assay
Transient transfection experiments for the analysis of nuclear receptor activation were performed in Caco-2 subclone TC7 cells, a colon adenocarcinoma cell line, in 96-well plates. For batch transfections, cells were seeded at a concentration of 4.0 × 106 cells/225 cm2 and incubated for 24 h in medium containing DMEM, 10% charcoal/dextran-treated fetal bovine serum (FBS), non-essential amino acids (0.1 mmol/l) and l-glutamine (4 mmol/l) (Invitrogen, San Diego, CA, USA). Cells were co-transfected with 5 μg pCMXGal4-receptor plasmid (where the ligand binding of the respective receptor was fused to the Gal4 DNA-binding domain) and 50 μg 4* Gal4re-luciferase reporter plasmid using FuGENE-6 (Roche, Basel, Switzerland) at 6 μl/μg DNA. Cells were seeded at a concentration of 25,000 cells per well and incubated for ∼5 h and subsequently treated with compounds (30 μmol/l to 50 nmol/l) in serum-free medium. The final concentration of charcoal/dextran FBS during induction was 5%. After 24 h, cells were harvested in buffer (0.1 mol/l Tris-HCl, 2 mol/l EDTA, 0.25% Triton X-100) and analysed for luciferase activity using a Luciferase Assay Kit (Bio Thema AB, Handen, Sweden). For curve fitting, Xlfit version 3.0.2 was used.
3T3-L1 cell differentiation and glucose uptake
3T3-L1 pre-adipocytes were maintained at ∼70% confluence in DMEM supplemented with 10% FBS, 25 mmol/l glucose and antibiotics (DMEM/FBS). Cells were grown for 2 days post-confluence and cultured in DMEM/FBS supplemented with 1 μg/ml insulin, 1 μmol/l dexamethasone and 0.5 mmol/l 3-isobutyl-1-methylxanthine for 3 days. Medium was replaced with DMEM/FBS supplemented with only 1 μg/ml insulin for 3 days and then DMEM/FBS alone for 2 days. Cytoplasmic triacylglycerol droplets were visible on day 5 after initiation of differentiation. Differentiated cells were used when ∼90% of the cells showed an adipocyte phenotype.
3T3-L1 adipocytes were incubated with T33 or rosiglitazone for 6 h or 4 days. Medium containing 0.1% DMSO was used as vehicle. Glucose uptake was determined after T33 or rosiglitazone treatment. Cells were washed twice with pre-warmed (37°C) PBS and incubated with Krebs–BSA buffer (140 mmol/l NaCl, 5 mmol/l KCl, 2.5 mmol/l MgSO4, 1 mmol/l CaCl2, 20 mmol/l HEPES, 0.5% BSA, pH 7.4) with or without insulin (basal state) for 15 min, followed by addition of 0.05 mmol/l 2-deoxy-d-glucose and 1.85 × 104 Bq/ml 2-deoxy-d-[1,2-3H]glucose for 5 min. The assay was terminated by washing the cells three times with ice-cold PBS. Cells were solubilised with 0.1% Triton X-100 and radioactivity was determined in a liquid scintillation counter (Beckman LS6500, Fullerton, CA, USA). Total cellular protein concentration was measured by the Bradford method (BioRad, Richmond, CA, USA). Non-specific glucose uptake was determined by treating cells with 10 μmol/l cytochalasin-B, and this value was subtracted from all data. Glucose uptake assays were performed in triplicate at least three times.
L6 cells differentiation and glucose uptake
L6 myoblasts were cultured in DMEM supplemented with 10% FBS. Confluent cells were differentiated to myotubes by culturing with DMEM supplemented with 2% FBS for 6 days. L6 myotubes were treated with T33 or rosiglitazone for 4 days. Glucose uptake was performed in 12-h serum-starved cells. Cells were washed with Krebs–Ringer phosphate-HEPES buffer (KRHB; 130 mmol/l NaCl, 5 mmol/l KCl, 1.3 mmol/l MgSO4, 1.3 mmol/l CaCl2, 25 mmol/l HEPES, pH 7.4) and incubated in KRHB containing 0.1% BSA with or without 100 nmol/l insulin for 40 min. Cells were incubated with 0.05 mmol/l 2-deoxy-d-glucose and 1.85 × 104 Bq/ml 2-deoxy-d-[1,2-3H]glucose for 10 min. The assay was terminated by washing the cells three times with ice-cold PBS. Cells were solubilised with 0.05 mol/l NaOH and radioactivity was determined by liquid scintillation counting (Beckman LS6500). Total cellular protein concentration was measured by the Bradford method (BioRad). Glucose uptake was performed in triplicate at least three times.
Animal treatment and muscle isolation
Six- to seven-week-old mice (ob/ob and lean control mice) were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and maintained under a 12-h light-dark cycle with free access to water and food. Animal experiments were approved by the local ethics board.
Based on fasting blood glucose values (first criterion) and initial body weights (second criterion), ob/ob mice were assigned to three groups and subjected to gavage-treatment once daily with vehicle (1% carboxymethylcellulose; CMC), T33 (8 mg/kg) or rosiglitazone (4 mg/kg), respectively. Lean mice were treated with vehicle (1% CMC). After 20 days, 5-h fasted mice were anaesthetised with an i.p. injection of sodium pentobarbital (40 mg/kg body weight). Soleus and extensor digitorum longus (EDL) muscles were isolated for 2-deoxyglucose uptake and protein phosphorylation measurements, respectively.
T33 treatment in diet-induced obese (DIO) mice
C57BL/6J male mice (3–4 weeks) were purchased from Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China) and maintained under a 12-h light-dark cycle with free access to water and food. Mice were fed a formulated research diet containing 60% energy as fat (no. D12492i; Research Diets, New Brunswick, NJ, USA) for 12 weeks before and throughout the duration of the experiment. DIO mice were assigned to four groups and subjected to gavage treatment once per day with vehicle (1% CMC), T33 (4 or 8 mg/kg) or rosiglitazone (4 mg/kg), respectively, for 28 days. Fasting blood glucose values and initial body weights were comparable between groups. Fasting blood glucose values were measured using a One-Touch Basic Glucose Monitor (Lifescan, Milpitas, CA, USA). Glucose tolerance, insulin tolerance and lipid tolerance were determined on day 18, day 22 and day 25 of the treatment, respectively. Glucose tolerance was determined in fasted mice (1 g/kg glucose administered by i.p. injection). Insulin tolerance was determined in 5-h fasted mice (0.5 U/kg insulin administered by an i.p. injection). Lipid tolerance was determined in overnight-fasted mice that were orally challenged with 10 ml/kg 20% intralipid. Serum triacylglycerol was measured before and after administration of the lipid load. Serum was collected for determination of insulin, triacylglycerol and NEFA concentration.
2-Deoxyglucose uptake in isolated soleus muscle
Incubation media were prepared from pre-gassed (95% O2/5% CO2) Krebs–Henseleit buffer (KHB) (NaCl 118.5 mmol/l, KCl 4.7 mmol/l, KH2PO4 1.2 mmol/l, NaHCO3 25.0 mmol/l, CaCl2.2H2O 2.5 mmol/l, MgCl2.7H2O 1.2 mmol/l) supplemented with 0.1% RIA-grade BSA and 5 mmol/l HEPES. The gas phase (95% O2/5% CO2) and temperature (30°C) were maintained during the muscle incubations. Soleus muscles were pre-incubated in KHB buffer containing 5 mmol/l glucose and 15 mmol/l mannitol, with or without 12 nmol/l insulin for 30 min and then rinsed in glucose-free KHB buffer with or without insulin for 10 min. Muscles were transferred to vials containing 1 mmol/l 2-deoxy-d-[1,2-3H]glucose (9.25 × 104 Bq /ml) and 19 mmol/l [14C]mannitol (2.59 × 104 Bq/ml) with or without 12 nmol/l insulin and incubated for 20 min. 2-Deoxyglucose uptake directly reflects glucose transport and not metabolism in mouse skeletal muscle when used under the present condition [17]. Muscles were frozen in liquid nitrogen and processed as described [18]. Glucose transport activity is expressed as μmol glucose analogue accumulated ml−1 intracellular water h−1.
Protein phosphorylation in isolated EDL muscle
EDL muscles were pre-incubated in KHB buffer containing 5 mmol/l glucose and 15 mmol/l mannitol for 30 min, followed by incubation in the same buffer with or without 120 nmol/l insulin for 20 min. EDL muscles were frozen in liquid nitrogen for subsequent analysis.
Protein preparation
After treatment with T33 or rosiglitazone, 3T3-L1 adipocytes were washed once in ice-cold PBS and harvested directly by scraping into ice-cold lysis buffer (137 mmol/l NaCl, 2.7 mmol/l KCl, 1 mmol/l MgCl2, 0.5 mmol/l Na3VO4, 1% Triton X-100, 10% glycerol, 20 mmol/l Tris-HCl [pH 7.8], 1 μg/ml leupeptin, 0.2 mmol/l phenylmethylsulfonyl fluoride, 10 mmol/l NaF, 1 μg/ml aprotinin, 1 mmol/l EDTA, 1 mmol/l dithiothreitol, 5 mmol/l Na pyrophosphate, 1 mmol/l benzamidine). Homogenates were sonicated (twice for 6 s), rotated for 40 min, and centrifuged (12,000 × g for 10 min at 4°C). Supernatant fractions were collected, and protein concentrations were determined using the Bradford method. EDL muscles were homogenised on ice in 0.3 ml lysis buffer and protein was determined as described above.
Western blot analysis
Equal amounts of proteins were separated by SDS-PAGE (7.5% resolving gel), transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA), and blocked with 7.5% non-fat milk. Membranes were incubated with a phospho-specific antibody overnight (4°C), and washed with Tris-buffered saline with Tween (10 mmol/l Tris, 140 mmol/l NaCl, 0.02% Tween 20 [pH 7.6]). The following phospho-specific antibodies (1:1,000) were used: phospho-Akt antibody, recognising Akt phosphorylation at Ser473; phospho-p38 mitogen-activated protein kinase (MAPK) antibody, recognising p38MAPK phosphorylation at Thr180 and Tyr182; and phospho-AMP-activated protein kinase (AMPK) antibody, recognising AMPK phosphorylation at Thr172. Membranes were probed with horseradish peroxidase-conjugated secondary antibody (1:3,000; BioRad) and washed in Tris-buffered saline with Tween. Antibodies were from Cell Signaling (Beverly, MA, USA). Phosphorylated proteins were detected using an ECL Plus Western Blotting Detection System (Amersham, Arlington Heights, IL, USA) and quantified by densitometry. Membranes were incubated in stripping buffer (1 mmol/l Tris [pH 6.7], 10% SDS and β-mercaptoethanol), extensively washed, and subjected to immunoblot analysis using antibodies that recognise Akt (1:1,000), p38MAPK (1:1,000) or AMPK (1:1,000) protein, respectively.
Statistics
Results are expressed as means±SEM. The statistical analysis was performed using a two-tailed unpaired t test for dual samples. p < 0.05 was considered to be statistically significant.
Results
Cell-based reporter gene assays
The potency and selectivity of T33 on the activation of human PPARα, PPARγ, PPARδ, liver X receptor (LXR)-α and retinoid X receptor (RXR)-α were determined using a reporter gene assay. Rosiglitazone and GW2313 were synthesised by Biovitrum, AB (Stockholm, Sweden) and used as positive controls for agonist action on PPARγ and PPARα, respectively. EC50 values are presented in Table 1. T33 activated PPARα and PPARγ. The EC50 of T33 on the activation of PPARγ was 19 nmol/l, whereas the EC50 of rosiglitazone was 23 nmol/l. T33 showed less potency for PPARα, compared with GW2313. The EC50 of T33 was 148 nmol/l, whereas the EC50 of GW2313 was 38 nmol/l. T33 did not show any agonistic effect against PPARδ, LXRα or RXRα. Therefore, T33 is a dual agonist against PPARα and PPARγ.
Glucose uptake in 3T3-L1 adipocytes
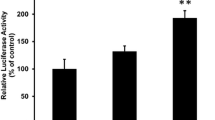
3T3-L1 adipocytes were incubated with different concentrations of T33 for 6 h (short-term) or 4 days (long-term). Basal and insulin-stimulated glucose uptake was unaltered after a 6-h incubation with 0.01, 0.1 or 1 μmol/l T33 or rosiglitazone (data not shown). After 4 days of incubation with T33, basal and insulin-stimulated glucose uptake in 3T3-L1 adipocytes was increased (Fig. 1). Basal glucose uptake increased 82% (p < 0.05) after treatment with 0.1 μmol/l T33. Insulin-stimulated glucose uptake was increased 32% (0.1 nmol/l insulin) and 48% (1 nmol/l insulin) following exposure to 0.1 μmol/l T33 (p < 0.05). Basal and insulin-stimulated 2-deoxyglucose uptake was also increased after a 4-day exposure to 1 μmol/l T33 (p < 0.5).

Long-term effect of T33 on glucose uptake in 3T3-L1 adipocytes. a Differentiated 3T3-L1 adipocytes were incubated with 0.01 μmol/l T33 or rosiglitazone for 4 days. b Differentiated 3T3-L1 adipocytes were incubated with 0.1 μmol/l T33 or rosiglitazone for 4 days. c Differentiated 3T3-L1 adipocytes were incubated with 1 μmol/l T33 or rosiglitazone for 4 days. The vehicle control contained 0.1% DMSO. 2-Deoxyglucose uptake was measured under basal or insulin-stimulated conditions. Values are expressed as fold over basal vehicle control. n = 3–4. Means±SEM. *p < 0.05, **p < 0.01 vs vehicle control group of each condition. Open bars, control; black bars, T33; hatched bars, rosiglitazone
Glucose uptake in L6 myotubes
L6 myoblasts were differentiated into myotubes and incubated in the presence of different concentrations of T33 for 4 days. Insulin increased glucose uptake in L6 myotubes 1.2-fold (p < 0.05). A 40-min exposure of myotubes to 100 nmol/l insulin increased glucose uptake by 25% (p < 0.05). Conversely, a 4-day treatment of myotubes with 0.1 or 1 μmol/l T33 or rosiglitazone was without effect on either basal or insulin-stimulated glucose uptake (Fig. 2).

Long-term effect of T33 on the glucose uptake in L6 myotubes. a Differentiated L6 myotubes were incubated with 0.1 μmol/l T33 or rosiglitazone for 4 days. b Differentiated L6 myotubes were incubated with 1 μmol/l T33 or rosiglitazone for 4 days. The vehicle control contained 0.1% DMSO. 2-Deoxyglucose uptake was measured under basal or insulin-stimulated conditions. Values are expressed as fold over basal vehicle control. n = 3. Means±SEM. *p < 0.05 vs vehicle control group of each condition. Open bars, control; black bars, T33; hatched bars, rosiglitazone
Glucose uptake in isolated soleus muscle from T33- or rosiglitazone-treated ob/ob mice
Mice were treated with T33, rosiglitazone or vehicle for 20 days and isolated skeletal muscle was dissected for glucose uptake and protein phosphorylation. Basal and insulin-stimulated 2-deoxyglucose uptake was significantly impaired in soleus muscles from ob/ob compared with lean mice (Fig. 3). Insulin-stimulated glucose uptake was reduced by 54% in soleus muscle from ob/ob mice, suggesting severe insulin resistance. T33 treatment improved basal and insulin-stimulated glucose uptake in soleus muscle from ob/ob mice. Basal and insulin-stimulated glucose uptake was increased by 77 and 78%, respectively, following T33 treatment. Rosiglitazone had a similar effect to improve basal and insulin-stimulated glucose transport in ob/ob mice.

Glucose uptake in isolated soleus muscle from in vivo T33- or rosiglitazone (Ros)-treated ob/ob mice. ob/ob mice were treated as described in Materials and methods. Soleus muscle was isolated and 2-deoxyglucose uptake was determined under basal or insulin-stimulated (12 nmol/l) conditions. n = 6 muscles. Means±SD. *p < 0.05, **p < 0.01 vs vehicle-treated ob/ob mice under basal conditions. # p < 0.05, ## p < 0.01 vs vehicle-treated ob/ob mice under insulin-stimulated conditions. Black bars, basal; grey bars, insulin-stimulated
Effects of T33 on the protein phosphorylation in 3T3-L1 adipocytes
Insulin exposure increased Akt phosphorylation, but had no effect on the phosphorylation of p38MAPK and AMPK in 3T3-L1 adipocytes (Fig. 4). After 4 days of treatment of 3T3-L1 adipocytes with 0.1 μmol/l T33, Akt phosphorylation was markedly enhanced under basal and insulin-stimulated conditions (2.7- and 1.4-fold, respectively), whereas phosphorylation of p38MAPK and AMPK was unaltered. T33-mediated glucose uptake in 3T3-L1 cells was increased in concert with increased Akt phosphorylation.

Effects of a 4-day T33 treatment on phosphorylation of Akt, p38MAPK and AMPK in 3T3-L1 adipocytes. Cells were incubated in the absence or presence of T33 for 4 days. Thereafter, cells were washed and treated with or without 100 nmol/l insulin for 15 min. Akt phosphorylation (a), p38MAPK phosphorylation (b) and AMPK phosphorylation (c) were determined. Lane 1, basal; lane 2, insulin; lane 3, basal plus T33; lane 4, insulin plus T33. Protein phosphorylation is presented as relative ratio to control cells (lane 1). n = 5. Means±SEM. **p < 0.01 vs control (lane 1). # p < 0.05 vs cells treated with insulin alone (lane 2)
Protein phosphorylation in isolated EDL muscle from T33-treated ob/ob mice
After a 20-day treatment with T33, rosiglitazone or vehicle, EDL muscles were isolated and phosphorylation of Akt, p38MAPK and AMPK was determined (Fig. 5). Compared with lean mice, the phosphorylation of Akt in EDL muscles from ob/ob mice was reduced by 62 and 56% under basal and insulin-stimulated conditions, whereas the Akt protein level was unchanged. p38MAPK phosphorylation was reduced by 27 and 26% in EDL muscle of ob/ob mice compared with lean mice, whereas p38MAPK protein levels were similar between lean and ob/ob mice. Phosphorylation and protein levels of AMPK were similar in EDL muscles from lean and ob/ob mice. In vivo T33 treatment increased Akt and p38MAPK phosphorylation under basal and insulin-stimulated conditions, whereas AMPK phosphorylation was unaltered. Basal and insulin-stimulated Akt phosphorylation was increased 2.1- and 1.9-fold and p38MAPK phosphorylation increased 1.5- and 1.3-fold, respectively, following T33 treatment.

Phosphorylation of Akt, p38MAPK and AMPK in isolated EDL muscles of in vivo T33-treated ob/ob mice. Muscles were incubated in the absence or presence of 120 nmol/l insulin for 20 min. Akt phosphorylation (a), p38MAPK phosphorylation (b) and AMPK phosphorylation (c) was determined. Protein phosphorylation is presented as a relative ratio to control ob/ob mice. n = 8 muscles. Means±SEM. *p < 0.05, **p < 0.01 vs ob/ob mice under basal conditions. # p < 0.05, ## p < 0.01 vs ob/ob mice under insulin-stimulated conditions. Black bars, basal; grey bars, insulin-stimulated. Ros, rosiglitazone
Effect of T33 on metabolic abnormalities of DIO mice
C57BL/6J mice fed a high-fat diet develop moderate obesity, mild hyperglycaemia, dyslipidaemia and insulin resistance. T33 treatment lowered fasting blood glucose and improved glucose tolerance in DIO mice (Fig. 6). Rosiglitazone showed a similar effect. Insulin tolerance was improved in DIO mice after T33 treatment. Rosiglitazone treatment improved insulin tolerance, but this effect was less pronounced compared with T33 treatment. Serum triacylglycerol, NEFA and insulin levels were improved after either T33 or rosiglitazone treatment. T33 and rosiglitazone treatment significantly improved serum triacylglycerol clearance. T33 showed a better reduction in reducing triacylglycerol level than rosiglitazone (61% for T33-treated vs 30% for rosiglitazone-treated). T33 and rosiglitazone did not alter body weight and the food intake in DIO mice. However, 8 mg/kg T33 treatment lowered the weight of subcutaneous fat significantly, whereas rosiglitazone was without effect on adipose tissue mass.

Effect of T33 on metabolic abnormalities of diet-induced obese mice. DIO mice were treated as described in Materials and methods. Fasting blood glucose concentration (a) and body weight (b) was measured before and after treatment. Different fat pads (c) were also weighed at the end of the experiment. Glucose tolerance (d), insulin tolerance (e) and lipid tolerance (f) were determined on days 18, 22 and 25 of the treatment, respectively. Serum triacylglycerol (g), NEFA (h) and insulin (i) concentrations were evaluated at the end of the treatment period. Data are means±SEM for n = 8 mice. *p < 0.05 and **p < 0.01 vs vehicle-treated mice. Open bars, control; black bars, T33 4 mg/kg; single-hatched bars, T33 8 mg/kg; cross-hatched bars, rosiglitazone 4 mg/kg. Filled circles, control; open circles, T33 4 mg/kg; filled triangles, T33 8 mg/kg; open triangles, rosiglitazone 4 mg/kg. Ros, rosiglitazone
Discussion
PPARs are ligand-activated transcription factors which play a key role in maintaining glucose and lipid homeostasis. Fibrates and TZDs used in the treatment of dyslipidaemia are ligands for PPARα and PPARγ, respectively. PPAR agonists, especially dual PPARα/γ agonists, have emerged as an important target for the treatment of insulin resistance and dyslipidaemia [19–21]. In our previous study [16], the insulin-sensitising effect of a series of benzopyran derivatives revealed T33 exhibited more potential ability to promote differentiation in 3T3-L1 cells. Moreover, T33 treatment has potent and efficacious hypoglycaemic, hypolipidaemic and insulin-sensitising effects in ob/ob mice [22]. We evaluated the activation of T33 on different PPAR isoforms and determined the effect on glucose uptake in adipocyte and skeletal muscle. We also determined the effect of T33 on signalling cascades known to regulate glucose uptake.
In the reporter gene assay, we used rosiglitazone and GW2313 as positive controls for PPARγ and PPARα, respectively. These results showed that rosiglitazone is a selective PPARγ agonist with an EC50 of 23 nmol/l, and GW2313 is a selective PPARα agonist with an EC50 of 38 nmol/l, which suggested our assay procedure is valid. T33 showed slightly higher activation potential on PPARγ with an EC50 of 19 nmol/l compared with rosiglitazone, whereas it exhibited less potency on the activation of PPARα with an EC50 of 148 nmol/l, compared with GW2313. T33 was without effect on the activation of human PPARδ, LXRα and RXRα. Therefore, we conclude T33 is a dual PPARα/γ agonist.
PPARγ is highly expressed in adipose tissue [3–8, 23]. Several investigators have demonstrated glucose uptake and glucose transporter levels are enhanced in 3T3-L1 and 3T3-F442A adipocytes treated with a variety of TZDs [24–26]. However, most of these studies were performed during fibroblast differentiation into adipocytes, and it is unclear whether the increase in glucose uptake is due to augmented differentiation, or a direct effect on fully differentiated adipocytes, which were more relevant to the therapeutic situation. Therefore, in the present study, we evaluated the effect of T33 on glucose uptake in differentiated 3T3-L1 adipocytes to eliminate confounding direct effects of T33 on the adipogenic process. T33 enhanced basal and insulin-stimulated glucose uptake after long-term treatment (4 days), whereas no effect was observed after short-term treatment (6 h). Rosiglitazone also exhibited a positive effect on glucose uptake after long-term, but not short-term treatment, which is similar to previous reports [27]. Since glucose uptake is a rate-limiting step in glucose metabolism, the positive effect of T33 on glucose uptake suggests this compound has an insulin-sensitising effect primarily in adipose tissues.
In even the most obese patients or animals, the disposal of glucose in adipose tissue is only a small fraction of whole-body glucose utilisation [4, 5]. Glucose uptake into skeletal muscle is the major component of whole-body insulin-stimulated glucose uptake, and also a key site of insulin resistance in type 2 diabetes mellitus. Since production of PPARγ in skeletal muscles is comparatively lower than that in adipose tissue, whether PPARγ agonists act directly in skeletal muscle or indirectly at the level of adipose tissue is unclear. Troglitazone stimulates glucose uptake in L6 mytotubes [28], indicating this PPARγ agonist has a direct effect on skeletal muscle. Another study showed the lack of a direct effect on skeletal muscle after a 5-h in vitro TZD treatment, which suggested that the improved skeletal muscle insulin sensitivity might be due to an indirect in vivo effect of PPARγ activation [13]. We determined the effect of T33 on glucose uptake in L6 myotubes to evaluate whether there was a direct or indirect effect of T33 on glucose metabolism in skeletal muscle. Glucose uptake was also determined in isolated soleus muscle from T33-treated ob/ob mice to elucidate whether T33 has an in vivo ability to enhance glucose homeostasis. A 4-day treatment of L6 myotubes with T33 did not alter basal or insulin-stimulated glucose uptake, indicating T33 does not have a direct effect in the cultured skeletal muscle. In ob/ob mice, a 20-day treatment with T33 normalised basal and insulin-stimulated glucose uptake in soleus muscles. T33 could improve insulin sensitivity in skeletal muscle, although this effect is probably via indirect mechanisms, presumably at the level of adipose tissue.
GLUT4 (also known as SLC2A4) has been shown to exist exclusively in insulin-sensitive tissue such as skeletal muscle and adipose tissue, and is one of the major proteins responsible for insulin-stimulated whole-body glucose uptake. In skeletal muscle, insulin and exercise independently increase glucose uptake via GLUT4 translocation to the plasma membrane [29]. Insulin-mediated glucose uptake involves activation of the phosphatidylinositol 3-kinase (PI3-kinase) pathway and to lesser degree, members of the MAPK pathway, whereas exercise-stimulated glucose transport is mediated by AMPK. Therefore, the effect of T33 on phosphorylation of Akt, p38MAPK and AMPK was determined in adipocytes and skeletal muscles to illuminate mechanisms underlying the enhanced glucose uptake. In 3T3-L1 adipocytes, Akt phosphorylation was increased after 4 days of treatment with T33, whereas phosphorylation of p38MAPK and AMPK was unaltered. The canonical insulin-signalling pathway involving PI3-kinase might be involved in mechanisms by which T33 stimulates glucose uptake in mature adipocytes. In EDL muscle of ob/ob mice, the phosphorylation of Akt and p38MAPK under basal and insulin-stimulated states was decreased as compared with lean mice, suggesting that insulin signal transduction was impaired. This finding is consistent with the insulin-resistant phenotype described for the ob/ob mouse model [22]. Twenty days of treatment with T33 enhanced phosphorylation of Akt and p38MAPK in skeletal muscle from ob/ob mice. Interestingly, rosiglitazone showed similar effects. In contrast, AMPK phosphorylation was unaltered in skeletal muscle of lean, T33-treated and untreated ob/ob mice. In vivo T33 treatment normalised impairments in insulin signalling. Furthermore, the insulin signalling appears to promote skeletal muscle glucose uptake, whereas AMPK signalling was dispensable.
T33 treatment improved the lipid profile and enhanced insulin sensitivity in diet-induced obese mice. When compared with the same dose of rosiglitazone, T33 elicited a similar blood glucose-lowering effect in DIO mice. T33 was more efficacious than rosiglitazone in reducing the serum triacylglycerol level and improving lipid tolerance in DIO mice. Since PPARα agonists have an insulin-sensitising effect independently of PPARγ activity [30, 31], the insulin-sensitising effect of T33, a PPARα/γ agonist, in DIO mice might be attributed to the combined activation of both PPARα and PPARγ in response to treatment with this compound. By virtue of its dual PPARα- and PPARγ-activating properties, T33 had a better effect than rosiglitazone on improving the lipid profile and insulin sensitivity in ob/ob and insulin-resistant DIO mice. Thus, PPARα/γ dual agonists may be more beneficial than PPARγ agonists in the treatment of metabolism disorders such as type 2 diabetes.
In summary, T33 is a new non-thiazolidinedione PPARα/γ dual agonist which increases basal and insulin-stimulated glucose uptake in 3T3-L1 adipocytes. T33 also improves whole-body insulin sensitivity and skeletal muscle glucose uptake, although this effect is indirect. Insulin-dependent signalling pathways may mediate T33-induced glucose uptake.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- CMC:
-
carboxymethylcellulose
- DIO:
-
diet-induced obese
- EDL:
-
extensor digitorum longus
- FBS:
-
fetal bovine serum
- KHB:
-
Krebs–Henseleit buffer
- LXR:
-
liver X receptor
- MAPK:
-
mitogen-activated protein kinase
- PPAR:
-
peroxisome proliferator-activated receptor
- RXR:
-
retinoid X receptor
- TZD:
-
thiazolidinedione
References
Kiberstis PA (2005) A surfeit of suspects. Science 21:369
DeFronzo RA, Bondonna RC, Ferranini E (1992) Pathogenesis of NIDDM: a balanced overview. Diabetes Care 15:318–367
DeFronzo RA, Ferrannnini E (1991) Insulin resistance: a multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 14:173–194
DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP (1981) The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30:1000–1007
Marin P, Rebuffe-Scrive M, Smith U, Bjorntorp P (1987) Glucose uptake in human adipose tissue. Metabolism 36:1154–1160
Ren JM, Marshall BA, Gulve EA et al (1993) Evidence from transgenic mice that glucose transport is rate-limiting for glycogen deposition and glycolysis in skeletal muscle. J Biol Chem 268:16113–16115
Leng Y, Karlsson HK, Zierath JR (2004) Insulin signaling defects in type 2 diabetes. Rev Endocr Metab Disord 5:111–117
Kliewer SA, Xu HE, Lambert MH, Willson TM (2001) Peroxisome proliferator-activated receptor: from genes to physiology. Recent Prog Horm Res 56:239–265
Schoonjans K, Staels B, Auwerx J (1996) The peroxisome proliferator activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta 1302:93–109
Schoonjans K, Peinado-Onsurbe J, Lefebvre AM et al (1996) PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J 15:5336–5348
Beeson M, Sajan MP, Dizon M et al (2003) Activation of protein kinase C-zeta by insulin and phosphatidylinositol-3,4,5(PO4)3 is defective in muscle in type 2 diabetes and impaired glucose tolerance: amelioration by rosiglitazone and exercise. Diabetes 52:1926–1934
Miyazaki Y, He H, Mandarino LJ, DeFronzo RA (2003) Rosiglitazone improves downstream insulin receptor signaling in type 2 diabetic patients. Diabetes 52:1943–1950
Zierath JR, Ryder JW, Doebber T et al (1998) Role of skeletal muscle in thiazolidinedione insulin sensitizer (PPARgamma agonist) action. Endocrinology 139:5034–5041
Robins SJ, Collins D, Wittes JT, VA-HIT Study Group, Veterans Affairs High-Density Lipoprotein Intervention Trial et al (2001) Relation of gemfibrozil treatment and lipid levels with major coronary events. VA-HIT: a randomized controlled trial. JAMA 285:1585–1591
Diabetes Atherosclerosis Intervention Study Investigators (2001) Effect of fenofibrate on progression of coronary artery disease in type 2 diabetes: the Diabetes Atherosclerosis Intervention Study, a randomised study. Lancet 357:905–910
Tang L, Yu J, Leng Y, Feng Y, Yang Y, Ji R (2003) Synthesis and insulin-sensitizing activity of a novel kind of benzopyran derivative. Bioorg Med Chem Lett 13:3437–3440
Hansen P, Gulve E, Gao J, Schluter J, Mueckler M, Holloszy JO (1995) Kinetics of 2-deoxyglucose transport in skeletal muscle: effects of insulin and contractions. Am J Physiol 259:C30–C35
Wallberg-Henriksson H, Zetan N, Henriksson J (1987) Reversibility of decreased insulin-stimulated glucose transport capacity in diabetic muscle with in vitro incubation. J Biol Chem 262:7665–7671
Chakrabarti R, Misra P, Vikramadithyan RK et al (2004) Antidiabetic and hypolipidemic potential of DRF 2519—a dual activator of PPAR-alpha and PPAR-gamma. Eur J Pharmacol 491:195–206
Ljung B, Bamberg K, Dahllof B et al (2002) AZ 242, a novel PPARalpha/gamma agonist with beneficial effects on insulin resistance and carbohydrate and lipid metabolism in ob/ob mice and obese Zucker rats. J Lipid Res 43:1855–1863
Harrity T, Farrelly D, Tieman A et al (2006) Muraglitazar, a novel dual (alpha/gamma) peroxisome proliferator-activated receptor activator, improves diabetes and other metabolic abnormalities and preserves beta-cell function in db/db mice. Diabetes 55240–55248
Hu X, Feng Y, Shen Y et al (2006) Antidiabetic effect of a novel non-thiazolidinedione PPAR γ/α agonist on ob/ob mice. Acta Pharmacol Sin 27:1346–1352
Kersten S, Desvergne B, Wahli W (2000) Roles of PPARs in health and disease. Nature 405:421–424
Kreutter DK, Andrews KM, Gibbs EM, Hutson NJ, Stevenson RW (1990) Insulin like activity of new antidiabetic agent CP 68722 in 3T3-L1 adipocytes. Diabetes 39:1414–1419
Sandouk T, Reda D, Hofmann C (1993) The antidiabetic agent pioglitazone increases expression of glucose transporters in 3T3-F442A cells by increasing messenger ribonucleic acid transcript stability. Endocrinology 133:352–359
Tafuri S (1996) Troglitazone enhances differentiation, basal glucose uptake, and Glut1 protein levels in 3T3-L1 adipocytes. Endocrinology 137:4706–4712
Nugent C, Prins JB, Whitehead JP et al (2001) Potentiation of glucose uptake in 3T3-L1 adipocytes by PPARγ agonists is maintained in cells expressing a PPARγ dominant-negative mutant: evidence for selectivity in the downstream responses to PPARγ activation. Mol Endocrinol 15:1729–1738
Yonemitus S, Nishimura H, Shintani M et al (2001) Troglitazone induces GLUT4 translocation in L6 myotubes. Diabetes 50:1093–1101
Wallberg-Henriksson H, Rincon J, Zierath JR (1998) Exercise in the management of non-insulin-dependent diabetes mellitus. Sports Med 25:25–35
Guerre-Millo M, Gervois P, Raspe E et al (2000) Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J Biol Chem 275:16638–16642
Koh EH, Kim MS, Park JY et al (2003) Peroxisome proliferator-activated receptor (PPAR)-α activation prevents diabetes in OLETF rats. Diabetes 52:2331–2337
Acknowledgements
This study was supported by the Swedish Research Council, Swedish Diabetes Association, Strategic Research Foundation (INGVAR), National Nature Science Foundation of China (Grant 30472046), Shanghai Rising-Star Foundation (Grant 04QMX1465) and State Key Program of Basic Research of China (Grant 2004CB518905). We thank M. Gilbert for valuable comments.
Duality of interest
None of the authors have any duality of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Hu, X., Feng, Y., Liu, X. et al. Effect of a novel non-thiazolidinedione peroxisome proliferator-activated receptor α/γ agonist on glucose uptake. Diabetologia 50, 1048–1057 (2007). https://doi.org/10.1007/s00125-007-0622-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0622-3