
Abstract
Aims/hypothesis
Glucose and the peptide growth factors insulin, IGF-I and IGF-II strongly regulate beta cell mass. Furthermore, beta cell expression of IGF-I receptor (Igf1r) and insulin receptor (Insr) is mandatory for several steps of insulin secretion.
Materials and methods
We hypothesised that glucose concentration might regulate expression of Igf1r, Insr and insulin receptor-related receptor (Insrr) in islets and beta cells. Moreover, since the ratio of ATP:ADP is the most important intracellular mechanism involved in insulin secretion, and since depletion of ATP leads to AMP accumulation, we evaluated the role of AMP-activated protein kinase (AMPK) in glucose-dependent receptor regulation.
Results
In rat islets, high glucose exposure (25 mmol/l) increased gene expression of Igf1r, Insr and Insrr but also of the metabolic glycolysis gene liver-type pyruvate kinase (Pklr) compared with intermediate (6.2 mmol/l) or low glucose concentration (1.6 mmol/l) after 24 h. In rat INS-1E beta cells, only Pklr expression was suppressed by low glucose as in islets, while Insr and Insrr were suppressed by high and increased by low glucose levels. Igf1r expression was suppressed by both high- and low- glucose concentration. Activation of AMPK by 5-amino-imidazolecarboxamide riboside (AICAR, 0.5 mmol/l) suppressed Pklr expression, but strongly stimulated gene expression of Igf1r, Insr and Insrr. Protein expression of IR and IGF-IR reflected glucose and AICAR-regulated mRNA expression of both receptors in INS-1E cells.
Conclusions/interpretation
We conclude that glucose directly interacts with islet and beta cell expression of growth factor receptors that are mandatory for both beta cell growth and insulin secretion. Stimulation of Igf1r and Insr gene expression by the AMPK-activator AICAR might indicate involvement of AMPK in the regulation of Igf1r, Insr and Insrr expression in beta cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glucose is a major factor for proliferation of pancreatic beta cells in vivo and in vitro [1, 2]. However, the pathways and mechanisms behind glucose-dependent regulation of growth and replication of pancreatic islets are largely unknown [3]. Among potent beta cell growth factors are nutrients like glucose or peptides like insulin, IGF-I or IGF-II. Furthermore, in situations where proliferation of pancreatic beta cells has been induced by IGF-I or growth hormone, this was strongly glucose dependent [2, 4, 5]. Increased glucose concentrations of 10–20 mmol/l were found to inhibit apoptosis in single beta cell cultures, compared with lower glucose levels [6]. Therefore characterisation of existing and new molecular mechanisms of nutrient- and growth-factor-induced proliferation is of importance to understand survival, regeneration and apoptosis of islets and beta cells in physiology and diabetes.
The insulin receptor (IR), IGF-I receptor (IGF-IR) and insulin-receptor-related receptor (IRR) are transmembrane heterotetramers formed by two identical extracellular α-subunits and two transmembrane and receptor-tyrosine-kinase-containing β-subunits. They are highly homologous in terms of amino acid sequence and structure and are in the IR family [2, 5, 7, 8]. Almost certainly, these receptors form chimeric receptor hybrids of IGF-IR (αIRβIR,αIGFIRβIGFIR), IR-IRR (αIRβIR,αIRRβIRR) and IGF-IR-IRR (αIGFIRβIGFIR,αIRRβIRR) tetramers that depend on specific expression levels for each receptor type [9]. Not belonging to the IR family, the IGF-II/mannose-6-phosphate receptor (IGF-II/M6PR) is a single-chain polypeptide without a tyrosine kinase region. IGF-II/M6PR binds predominantly IGF-II and is mainly involved in intracellular trafficking of lysosomal enzymes and decay of IGF-II [10]. All members of the IR family and IGF-II/M6PR are expressed in islets and beta cells [11–13].
In vitro studies and in vivo mutant mice models give further insights into the role of insulin and IGF signalling in pancreatic islets and beta cells. In INS-1E rat beta cells, IGF-I and IGF-II inhibit apoptosis induced by the cytokines IFN-γ and TNF-α or by high glucose concentrations [5]. In transgenic mice, beta cell expression of IGF-I leads to recovery from streptozotocin-induced diabetes [14] while overexpression of IGF-II leads to an increased beta cell mass at birth [15]. On the other hand, beta-cell-specific knock-out of IGF-IR or IR in mice leads to altered glucose sensing and glucose intolerance in adult mice but only the IR knock-out mice display a reduced beta cell mass in adult life [16–18]. Furthermore, in vitro knock-down studies applying a small-interfering-RNA technique allowed detailed comparison of the specific function that each receptor, IR and IGF-IR, plays in beta cell function [11]. Loss of either receptor in MIN-6 beta cells completely inhibited glucose-mediated insulin secretion but not insulin secretion mediated by depolarising agents. In detail, knock-down of IGF-IRs is associated with a dysregulation of cellular, ATP-sensitive insulin secretion, while knock-down of IRs inhibits expression of specific, glucose-regulated genes. These results underline the important role of both receptors in glucose-dependent insulin secretion and expression of glucose-regulated genes. Also a distinct functional role of hybrid IGF-IRs in mediating their own specific effects has been hypothesised, even if there was no precedence for this [9]. The IRR is an orphan receptor without any known ligand and might predominantly modulate IR or IGF-IR signalling through IRR-IGF-IR or IRR-IR hybrid formation [19, 20]. Less is known about IRR and beta cell function, but mutant IRR knock-out mice appear to have normal beta cell mass and beta cell function [21]. Nevertheless, IRR is specifically expressed in pancreatic beta cells and its intracellular tyrosine kinase domain is able to activate insulin receptor substrate (IRS)-1 and IRS-2 [19].
In summary, both IR and IGF-IR seem to be important in maintaining normal beta cell function in terms of glucose-stimulated insulin secretion. In contrast, they do not play a crucial role in pre- or postnatal regulation of beta cell mass but could play a role in adapting islet mass to hyperglycaemia or following pancreas injury [1, 17, 18]. Thus activation of the IGF/IGF-IR axis seems to protect beta cells against cell death and to increase beta cell mass, while during embryonic and fetal beta cell growth, both IGF-IR and IR appear to have overlapping functions and partly cover defects of either IGF-IR or IR signalling [14, 22].
In beta cells, glucose-dependent gene regulation was extensively investigated with respect to genes which modulate glucose metabolism and insulin secretion. The AMP-activated protein kinase (AMPK) is activated by high AMP and low ATP concentrations. In detail, 5′-AMP allosterically activates AMPK but also activates the upstream AMPK kinase and makes AMPK a worse substrate for protein phosphatases [23, 24]. A low cellular glucose concentration leads to depletion of ATP and cellular accumulation of AMP, which then leads to AMPK activation. For cellular experiments, AMPK can be activated by 5-amino-4-imidazolecarboxamide riboside (AICAR). AICAR is taken up by cells and converted to its monophosphate, ZMP, which than acts in an AMP-like way on all levels of AMP-dependent activation of AMPK [24]. In pancreatic beta cells, activation of AMPK by low glucose or by AICAR decreased the expression of metabolic genes, including solute carrier family 2 (facilitated glucose transporter), member 2 (SLC2A2), aldolase B (ALDOB), liver-type pyruvate kinase (PKLR) or preproinsulin (INS) [24, 25]. The nuclear transcription factor hepatocyte nuclear factor-4α (HNF4α), but also the newly discovered carbohydrate-response-element-binding protein, were found to be downstream targets of AMPK [26, 27]. Thus AMPK was claimed to have a pivotal role in the regulation of glucose-dependent metabolic genes [28].
Since glucose is the main source of islet function, we tested the hypothesis that glucose concentration regulates expression of receptors that participate in the transduction of growth and of metabolic function in beta cells. In detail, we investigated in rat islets and in INS-1E beta cells whether high or low glucose concentrations regulate mRNA and protein expression of several members of the IR family, most importantly the tyrosine kinase receptors IGF-IR, IR and IRR. Moreover, since the ratio of ATP:ADP is the most important intracellular mechanism involved in glucose-stimulated insulin secretion, and since depletion of ATP leads to AMP accumulation, we evaluated the role of AMPK in glucose-dependent receptor regulation.
Materials and methods
Cells and reagents
The rat insulinoma cell line INS-1E was a generous gift from Prof. Claes Wollheim, Geneva, Switzerland and represents a highly differentiated and glucose-sensitive clone of parental INS-1 cells. INS-1E cells were cultured in RPMI 1640 medium supplemented with 5% fetal calf serum, 10 mmol/l HEPES, 2 mmol/l l-glutamine, 1 mmol/l Na-pyruvate, 50 μmol/l β-mercaptoethanol, 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma Chemical, Taufkirchen, Germany) in 5% CO2 at 37°C. Standard cell culture medium contains 200 mg/dl (11.1 mmol/l) glucose concentration. Dextrose (d-glucose) and laevulose (l-glucose), serving as an osmotic control without metabolic activity, were from Sigma.
Glucose culture of INS-1E cells
INS-1E cells were seeded into six-well plates and kept in culture medium (containing 11.1 mmol/l glucose) for 3 days. Cells were then washed in serum-free incubation medium without glucose and then exposed to incubation medium containing glucose concentrations between 1.6 and 50 mmol/l glucose. Incubation medium was based on glucose-free RPMI 1640 medium, supplemented with 0.2% BSA, 10 mmol/l HEPES, 2 mmol/l l-glutamine, 1 mmol/l Na-pyruvate, 50 μmol/l β-mercaptoethanol, 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma). INS-1E cells remained viable for up to 72 h under these serum-free incubation conditions (data not shown).
Animals and preparation of isolated islets of Langerhans
Islets were isolated from the disease-resistant rat strain LEW.1W. Rats were bred and kept in the Animal Laboratories at the University of Greifswald in Karlsburg, Germany under strict hygienic conditions and were free of major pathogens as described [29]. Animals were treated in line with national and international guidelines on the care of laboratory animals. Rats were maintained at a 12 h light and darkness cycle (05:00/17:00 hours) and had free access to food (Ssniff; Soest, Germany) and acidulated water. Islets of Langerhans were isolated from pooled pancreases of 8-day-old male LEW.1W rats. Pancreatic islets were prepared by fractionated collagenase digestion (Serva, Heidelberg, Germany) followed by Dextran M70 density gradient centrifugation. After separation from exocrine tissue, islets were hand-picked under a stereomicroscope [30].
Glucose culture of islets
Following islet isolation, groups of 500 islets were incubated with glucose concentrations of 1.6, 6.2 and 25 mmol/l under sterile condition in RPMI 1640 medium supplemented with 10% fetal calf serum, 20 mmol/l l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma) for 24 h at 37°C under a humidified gas atmosphere (5% CO2:95% O2). Islet function was tested for each single experiment by islet insulin content before and immediately after incubation experiments and by insulin release into incubation medium during the incubation period.
RNA isolation and RT-PCR analysis
Total RNA was isolated from 500 islets for one experiment or from INS-1E cells grown in six-well plates at 106 cells per well using RNeasy mini columns (Qiagen, Hilden, Germany). DNA was digested with ribonuclease-free deoxyribonuclease (Qiagen) following the original protocol, and complete DNA removal was confirmed by PCR for rat Actb with an RNA template. For first-strand synthesis, 21 μl reaction mix containing 2 μg RNA, 1×RT buffer (Life Technologies, Karlsruhe, Germany), 200 μmol/l deoxy-NTPs, 200 ng random primers (p[dN]6) and 0.01 mol/l dithiothreitol were denatured at 65°C for 5 min and incubated on ice for 2 min. After adding 200 U Moloney murine leukaemia virus reverse transcriptase (Life Technologies) and 40 U ribonuclease inhibitor, the reaction was performed at 39°C for 60 min and was stopped by denaturation at 94°C for 2 min. Contamination controls with enzyme omitted in the reaction were performed. The integrity of each template was confirmed by control PCR for rat β-actin (Actb). PCR was performed in a thermal cycler (GeneAmp PCR system 2400; Perkin-Elmer, Uberlingen, Germany) using the Advantage cDNA Polymerase Mix (BD Biosciences, Clontech, Palo Alto, CA, USA). PCR primers and conditions are summarised in Table 1. In all PCR experiments amplification in the absence of cDNA was performed as a negative control. PCR products were stained by Gelstar nucleic acid staining (BMA, Rockland, ME, USA) and separated according to their length (bp) in a 1.5% agarose gel. Length of specific PCR products was estimated by a 100-bp ladder (Life Technologies).
Semi-quantitative analysis of PCR products
We selected rat Actb as control gene and all RT-PCR products were diluted according to their level of Actb gene expression. Correct dilution was confirmed by a final Actb PCR of templates. The optical density of each specific Actb band was compared with a standard dilution of pooled RT-PCR products from 1:5 down to 1:40 to create a standard curve, while PCR templates were originally diluted 1:10. Within this standard, the optical density of Actb PCR products finally differed with a CV of approximately 15%. Following final dilution and confirmation of Actb adjustment, final aliquots of each stimulation experiment were frozen at −20°C until PCRs with specific primer sets were performed. Optical density of specific PCR products was measured and quantified as relative density compared with one reference experiment at the start of incubation (time-dependent regulation) or to one reference glucose concentration (regulation by glucose concentration).
Quantitative analysis of PCR products
Gene expression of rat Pklr, Igf1r, Insr, Insrr, Actb and 36B4 was measured by quantitative real-time RT-PCR in a fluorescence temperature cycler (TaqMan; Applied Biosystems, Darmstadt, Germany) as described recently [31]. Briefly, total RNA was isolated from islets or INS-1E cells using TRIzol (Life Technologies) and 1–2 μg RNA was reverse transcribed with standard reagents (Life Technologies). Two microlitres of each RT reaction were amplified in a 26 μl PCR by using the Brilliant SYBR Green QPCR Core Reagent Kit from Stratagene (La Jolla, CA, USA) according to the manufacturer’s instructions. Samples were incubated in the Taqman for an initial denaturation at 95°C for 10 min, followed by 40 PCR cycles, each cycle consisting of 95°C for 15 s, 60°C for 1 min and 72°C for 1 min. Primers were used as for semi-quantitative RT-PCR (Table 1). SYBR Green I fluorescent emissions were monitored after each cycle. Expression of Pklr, Igf1r, Insr, Insrr, Actb and 36B4 mRNA was quantified by the second derivative maximum method of the TaqMan Software (Applied Biosystems) determining the crossing points of individual samples by an algorithm which identifies the first turning point of the fluorescence curve. Specific expression of genes was calculated relative to 36B4 in islets or Actb in INS-1E cells, respectively, which were used as an internal control due to their resistance to glucose-dependent regulation [32]. Amplification of specific transcripts was confirmed by melting curve profiles (cooling the sample to 68°C and heating slowly to 95°C with measurement of fluorescence) at the end of each PCR. The specificity of the PCR was further verified by subjecting the amplification products to agarose gel electrophoresis.
Immunoblotting of AMPK, Igf-IR and IR
Cells were washed twice in ice-cold PBS, scraped in ice-cold HEPES lysis buffer (HEPES 50 mmol/l, NaCl 100 mmol/l, EDTA 10 mmol/l, 1.0 mmol/l sodium ortho-vanadate, Triton X-100 1%, NaF 1 mmol/l, leupeptin 1 μg/ml), vortex-mixed and centrifuged (14,000×g; 3 min; 4°C). Protein content was assayed using a DC protein assay kit (Bio-Rad, Hercules, CA, USA), against BSA Type V (Sigma) standards. Total protein extracts (10–30 μg) were resolved by SDS-PAGE (10% v/v bisacrylamide:acrylamide, 37.5:1) and transferred to nitrocellulose membranes, followed by immunoblotting with specific antibodies or antisera. Secondary antibodies were revealed using chemiluminescence blotting substrate (Supersignal; Pierce/Perbio, Bonn, Germany). The following primary antibodies or polyclonal antisera were applied: anti-AMPK (dilution 1:1,000) and anti-phospho-AMPK-Thr172 (1:1,000), both polyclonal rabbit antisera (Cell Signaling Technology, Beverly, MA), and polyclonal goat antisera against IGF-IRβ (C-20, 1:2,000) and IRβ (C-19, 1:5,000), both from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Primary rabbit antibodies were detected by goat anti-rabbit immunoserum (Pierce) diluted 1:40,000, and primary goat antibodies by rabbit anti-goat immunoserum (DakoCytomation, Hamburg, Germany) diluted 1:40,000. Density analysis of specific protein bands was performed with Scion Image Beta 4.0.2 (Scion Corporation).
Data analysis
Statistical comparisons were carried out by paired Student’s t-test with Welch’s correction for unequal variances, where appropriate. Analysis and graphs were performed using the GraphPad Prism 3.0 software. Data are plotted as mean values and SE of the mean.
Results
RNA expression of IR family members in rat islets and INS-1E beta cells
To compare gene expression of the IR family members Igf1r, Insr and Insrr but also of Igf2r and Actb, specific rat primers (Table 1) were used to detect mRNA transcripts in rat islets, INS-1E insulinoma cells, and as controls in rat hypothalamus. Transcripts of Igf1r, Insr, Insrr and Igf2r were detected in rat islets, rat INS-1E beta cells, and in rat hypothalamus. Expression levels did not vary substantially (Fig. 1).

RT-PCR analysis of insulin receptor (Insr), IGF-I receptor (Igf1r), insulin receptor-related receptor (Insrr), IGF-II/mannose-6-phosphate receptor (Igf2r) and Actb transcripts in rat islets (Islets), INS-1E beta cells (INS1) and rat hypothalamus (RH). Negative controls (C) were an RT-PCR without template. RT-PCR was carried out using equal amounts of mRNA transcripts, 32 PCR cycles and the annealing temperatures indicated in Table 1. Data were representative of three independent experiments
Glucose-regulated gene expression in rat INS-1E beta cells
In rat INS-1E cells, gene expression of Insr and Insrr was strongly regulated by changes in glucose concentration after 24 h. Expression of Insr and Insrr was increased by low glucose concentration (1.6 mmol/l), while expression of Insrr but not of Insr was suppressed at high glucose concentration (25 mmol/l). Igf1r expression was suppressed at high or low glucose concentrations (Fig. 2). The metabolic gene Pklr was upregulated at high (25 mmol/l) glucose concentration and downregulated at low glucose concentration (1.6 mmol/l). This downregulation was already significant at 3 h of glucose incubation and thus Pklr was the earliest glucose-regulated gene in our experiments (Fig. 2d).

Glucose-induced expression of Igf1r (a), Insr (b), Insrr (c) and Pklr (d) in rat INS-1E beta cells. INS-1E cells were exposed to high (50 mmol/l, filled bars), intermediate (12.5 mmol/l; dotted bars) and low (1.6 mmol/l; open bars) glucose concentrations for the indicated time intervals. Bars represent means±SE and expression levels are calculated as relative expression compared with expression in culture medium with a glucose concentration of 11.1 mmol/l before glucose incubation, i.e. at 0 h (hatched bars). Plotted data are mean values from at least three independent cell stimulations and RT-PCR reactions. Significant differences (paired t-test compared with expression levels at an intermediate glucose concentration of 12.5 mmol/l) are indicated as *p<0.05, **p<0.01 and ***p<0.001
Moreover, to investigate clear dose–response effects and also regulations at extreme glucose levels, INS-1E cells were incubated at glucose concentrations from 50 to 0.8 mmol/l. First, expression of Igf1r, Insr and Insrr was regulated in a clear dose-dependent way within 24 h (Fig. 3). In detail, expression of Insr was stimulated by low glucose concentrations, while expression of Insrr was upregulated by glucose concentrations below 6.2 mmol/l and suppressed at high glucose levels above 12.5 mmol/l. Expression of Igf1r gene formed a bell-shaped curve with suppression at very low and very high glucose concentrations. In contrast, Igf2r was not regulated by changes in glucose concentration. Interestingly, extreme glucose concentrations at the very low (0.8 mmol/l) or very high (50 mmol/l) end regulated expression similarly to lower or higher glucose concentrations and thus we assume that gene regulation at these extreme values still followed the general mechanism observed at lower or higher glucose concentrations. Moreover, high osmolarity produced by 37.5 mmol/l l-glucose plus 12.5 mmol/l d-glucose was not different from gene expression levels at 12.5 mmol/l d-glucose alone. Therefore, the mechanism of gene regulation by high glucose concentrations was most probably due to glucose uptake and its intracellular metabolic activity, not to changes in osmolarity.

Glucose-regulated mRNA expression of Igf1r (a), Insr (b), Insrr (c) and Igf2r (d) at glucose concentrations from 0.8 up to 50 mmol/l in rat INS-1E cells. Cells were stimulated for 24 h with the indicated glucose concentrations. Plotted data are mean values from at least three independent cell stimulations and RT-PCR reactions. Bars represent means±SE. Specific RT-PCR templates had been adjusted to their level of Actb expression. l-glucose (37.5 mmol/l) plus a d-glucose concentration of 12.5 mmol/l served as an osmotic control for a d-glucose concentration of 50 mmol/l. Significant differences (paired t-test against a glucose level of 12.5 mmol/l) are indicated as *p<0.05, **p<0.01 and ***p<0.01
Glucose-regulated gene expression in rat islets
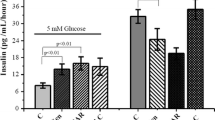
In rat islets, glucose-dependent gene expression of Igf1r, Insr and Insrr was different from that found in INS-1E cells (Fig. 4). First, in rat islets Igf1r was more highly expressed at a glucose concentration of 25 mmol/l than at 6.2 or 1.6 mmol/l. The upregulation by high glucose was found to be 83-fold higher than at 6.2 mmol/l and, furthermore, Igf1r expression was another 2.6-fold lower at 1.6 mmol/l glucose than at 6.2 mmol/l (Fig. 4a). Secondly, both Insr and Insrr were suppressed at low (1.6 mmol/l), but not significantly upregulated at high (25 mmol/l), glucose concentration. In contrast to these receptor genes, Pklr expression was upregulated by high glucose concentrations in rat islets and in INS-1E beta cells. Overall, regulation of the metabolic Pklr gene was the same in rat islets and INS-1E insulinoma cells, while expression of Igf1r, Insr and Insrr was different within a time range of up to 24 h. Islet function remained intact within the investigated 24 h, as measured in terms of insulin content and insulin secretion. Insulin content was reduced at 25 mmol/l glucose (mean 3.84 pmol/islet; p<0.01) but was not found to be different at 6.2 (mean 5.06 pmol/islet) or 1.6 mmol/l (mean 4.38 pmol/islet). Furthermore, insulin secretion was strongly increased at 25 mmol/l glucose concentration (mean 5.65 pmol ml−1 24 h−1, p<0.05) and was comparably low at 6.2 mmol/l (mean 0.43 pmol ml−1 24 h−1) and 1.6 mmol/l (mean 0.33 pmol ml−1 24 h−1).

Quantitative mRNA expression of Igf1r (a), Insr (b), Insrr (c) and Pklr (d) in response to glucose concentration in intact rat islets. Isolated rat islets were exposed for 24 h to high (25 mmol/l), intermediate (6.2 mmol/l) and low (1.6 mmol/l) glucose concentrations. Plotted data are mean values from three independent experiments including islet isolation, glucose exposure, RNA isolation, and quantitative real-time PCR using rat 36B4 as reference gene for quantification. Bars represent means±SE. Significant differences are indicated as *p<0.05
AMPK phosphorylation by AICAR and glucose
We tested whether AMPK protein is expressed in INS-1E beta cells and whether low glucose concentration or AICAR incubation leads to AMPK phosphorylation. We found that high glucose concentration (50 mmol/l) suppressed AMPK phosphorylation only within 10 min, while AICAR (0.5 mmol/l) induced phosphorylation of AMPK at 12.5 and 50 mmol/l glucose within 10 and 60 min (Fig. 5).

Low glucose concentration and stimulation with AICAR leads to phosphorylation of AMPK at threonine-172. a INS-1E cells were kept at 12.5 mmol/l glucose concentration and then exposed to 50, 12.5 and 1.6 mmol/l glucose with or without addition of AICAR at 0.5 mmol/l (black bars) for 10 and 60 min prior to cell lysis and immunoblotting with specific antibodies against AMPK and phosphorylated AMPK (P-AMPK Thr172). Suppression of AMPK phosphorylation by high glucose concentrations at 10 min could be compensated by addition of AICAR. Bars represent means±SE. b AMPK phosphorylation was quantified from three western blot experiments. *p<0.05 for the effects of AICAR; #p<0.05 for the effects of high glucose
Activation of AMPK by AICAR increased Igf1r, Insrr and Insr gene expression
Activation of AMPK by AICAR interacted with the glucose-dependent gene expression of Igf1r, Insr and Insrr in INS-1E beta cells (Fig. 6a–c). AICAR strongly stimulated expression of Igf1r after 12 and 24 h at all glucose concentrations. In addition, AICAR significantly increased gene expression of Insrr and Insr at 25 mmol/l glucose concentration. Expression of Pklr was found to be strongly stimulated at high glucose concentration in a dose-dependent way and AICAR suppressed the level of Pklr expression in a general way with the glucose-concentration-dependent effect still evident (Fig. 6d). In INS-1E beta cells, we found no significant gene regulation of Actb compared with 36B4 and finally Actb was selected as reference gene for all studies in INS-1E cells, including quantitative PCR experiments (Fig. 6e). Because of the limited source of rat islets, studies on whether AMPK is involved in glucose-regulated receptor mRNA and protein expression were performed in INS-1E cells only.

Quantitative PCR of Igf1r (a), Insr (b) and Insrr (c) and of Pklr (d) mRNA expression stimulated by high (25 mmol/l), intermediate (12.5 mmol/l) and low (1.6 mmol/l) glucose concentrations in INS-1E cells. To test whether AMP-kinase stimulation had an effect on glucose-induced regulation of mRNA expression, 0.5 mmol/l AICAR (black bars) was added. Pklr was known to be suppressed by AMPK stimulation and served as a control for effective AICAR stimulation. INS-1E cells were incubated at the indicated glucose concentrations with or without 0.5 mmol/l AICAR for 12 and 24 h. Plotted data are mean values from three independent experiments. e In INS-1E cells, Actb was selected as reference gene and displayed no regulation by glucose or AICAR compared with 36B4. Bars represent means±SE. Significant effects of AICAR are indicated as *p<0.05
Glucose and activation of AMP-kinase regulated protein expression of IGFIR and IR
Glucose concentration clearly regulated protein expression of IR and IGF-IR in a dose-dependent way (Fig. 7). The IGF-IR protein was decreased at low (3.1 mmol/l) and high (50 mmol/l) glucose concentrations; this regulation was found to be significant only for the pre-IGF-IR protein (Fig. 7c). Furthermore, activation of AMPK by AICAR resulted in a strong upregulation of pre-IGF-IR and IGF-IR β-subunit at all glucose concentrations with an exception at highest concentrations of 50 mmol/l glucose (Fig. 7a,c). The IR protein was suppressed at high glucose concentrations (50 mmol/l) and increased at low glucose levels (3.1 mmol/l). AICAR incubation upregulated IR expression significantly at high glucose levels, while high glucose concentration alone appeared to suppress receptor expression. In general, glucose-regulated and AICAR-modulated protein expression of both IGF-IR and IR paralleled mRNA expression of both receptors as investigated by PCR (Figs. 3 and 6). To investigate whether AICAR/ZMP-dependent stimulation of AMPK was still present after 24 h, we performed immunoblotting of AMPK and phosphorylated AMPK. Expression of AMPK was not changed after 24 h, but only if AICAR was present, continuous activation of AMPK by low glucose concentrations persisted (Fig. 7a).

a–c. Effects of glucose concentration and AICAR (0.5 mmol/l) on protein expression of Thr172-phosphorylated AMPK (P-AMPK), AMPK, insulin receptor prereceptor (Pre-IR), insulin receptor β-subunit (IR-β), IGF-I receptor pre-receptor (Pre-IGF-IR) and IGF-IR β-subunit (IGF-IR-β) in INS-1E cells (a). Cells were treated for 24 h with glucose concentrations from 50 to 1.6 mmol/l and AMP-kinase-dependent expression was tested by co-incubation with AICAR. Expression levels of insulin receptor (b) and IGF-I receptor (c) were quantified for full-length prereceptors (left, both panels) and β-subunits (right, both panels). Expression levels were quantified by densitometry and bars represent means±SE from three independent experiments, with one representative experiment depicted (a). AICAR effects are indicated as black bars for each glucose concentration (b, c). *p<0.05, **p<0.01 for the effects of AICAR; #p<0.05 for the effects of glucose concentration
Discussion
We have found evidence that glucose concentration on its own is sufficient to regulate the tyrosine kinase receptor genes Igf1r, Insr and Insrr in rat islets and in the rat beta cell line INS-1E. Both IGF-IR and IR strongly participate in metabolic and growth functions of pancreatic islets and beta cells. The fact that glucose concentration regulates mRNA and protein expression of these receptors might indicate a new and independent mechanism of glucose-stimulated islet growth and beta cell function.
Glucose concentration regulates expression of Igf1r, Insr and Insrr
In our study, rat islets increasingly expressed Igf1r, Insr and Insrr at high vs low glucose concentrations. In addition, expression of these receptors was paralleled by expression of the metabolic gene Pklr, a key enzyme of glycolysis. Until now, high glucose concentration has been thought to trigger specific beta cell functions, predominantly insulin secretion and preproinsulin expression [3, 27]. Furthermore, distinct roles of IGF-IR and IR signalling on glucose-stimulated insulin secretion were previously investigated by Da Silva Xavier et al. [11] using an RNA-silencing technique by small-interfering-RNAs in pancreatic MIN6 beta cells. They found that a reduction of IR expression strongly inhibited insulin secretion and blocked mRNA expression of the glucose-regulated genes INS, insulin promoter factor 1 (IPF1) and glucokinase (GCK). Moreover, loss of Igf1r inhibited glucose-dependent insulin secretion, glucose-dependent ATP accumulation, but not expression of the aforementioned glucose-regulated genes [2]. Therefore, beta cells require function of both IR and Igf-IR for the complete process of glucose-dependent insulin secretion. Our study indicates that glucose concentration sufficiently regulates expression of Igf1r and Insr and therefore increases expression of receptor proteins that are mandatory for glucose-dependent beta cell function. Furthermore, chronic hyperglycaemia reduced absolute levels of Insr expression in RIN beta cells and in human islets, but also increased relative abundance of its exon 11+ splicing variant that binds insulin with lower affinity. Specific insulin signalling was decreased and this specific regulation of IR expression and shift in alternative splicing was claimed to play a role in beta cell dysfunction caused by chronic hyperglycaemia [33]. In contrast to IR and IGF-IR, detailed function of IRR that is also strongly regulated by glucose remains unclear. The INSRR is highly expressed in pancreatic beta cells and might interact with IGF-IR and IR signalling either by direct signalling via IRS-1 or IRS-2 or by formation of heterotetramers with IR or IGF-IR [19, 20]. Nevertheless, until now no morphological or functional abnormalities had been found in islets of IRR knock-out mice and therefore its physiological function in pancreatic beta cells remains unclear [21].
Importantly, we found that gene expression of Igf1r, Insr and Insrr is differentially regulated in intact rat islets and in INS-1E beta cells. Most prominent was an upregulation of Igf1r, Insr and Insrr by high glucose concentrations in rat islets which was not found in INS-1E cells. Moreover, Pklr was upregulated by high glucose concentrations in both islets and INS-1E cells and therefore glucose-dependent regulation of this metabolic gene was not different.
Pathogenesis of newborns from diabetic mothers indicates that glucose is a strong regulator of beta cell mass in humans in vivo. These babies are exposed to chronic hyperglycaemia in utero, developing islet hyperplasia and hyperinsulinism with a high risk of displaying severe hypoglycaemia postnatally [34]. Moreover, in vivo animal experiments and in vitro studies indicate that IGF-I-, IGF-II- and growth-hormone-dependent proliferation of beta cells strongly depends on glucose concentration and requires glucose metabolism [4, 5]. It was found that high glucose concentration increased protein kinase B and protein kinase C activation, ERK1/2 phosphorylation and IGF-I-induced phosphatidylinositol-3 kinase association with IRS-1 and IRS-2. Thus, the IGF-I signalling network and finally IGF-I-induced beta cell proliferation was influenced at several levels by glucose concentration [35, 36]. Our findings point to a new mechanism by which glucose could regulate beta cell mass. Upregulation of gene and protein expression of IGF-IR, IR and maybe other beta cell growth factor receptors enhances signalling mechanisms that promote proliferation in response to high glucose concentrations in islets. By this, the pancreatic islet mass could adapt to higher insulin requirements in states of absolute or relative insulin deficiency, like high nutrient supply, diabetes or pancreas injury. This hypothesis was supported by the finding that in islets gene expression of Igf1r was strongly upregulated by high glucose concentration and thus could contribute to higher beta cell survival [1, 5]. Furthermore, Hoorens et al. [6] demonstrated that increased glucose levels prevent apoptosis in isolated beta cell cultures. Based on their findings and our new results, increased expression of Igf1r and Insr could represent signalling systems mediating these anti-apoptotic effects of high glucose in islets and differentiated beta cells.
AMPK-activation increases Igf1r, Insr and Insrr expression
Recent studies demonstrated that glucose depletion increases cellular AMP levels and activates AMPK in several beta cell lines, including HIT-T15 and MIN-6 insulinoma cells [24, 37, 38]. In contrast to some data obtained in these cell models showing a more permanent AMPK phosphorylation, we found only a short-term suppression of AMPK by high glucose in INS-1E cells. In contrast to glucose effects, AICAR permanently and strongly activated AMPK up to 24 h. We tested whether direct stimulation of AMPK by AICAR interferes with glucose-regulated receptor gene and protein expression. PKLR, ALDO-B or SLC2A2 are known glucose- and AICAR-regulated genes in beta cells and their respective gene products are involved in glucose transport and metabolism. Expression of these genes is regulated by the transcription factor HNF4α, which is a substrate of AMPK [27]. Phosphorylation of HNF4α by AMPK inhibits the formation of HNF4α homodimers and thus subsequently prevents HNF4α binding to DNA, while gene expression of HNF4α is not strongly regulated by AMPK activation [39]. Thus AMPK/HNF4α could represent an important axis of glucose-dependent gene regulation, not only for genes of glucose metabolism, but also for genes regulating beta cell growth, survival and differentiation. We found it remarkable that AMP-kinase activation by AICAR strongly increased mRNA and protein expression first of all of IGF-IR, but also of IR at high glucose concentrations. Thus, AMPK might not only regulate gene expression of proteins for glucose transport and metabolism like GLUT-2 or L-PK, but furthermore of growth factor receptors that are involved in regulation of beta cell mass, beta-cell-specific glucose sensing and insulin secretion. Nevertheless, AICAR and its respective monophosphate ZMP do not selectively activate AMPK but also other enzymes regulated by AMP, such as fructose-1,6-biphosphatase [24]. Therefore AICAR incubation mimics low cellular energy charge and high AMP content, which is strongly induced by low glucose levels. Nevertheless, glucose-dependent transcriptional regulation of IGF1R and INSR genes will be important in understanding how pancreatic islets respond to hyperglycaemia acutely by increasing insulin secretion, but also chronically by adapting islet cell mass to glucose levels.
Conclusions
These data indicate that in general expression of Igf1r, Insr and Insrr is regulated by glucose concentration in rat islets and in INS-1E insulinoma cells. Why expression of Igf1r, Insr and Insrr is regulated inversely in islets and INS-1E beta cells at high or low glucose concentrations, remains unexplained. INS-1E cells stem from clonal selection of radiation-induced insulinoma cells that slowly proliferate and are less glucose-sensitive and differentiated than beta cells within islets. The switch in glucose-dependent receptor expression might be part of their transformation and regulate tumour cell growth vs highly glucose-sensitive insulin secretion. We find that our descriptive and functional comparison will be helpful for developing new hypotheses of glucose-regulated beta cell growth and function. This will be important for both differentiated islets and transformed insulinoma cells, like INS-1E cells. AMPK activation by AICAR has been shown to increase expression of Igf1r, Insrr and Insrr in INS-1E cells. But whether AMPK is the key regulator of glucose-mediated receptor regulation remains to be investigated.
Abbreviations
- AICAR:
-
5-amino-imidazolecarboxamide riboside
- AMPK:
-
AMP-activated protein kinase
- HNF4:
-
hepatocyte nuclear factor-4
- IGF-II/M6PR:
-
IGF-II/mannose-6-phosphate receptor
- IGF-IR:
-
IGF-I receptor
- IR:
-
insulin receptor
- IRR:
-
insulin-receptor-related receptor
- IRS-1:
-
insulin receptor substrate-1
- IRS-2:
-
insulin receptor substrate-2
- L-PK:
-
liver-type pyruvate kinase
- ZMP:
-
AICAR monophosphate
References
Bonner-Weir S, Deery D, Leahy JL, Weir GC (1989) Compensatory growth of pancreatic beta-cells in adult rats after short-term glucose infusion. Diabetes 38:49–53
Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A (2003) Specific and combined effects of insulin and glucose on functional pancreatic beta-cell mass in vivo in adult rats. Endocrinology 144:2717–2727
Kulkarni RN (2004) The islet beta-cell. Int J Biochem Cell Biol 36:365–371
Cousin SP, Hugl SR, Myers MG Jr, White MF, Reifel-Miller A, Rhodes CJ (1999) Stimulation of pancreatic beta-cell proliferation by growth hormone is glucose-dependent: signal transduction via janus kinase 2 (JAK2)/signal transducer and activator of transcription 5 (STAT5) with no crosstalk to insulin receptor substrate-mediated mitogenic signalling. Biochem J 15:649–658
Raile K, Berthold A, Banning U, Horn F, Pfeifer G, Kiess W (2003) IGFs, basic FGF, and glucose modulate proliferation and apoptosis induced by IFNγ but not by IL-1β in INS-1E rat β-cells. Horm Metab Res 35:1–8
Hoorens A, Van de Casteele M, Kloppel G, Pipeleers D (1996) Glucose promotes survival of rat pancreatic beta cells by activating synthesis of proteins which suppress a constitutive apoptotic program. J Clin Invest 98:1568–1574
Hanze J, Berthold A, Klammt J et al (1999) Cloning and sequencing of the complete cDNA encoding the human insulin receptor related receptor. Horm Metab Res 31:77–79
Ullrich A, Gray A, Tam AW et al (1986) Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J 5:2503–2512
Shepherd PR (2004) Secrets of insulin and IGF-1 regulation of insulin secretion revealed. Biochem J 377:1–2
Kiess W, Blickenstaff GD, Sklar MM, Thomas CL, Nissley SP, Sahagian GG (1988) Biochemical evidence that the type II insulin-like growth factor receptor is identical to the cation-independent mannose 6-phosphate receptor. J Biol Chem 263:9339–9344
Da Silva Xavier G, Qian Q, Cullen PJ, Rutter GA (2004) Distinct roles for insulin and insulin-like growth factor-1 receptors in pancreatic beta-cell glucose sensing revealed by RNA silencing. Biochem J 377:149–158
Harbeck MC, Louie DC, Howland J, Wolf BA, Rothenberg PL (1996) Expression of insulin receptor mRNA and insulin receptor substrate 1 in pancreatic islet beta-cells. Diabetes 45:711–717
Zhang Q, Tally M, Larsson O et al (1997) Insulin-like growth factor II signaling through the insulin-like growth factor II/mannose-6-phosphate receptor promotes exocytosis in insulin-secreting cells. Proc Natl Acad Sci U S A 94:6232–6237
George M, Ayuso E, Casellas A et al (2002) Beta cell expression of IGF-I leads to recovery from type 1 diabetes. J Clin Invest 109:1153–1163
Hill DJ, Strutt B, Arany E et al (2000) Increased and persistent circulating insulin-like growth factor II in neonatal transgenic mice suppresses developmental apoptosis in the pancreatic islets. Endocrinology 141:1151–1157
Jackerott M, Baudry A, Lamothe B et al (2001) Endocrine pancreas in insulin receptor-deficient mouse pups. Diabetes 50:S146–S149
Kulkarni RN, Bruning JC, Winnay JN et al (1999) Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96:329–339
Kulkarni RN, Holzenberger M, Shih DQ et al (2002) Beta-cell-specific deletion of the IGF1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat Genet 31:111–115
Hirayama I, Tamemoto H, Yokota H et al (1999) Insulin receptor-related receptor is expressed in pancreatic beta-cells and stimulates tyrosine phosphorylation of insulin receptor substrate-1 and -2. Diabetes 48:1237–1242
Jui HY, Accili D, Taylor SI (1996) Characterization of a hybrid receptor formed by dimerization of the insulin receptor-related receptor (IRR) with the insulin receptor (IR): coexpression of cDNAs encoding human IRR and human IR in NIH-3T3 cells. Biochemistry 35:14326–14330
Kitamura T, Kido Y, Nef S et al (2001) Preserved pancreatic beta-cell development and function in mice lacking the insulin receptor-related receptor. Mol Cell Biol 21:5624–5630
Kido Y, Nakae J, Hribal ML et al (2002) Effects of mutations in the insulin-like growth factor signaling system on embryonic pancreas development and beta-cell compensation to insulin resistance. J Biol Chem 277:36740–36747
Hardie GD, Scott JW, Pan DA, Hudson ER (2003) Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett 546:113–120
Da Silva Xavier G, Leclerc I, Salt IP et al (2000) Role of AMP-activated protein kinase in the regulation by glucose of islet beta cell gene expression. Proc Natl Acad Sci U S A 97:4023–4028
Wang H, Maechler P, Antinozzi PA, Hagenfeldt KA, Wollheim CB (2000) Hepatocyte nuclear factor 4alpha regulates the expression of pancreatic beta-cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J Biol Chem 275:35953–35959
Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K (2002) Mechanism for fatty acid ‘sparing’ effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem 277:3829–3835
Leclerc I, Lenzner C, Gourdon L et al (2001) Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes 50:1515–1521
Kemp BE, Stapleton D, Campbell DJ et al (2003) AMP-activated protein kinase, super metabolic regulator. Biochem Soc Trans 31:162–168
Kloting I, Vogt L (1991) BB/O(ttawa)K(arlsburg) rats: features of a subline of diabetes-prone BB rats. Diabetes Res 18:79–87
Hehmke B, Kohnert KD, Odselius R (1986) The use of a new dextran gradient medium for rapid isolation of functionally intact neonatal rat pancreatic islets. Diabetes Res 3:13–16
Fasshauer M, Klein J, Neumann S, Eszlinger M, Paschke R (2001) Tumor necrosis factor alpha is a negative regulator of resistin gene expression and secretion in 3T3-L1 adipocytes. Biochem Biophys Res Commun 288:1027–1031
Krowczynska AM, Coutts M, Makrides S, Brawerman G (1989) The mouse homologue of the human acidic ribosomal phosphoprotein PO: a highly conserved polypeptide that is under translational control. Nucleic Acids Res 17:6408
Hribal ML, Perego L, Lovari S et al (2003) Chronic hyperglycemia impairs insulin secretion by affecting insulin receptor expression, splicing, and signaling in RIN beta cell line and human islets of Langerhans. FASEB J 17:1340–1342
Schwartz R, Teramo KA (1999) What is the significance of macrosomia? Diabetes Care 22:1201–1205
Hugl SR, White MF, Rhodes CJ (1998) Insulin-like growth factor I (IGF-I)-stimulated pancreatic beta-cell growth is glucose-dependent. Synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J Biol Chem 273:17771–17779
Lingohr MK, Dickson LM, McCuaig JF, Hugl SR, Twardzik DR, Rhodes CJ (2002) Activation of IRS-2-mediated signal transduction by IGF-1, but not TGF-alpha or EGF, augments pancreatic beta-cell proliferation. Diabetes 51:966–976
Salt IP, Johnson G, Ashcroft SJ, Hardie DG (1998) AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic beta cells, and may regulate insulin release. Biochem J 335:533–539
Kefas BA, Cai Y, Ling Z, Heimberg H, Hue L, Pipeleers D, Van de Casteele M (2003) AMP-activated protein kinase can induce apoptosis of insulin-producing MIN6 cells through stimulation of c-Jun-N-terminal kinase. J Mol Endocrinol 30:151–161
Leff T (2003) AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Biochem Soc Trans 31:224–231
Acknowledgements
This study was supported by the International Eli Lilly Foundation (to K. Raile), the German Diabetes Association (DDG) (to K. Raile and A. Garten), and in part grants from BMBF, Germany (IZKF Leipzig, grant B12, to W. Kiess).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Raile, K., Klammt, J., Laue, S. et al. Glucose concentration and AMP-dependent kinase activation regulate expression of insulin receptor family members in rat islets and INS-1E beta cells. Diabetologia 48, 1798–1809 (2005). https://doi.org/10.1007/s00125-005-1860-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-1860-x