
Abstract
The colonic mucosa provides a vital defensive barrier separating the body from the microbial populations residing in the intestinal lumen. Indeed, growing evidence shows that loss of this barrier may cause disease or exacerbate disease progression. The loss of barrier integrity increases the translocation of bacterial antigens and stimulates inflammation in the intestinal mucosa, which is the central pathological feature of inflammatory bowel diseases (IBDs). This review focuses on how intestinal mucus and intercellular tight junctions (TJs) act together to maintain the integrity of the colonic barrier and how barrier integrity is dysregulated in IBD.
Similar content being viewed by others

Introduction
Inflammatory bowel diseases (IBD), encompassing Crohn’s disease (CD) and ulcerative colitis (UC), are characterized by chronic or intermittent inflammation of the intestinal mucosa [1]. IBD is an idiopathic and multifactorial condition, and recent studies indicate that both genetic and environmental factors contribute to the disease [2, 3]. During the etiology of IBD, aberrant immune responses triggered by the microbiota itself or excessive leakage of bacterial antigens into the mucosa cause inflammatory responses that progressively degrade the intestinal epithelia. This in turn permits more antigen leak and exacerbates inflammation, further compromising barrier integrity. The mucosal barrier integrity, therefore, is a key determinant of disease initiation, progression, and severity in IBD patients. Disrupting the inflammatory feedback cycle is a fundamental goal of IBD therapy. In experimental models, exposure of epithelial cells to proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) causes cell death, alters the production of secreted mucins, and disrupts the epithelial barriers) [4,5,6,7,8]. Notably, the anti-TNF-α monoclonal antibody, infliximab, a treatment for advanced IBD, short-circuits this inflammatory feedback loop by limiting immune activation and mitigating cytotoxic effects [5].
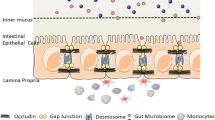
Epithelial tissue acts not only to separate essential internal organ systems from the outside world but also facilitates communication with the microbiota and absorbs nutrients. Additional features of the colonic barrier include secreted mucus and water absorption. The epithelial layer is composed of a simple monolayer of around 20 billion contiguous cells [9], which, like the skin, continually regenerates. Stem cells, located at the base of the crypts of Lieberkühn, produce daughter cells that differentiate. As the progeny cells migrate toward the luminal surface, they lose their proliferative capacity and differentiate into specialized cells, which include colonocytes and mucin producing goblet cells (Fig. 1) [10, 11]. Cells at the luminal surface eventually undergo programmed cell death (apoptosis) and are shed (Fig. 1). From birth to death, the regenerative process is complete within a few days [12].

Protective layers of the colonic mucosal barrier. The outer mucus layer, composed of Muc2 and various carbohydrate modifications, interacts with colonic microflora, while the density of the inner layer prevents bacterial penetration. The innermost mucus layer, the glycocalyx, is attached to the plasma membrane. Together, the inner and outer mucus layer limit abrasion and trap bacteria, thereby restricting their contact with the epithelia. In general, the mucus limits contact of bacteria with underlying epithelial cells but does not restrict access of bacterial metabolites. The epithelial paracellular barrier is composed of intercellular contacts called tight junctions (TJs). TJ strands, composed of proteins called claudins, connect apposing cells and occlude the paracellular space. Some claudins form ion pores within the TJ (pore pathway, inset), which selectively permit ion and water exchange. However, bacterial products may breach TJ defenses upon separation (a) or rupture of the TJ strands (b). Epithelial cell turnover likely helps to remove attached bacteria, and mucus flux ensures trapped bacteria are eliminated. Stem cells residing in the crypt bases produce progeny that migrate toward the lumen surface. While doing so they differentiate, producing goblet cells that secrete mucins. Differentiating cells also change the complement of claudins that they express, such that pore forming claudins are more highly expressed in the crypt-base compared to the luminal surface [54]
Mature cells facing the lumen, termed surface cells, are in close apposition to layers of mucus. The cells and the mucus together form a layered barrier separating the body from the colonic microflora and limiting influx of bacteria and bacterial antigens. The combination of the epithelial sheet and the mucus layers forms a biologically flexible and environmentally responsive barrier to luminal contents. Indeed, growing evidence shows that loss of barrier integrity may cause disease or exacerbate disease progression (reviewed in [13, 14]). This review focuses on how the mucus and TJs together provide a layered defensive barrier in the colon and how their dysregulation contributes to IBD.
Mucins separate the epithelia from the intestinal lumen
The colonic mucosa secretes copious amounts of mucus, which is composed of complex and extensive O-linked oligosaccharide modifications on a mucin protein backbone. These glycoproteins form large disulfide-linked macromolecules, and upon release from goblet cell granules into the lumen, become hydrated and expand to form a net-like gel [15]. Whereas the small intestine contains a single layer of loose unattached mucus, mucus in the colon is organized into three distinct layers. Membrane-anchored mucins associated with the colonic epithelial cells form the glycocalyx. The glycocalyx gives way to a second tightly crosslinked inner layer primarily composed of the mucin protein MUC2. The outermost layer, generated by proteolysis of the inner layer, is less dense and less viscous. A more detailed description of mucin organization in the colon is reviewed elsewhere [16,17,18].
These mucus layers serve to separate bacteria in the lumen from the epithelia in several ways. First, the mucus layers achieve different densities, such that commensal flora or even pathogens can reside within the low density outer layer [16, 18] but are generally excluded from the high density inner layer and glycocalyx [19]. The outer mucus layers contain diverse carbohydrate motifs, immunoglobulins, and other proteins that serve as binding sites for bacteria [20, 21]. A second means of separating the bacteria is mucus turnover. As secreted mucus is extruded, proteolyzed in the inner layer, and ultimately secreted into the lumen, bound bacteria are expelled and removed from the body by intestinal peristalsis. The mucus layers turn over in a matter of hours, providing dynamic removal of bacteria and limiting their access to the epithelium [22]. Finally, the mucus serves as a lubricant to prevent feces from abrading and tearing the epithelia [22]. Thus, the mucus serves to limit access of bacteria to the epithelia. It is important to note that the density of the mucus does not preclude access of secreted bacterial metabolites or toxins to the epithelia based on size alone. Given the importance of these molecules in epithelial barrier integrity, a role for the mucus in regulating their access promises to be an increasingly important area of investigation.
Defective mucin production or processing has been linked to human IBD. UC patients have fewer goblet cells and decreased synthesis and secretion of MUC2, especially during episodes of severe disease. This allows direct contact of the colonic microbiota with the epithelial barrier [23]. In addition, lower levels of the goblet cell differentiation factors HATH1 and KLF4 were evident in biopsies from patients with active UC [24]. Furthermore, genome-wide association studies (GWAS) have implicated mutations in the MUC genes in IBD pathogenesis, including the membrane-anchored MUC3 and MUC19 [2, 25]. Moreover, variants of MUC2, the major secreted mucin in the human colon, have been found in IBD cases [26].
Unlike UC patients, CD patients exhibit increased mucus production [24, 27]. However, despite the increased levels, the mucus barrier still fails to restrict bacterial access to the epithelia [28]. These observations suggest that secondary structural modifications, in addition to mucin levels, are critical for function of the mucus barrier. In this regard, it has been proposed that increased bacterial access is due to impaired mucin processing in CD patients, which may affect the length of glycans attached by O-glycosylation [8, 23]. Notably, proper glycosylation, sulfation, and sialylation are essential for the viscoelastic functions of mucus. Humans and mice with IBD have been found to have higher levels of sulfide, a product of sulfate reducing bacteria, that could reduce the disulfide bond between mucins and degrade the mucus network, thus allowing increased microbial contact with the host [29, 30]. Accordingly, glycosylation and sulfation defects have also been found in a UC cohorts, indicating that mucus modification may play a role in limiting barrier function in disease [31,32,33]. More specifically, mutations in core 1 β3GalT-specific molecular chaperone (Cosmc), a chaperone for the T-synthase glycosyltransferase responsible for the synthesis of the O-glycans on mucin proteins, have also been associated with IBD in GWAS studies [34]. The location of Cosmc on X chromosome may provide an explanation for the male gender bias of IBD [35].
Experimental disease models in mice further strengthen the role the mucus barrier and mucins can play in the prevention or pathogenesis of IBD. For example, 5 weeks after birth, Muc2 knockout animals (Muc2−/−) develop spontaneous colitis and display increased susceptibility to experimental DSS colitis, presumably due to the direct contact of intestinal microbiota with the epithelia [36]. Animal models also demonstrate that glycoprotein modification is crucial to intestinal homeostasis. Mice lacking core 1-derived O-glycans, recapitulating defects in humans with mutations in Cosmc, show loss of mucus complexity and rapid spontaneous colitis [31]. It is important to note that the outer and inner mucus layers of the colon are almost entirely composed of Muc2, and it is expected that Muc2−/− strains would then depend only on the glycocalyx for fecal lubrication [21]. In summary, the secreted mucus barrier appears to function as a means of preventing or limiting contact of bacteria and bacterial antigens with epithelial cells.
The outer layer of colonic mucus is a habitat for both indigenous and transient microorganisms, called autochthonous and allochthonous, respectively. Changes in the resident autochthonous bacteria appear to have more impact on the host’s health than do changes in transient luminal allochthonous bacteria found in the fecal matter [37]. An imbalance of the microbiota, a state referred to as dysbiosis, is characteristic of IBD, indicating the importance of maintaining the appropriate bacteria in the intestinal mucus [38]. Mucus provides nutrients to bacteria, including amino acids and sugars, which are especially important for those bacteria capable of degrading the glycans on the mucin backbone [39, 40]. Akkermansia muciniphila is a mucus-degrading bacterium underrepresented in many disease states including IBD, obesity, and type 2 diabetes, and numerous studies have correlated the presence of A. muciniphila with a healthy mucosa [41, 42]. Additionally, providing mice with A. muciniphila during high-fat diet (HFD) feeding, which normally results in decreased barrier integrity, led to a restored barrier, increased goblet cell numbers, and prevention of metabolic endotoxemia [42] [43] [44]. Interestingly, bacteriophages within the mucus layer may also dictate the abundance and diversity of bacteria found in the intestine [38]. One study of an IBD cohort demonstrated an inverse correlation between bacteriophage expansion and diversification on the one hand and bacterial diversity on the other, raising the possibility that bacteriophages may contribute to the dysbiotic state in IBD [45]. Taken together, these studies demonstrate the importance of intestinal mucus in supporting growth of protective commensal bacteria as well as facilitating repopulation and maintaining commensal homeostasis to prevent dysbiosis. Accordingly, disruption of the mucus barrier may result in dysbiosis.
TJs form a paracellular seal
Epithelial cells are networked together through proteinaceous adhesive contacts called junctions, which both join cells together and provide a paracellular seal. The seal between cells requires tight junctions (TJs), a specialized multipurpose adhesion that simultaneously occludes the paracellular space, dictates ion flux across the tissue, and maintains cellular polarity. The TJs are positioned at the boundary of the apical and lateral membrane surfaces of adjacent epithelial cells in the colon and consist of 5–7 membrane fusion sites called “kissing points” [46, 47]. The entire circumference of each cell is joined to apposing cells via an adhesive TJ band, called a strand. TJ strands in the colon are not linear but rather highly branched structures that form anastomosed webs that extend several hundred nanometers laterally from the apex of the cell. All epithelial cells that line the intestine are joined in this manner.
TJs regulate the flux of ions and solutes on the one hand, termed the “gate function,” and maintain cell polarity on the other, termed the “fence function” [48, 49]. Thus, TJs serve as a barrier to bacteria and bacterial products while also corralling apical plasma membrane proteins, and presumably the glycocalyx mucins, at the lumen-facing domain of the epithelial cell.
The claudin family proteins are essential components of TJs. Claudins form TJ strands by polymerizing within the plasma membrane and dimerizing with claudins on apposing cells, across the extracellular space, to generate the paracellular seal. There are 24 claudin genes in humans, with multiple claudins expressed within any given cell [50]. Importantly, several claudin proteins dimerize to form charge and size-selective ion pores that are vital for ionic homeostasis in epithelial tissues. For example, mice deficient in both claudin 2 and 15 mice fail to equilibrate sodium levels in the luminal space of the small bowel, which leads to low nutrient absorption, wasting disease, and premature death [51]. Other claudins, such as claudin 4, appear to promote a tighter seal; claudin 4 does not form ion pores within the TJ but appears to exclude pore-forming claudin 2 [52]. The permeability of the TJ is thought to derive at least in part from the relative amounts of amounts of pore forming or sealing claudins within the stands, as well as the architecture of the strands, particularly the complexity and numbers of TJ strands [49, 53].
Different complements of claudins are expressed at different levels in epithelia along the length of the intestinal tract, as well as within the intestinal crypts themselves [52] (Fig. 1). For example, our recent studies in mice indicate that 11 claudins are expressed as a gradient within the crypts (Fig. 1) [54]. In general, “leaky” pore forming claudins are restricted to the colonic crypt base, whereas “tight” sealing claudins are more prominently expressed in surface cells facing the lumen.
At the molecular level, the TJ is a highly diverse structure composed of both transmembrane and cytoplasmic proteins [55, 56]. Besides claudins, there exist three additional classes of transmembrane proteins in the TJ: occludin, tricellulin, and junctional adhesion molecules (JAMs) [53, 57,58,59]. A dense “plaque” of scaffolding molecules is anchored to the transmembrane proteins, which include the Zonula Occludins (ZO) and MAGUK family proteins (reviewed in [49]). These scaffolding proteins link the transmembrane proteins to kinases and signaling molecules that localize at the junction. These molecules in turn control not only cell-cell contacts but also the actin polymerization machinery and contractility apparatus of apically situated actin and myosin [49, 60]. Scaffold proteins have different affinities for claudins and may regulate the types of claudins in the TJs [61]. Likewise, the contractile machinery appears to regulate localization of claudins within the TJs [62,63,64,65]. In summary, this molecular signaling apparatus controls claudin localization and function, and thus the permeability of the epithelial barrier.
In addition to forming ion pores, claudin strands have a poorly understood mechanism that permits small molecules to traverse the TJ, termed the paracellular “leak pathway” [66,67,68]. There is accumulating evidence that TJ strands themselves are dynamic and frequently break, reform, and exchange claudin proteins in response to physiological, environmental, and pathogenic stimuli [52, 63]. However, it remains to be established whether the paracellular leakage results from separation of transcellular dimers, strand breakage, or some other unknown mechanism. Interestingly, several pathogens, including both bacteria and viruses, have evolved means to traverse the paracellular junctions by disrupting claudins, or the actin structures that provide structural integrity to the cell and the TJs (reviewed in [69]). For example, the bacterium Clostridium perfringes secretes a toxin that binds claudins 3 and 4, whereas Hepatitis C virus interacts with claudin 1 [69]. In summary, the composition and numbers of the TJ strands, the type of claudins that compose them, their localization within the intestinal tract, and the intracellular signaling apparatus all contribute to the permeability of the intestinal barrier.
Several lines of evidence implicate dysregulation of the mucosal barrier, and of TJ architecture and claudin expression in particular, in the etiology of IBD [47, 70]. GWAS studies have identified several genes that link TJ function to IBD [2, 71]. Of the IBD implicated genes, one of the best understood is hepatocyte nuclear factor alpha (HNF4a) [72]. HNF4a is a transcription factor involved in the maturation of colonocytes as they migrate out of the crypts. HNF4a regulates claudin expression, including claudin 7 [73]. Moreover, multiple studies have demonstrated a change in TJ transmembrane proteins in IBD patients. Tricellulin, a specialized occludin-like molecule responsible for sealing the TJ at tricellular contacts, is decreased in UC [74, 75]. Moreover, expression of the sealing claudins 1 and 4 is suppressed in IBD patients [70, 76]. Furthermore, upregulation of claudin 2 expression and downregulation of claudin 5 and 8 correlate with barrier dysfunction and active CD [77]. A more comprehensive review of the TJ and TJ-associated proteins dysregulated in IBD can be found elsewhere [75, 78, 79].
Disease phenotypes of human IBD are recapitulated in mice with genetic deficiencies similar to those found in human patients and confirm the importance of altering TJs and barrier integrity in IBD pathogenesis. For example, mice deficient in claudin 7 in the colon develop lethal colitis soon after birth [80, 81]. Notably, based on freeze fracture EM analysis, TJs of wild type and claudin 7 knockout animals have nearly identical structure, yet the character and function of the TJ appears compromised. Without claudin 7, the epithelial barrier is more permeant to small molecules (~400 Da), although the flux of larger molecules (~4 kDa) and the overall balance of Na+ and Cl− were unchanged [80]. Therefore, claudin 7 appears to function to selectively regulate permeability of small molecules, and its dyregulation is sufficient to cause disease. The observation that loss of claudin 7 disrupts the TJ seal raised the possibility that this claudin might also limit flux of bacterial antigens. Accordingly, antibiotics ameliorated colitis in mice with claudin 7 deficiency, whereas addition of bacterial antigen (fMLF) reversed this protective effect [80]. Together, these data support a model of IBD pathogenesis in which dysregulation of TJs facilitates leakage of luminal antigens across the epithelial barrier that trigger inflammation and initiate colitis. Master regulators of claudin expression have also been implicated in mouse models of IBD. As in humans, Hnf4a regulates expression of claudin genes in mice [82, 83]. Accordingly, loss of function alleles of Hnf4a in mice results in dysregulation of claudin expression and spontaneous colitis [84].
The mucosal barrier, inflammation, and IBD
The mucosal barrier is not a static structure, and epithelial tight junctions and mucus production both respond to inflammatory stimuli. For example, upon infection, the inflammatory cytokine TNF-α increases epithelial permeability through alterations in TJ function, structure, and dynamics [52, 85]. Yet, TNF-α also increases mucus production by goblet cells to limit the inflammatory response by stemming influx of bacteria through the mucus layers.
Importantly, sustained inflammation or protracted dysregulation of barrier integrity initiates or exacerbates IBD. In this regard, loss or dysregulation of either the mucus or the TJs suffice to cause colitis, an effect that depends upon bacterial or antigen translocation. Thus, mice lacking claudin 7, Hnf4a, and Muc2 all develop spontaneous colitis [72, 80, 86]. The observation that symptoms are relieved under germ-free conditions or after treatment with antibiotics highlights the non-redundant role of the mucus as well as TJs in limiting access of microbes and microbial products to the body and the contribution of bacterial antigens to colitis. Additionally, the mucus barrier and TJs are interdependent such that loss of one diminishes the other. In this regard, Muc2−/− mice display increased epithelia barrier permeability and dysregulated claudin gene expression in addition to defects in the mucus [86]. Likewise, numbers of mucin-producing goblet cells and mucus are diminished in Hnf4a−/− mice [84]. Such interdependence could result from dysregulation of signals that coordinate both the mucus and TJs. Alternatively, inflammation associated with either knockout phenotype could damage the remaining barrier. In any case, such interdependence may facilitate feedback inflammatory responses that perpetuate IBD.
Summary and future directions
The epithelial TJs and the three mucus layers cooperate to form a highly integrated barrier system that together limit access of luminal contents to the body proper. The molecular constituents of this barrier are still being identified and their functions elucidated. Nevertheless, through in vivo and in vitro studies in experimental models, as well as studies of human IBD, an integrated understanding of mucosal barrier function is beginning to emerge. The capacity of the mucus to prevent abrasion and trap bacteria represents the first line of defense, while the paracellular TJ barrier prevents leakage of bacterial antigens from the lumen into the body. Furthermore, the rapid turnover of both mucus and lumen-facing epithelial cells ensures that bacteria that do interact with these structures are constantly being evicted. This multilayer system thus functions to limit bacterial contact with the host while still permitting access of small molecules, including microbial metabolites.
A comprehensive understanding of the epithelial barrier system and its relationship to commensal microbes and to the immune system may provide a more integrated approach to treatment of IBDs. For example, strategies aimed at strengthening both the mucus and epithelia barriers and thereby reducing exposure to inflammatory antigens, coupled with therapeutics that reduce susceptibility of the mucosa to damage caused by bacteria or by inflammatory responses, may restore the intestinal tract of IBD patients to a more benevolent state.
References
de Souza HS, Fiocchi C (2016) Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol 13:13–27
McCole DF (2014) IBD candidate genes and intestinal barrier regulation. Inflamm Bowel Dis 20:1829–1849
Basson A, Trotter A, Rodriguez-Palacios A, Cominelli F (2016) Mucosal interactions between genetics, diet, and microbiome in inflammatory bowel disease. Front Immunol 7:290
Capaldo CT, Nusrat A (2009) Cytokine regulation of tight junctions. Biochim Biophys Acta 1788:864–871
Blander JM (2016) Death in the intestinal epithelium-basic biology and implications for inflammatory bowel disease. FEBS J 283:2720–2730
Kalliolias GD, Ivashkiv LB (2016) TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol 12:49–62
Enss ML, Cornberg M, Wagner S, Gebert A, Henrichs M, Eisenblatter R, Beil W, Kownatzki R, Hedrich HJ (2000) Proinflammatory cytokines trigger MUC gene expression and mucin release in the intestinal cancer cell line LS180. Inflamm Res 49:162–169
Smirnova MG, Birchall JP, Pearson JP (2000) TNF-alpha in the regulation of MUC5AC secretion: some aspects of cytokine-induced mucin hypersecretion on the in vitro model. Cytokine 12:1732–1736
Nguyen H, Loustaunau C, Facista A, Ramsey L, Hassounah N, Taylor H, Krouse R, Payne CM, Tsikitis VL, Goldschmid S et al (2010) Deficient Pms2, ERCC1, Ku86. CcOI in field defects during progression to colon cancer J Vis Exp DOI. doi:10.3791/1931
Noah TK, Shroyer NF (2013) Notch in the intestine: regulation of homeostasis and pathogenesis. Annu Rev Physiol 75:263–288
Biswas S, Davis H, Irshad S, Sandberg T, Worthley D, Leedham S (2015) Microenvironmental control of stem cell fate in intestinal homeostasis and disease. J Pathol 237:135–145
Barker N (2014) Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol 15:19–33
Nalle SC, Turner JR (2015) Intestinal barrier loss as a critical pathogenic link between inflammatory bowel disease and graft-versus-host disease. Mucosal Immunol 8:720–730
Weber CR, Turner JR (2007) Inflammatory bowel disease: is it really just another break in the wall? Gut 56:6–8
Johansson ME, Sjovall H, Hansson GC (2013) The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol 10:352–361
Linden SK, Sutton P, Karlsson NG, Korolik V, McGuckin MA (2008) Mucins in the mucosal barrier to infection. Mucosal Immunol 1:183–197
Perez-Vilar J, Hill RL (1999) The structure and assembly of secreted mucins. J Biol Chem 274:31751–31754
McGuckin MA, Linden SK, Sutton P, Florin TH (2011) Mucin dynamics and enteric pathogens. Nat Rev Microbiol 9:265–278
Johansson ME, Larsson JM, Hansson GC (2011) The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A 108(Suppl 1):4659–4665
Lecuit M, Dramsi S, Gottardi C, Fedor-Chaiken M, Gumbiner B, Cossart P (1999) A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J 18:3956–3963
Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC (2008) The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A 105:15064–15069
Johansson ME (2012) Fast renewal of the distal colonic mucus layers by the surface goblet cells as measured by in vivo labeling of mucin glycoproteins. PLoS One 7:e41009
Sun J, Shen X, Li Y, Guo Z, Zhu W, Zuo L, Zhao J, Gu L, Gong J, Li J (2016) Therapeutic potential to modify the mucus barrier in inflammatory bowel disease. Nutrients 8
Gersemann M, Becker S, Kubler I, Koslowski M, Wang G, Herrlinger KR, Griger J, Fritz P, Fellermann K, Schwab M et al (2009) Differences in goblet cell differentiation between Crohn’s disease and ulcerative colitis. Differentiation 77:84–94
Kyo K, Muto T, Nagawa H, Lathrop GM, Nakamura Y (2001) Associations of distinct variants of the intestinal mucin gene MUC3A with ulcerative colitis and Crohn’s disease. J Hum Genet 46:5–20
Visschedijk MC, Alberts R, Mucha S, Deelen P, de Jong DJ, Pierik M, Spekhorst LM, Imhann F, van der Meulen-de Jong AE, van der Woude CJ et al (2016) Pooled resequencing of 122 ulcerative colitis genes in a large Dutch cohort suggests population-specific associations of rare variants in MUC2. PLoS One 11:e0159609
Pullan RD, Thomas GA, Rhodes M, Newcombe RG, Williams GT, Allen A, Rhodes J (1994) Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 35:353–359
Johansson ME, Gustafsson JK, Holmen-Larsson J, Jabbar KS, Xia L, Xu H, Ghishan FK, Carvalho FA, Gewirtz AT, Sjovall H et al (2014) Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 63:281–291
Ijssennagger N, Belzer C, Hooiveld GJ, Dekker J, van Mil SW, Muller M, Kleerebezem M, van der Meer R (2015) Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc Natl Acad Sci U S A 112:10038–10043
Ijssennagger N, van der Meer R, van Mil SW (2016) Sulfide as a mucus barrier-breaker in inflammatory bowel disease? Trends Mol Med 22:190–199
Fu J, Wei B, Wen T, Johansson ME, Liu X, Bradford E, Thomsson KA, McGee S, Mansour L, Tong M et al (2011) Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J Clin Invest 121:1657–1666
Raouf AH, Tsai HH, Parker N, Hoffman J, Walker RJ, Rhodes JM (1992) Sulphation of colonic and rectal mucin in inflammatory bowel disease: reduced sulphation of rectal mucus in ulcerative colitis. Clin Sci (Lond) 83:623–626
Larsson JM, Karlsson H, Crespo JG, Johansson ME, Eklund L, Sjovall H, Hansson GC (2011) Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm Bowel Dis 17:2299–2307
Chang D, Gao F, Slavney A, Ma L, Waldman YY, Sams AJ, Billing-Ross P, Madar A, Spritz R, Keinan A (2014) Accounting for eXentricities: analysis of the X chromosome in GWAS reveals X-linked genes implicated in autoimmune diseases. PLoS One 9:e113684
Kudelka MR, Hinrichs BH, Darby T, Moreno CS, Nishio H, Cutler CE, Wang J, Wu H, Zeng J, Wang Y et al (2016) Cosmc is an X-linked inflammatory bowel disease risk gene that spatially regulates gut microbiota and contributes to sex-specific risk. Proc Natl Acad Sci U S A 113:14787–14792
Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Buller HA, Dekker J, Van Seuningen I, Renes IB et al (2006) Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131:117–129
Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M et al (2014) The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15:382–392
Weiss GA, Hennet T (2017) Mechanisms and consequences of intestinal dysbiosis. Cell Mol Life Sci DOI. doi:10.1007/s00018-017-2509-x
Tailford LE, Crost EH, Kavanaugh D, Juge N (2015) Mucin glycan foraging in the human gut microbiome. Front Genet 6:81
Derrien M, van Passel MW, van de Bovenkamp JH, Schipper RG, de Vos WM, Dekker J (2010) Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 1:254–268
Rajilic-Stojanovic M, Shanahan F, Guarner F, de Vos WM (2013) Phylogenetic analysis of dysbiosis in ulcerative colitis during remission. Inflamm Bowel Dis 19:481–488
Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin TH (2010) Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol 105:2420–2428
Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM et al (2013) Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A 110:9066–9071
Shin NR, Lee JC, Lee HY, Kim MS, Whon TW, Lee MS, Bae JW (2014) An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 63:727–735
Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, Kambal A, Monaco CL, Zhao G, Fleshner P et al (2015) Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160:447–460
Farquhar MG, Palade GE (1963) Junctional complexes in various epithelia. J Cell Biol 17:375–412
Schmitz H, Barmeyer C, Fromm M, Runkel N, Foss HD, Bentzel CJ, Riecken EO, Schulzke JD (1999) Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology 116:301–309
Diamond JM (1977) Twenty-first Bowditch lecture. The epithelial junction: bridge, gate, and fence. Physiologist 20:10–18
Zihni C, Mills C, Matter K, Balda MS (2016) Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol 17:564–580
Mineta K, Yamamoto Y, Yamazaki Y, Tanaka H, Tada Y, Saito K, Tamura A, Igarashi M, Endo T, Takeuchi K et al (2011) Predicted expansion of the claudin multigene family. FEBS Lett 585:606–612
Wada M, Tamura A, Takahashi N, Tsukita S (2013) Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 144:369–380
Capaldo CT, Nusrat A (2015) Claudin switching: physiological plasticity of the tight junction. Semin Cell Dev Biol 42:22–29
Tsukita S, Furuse M, Itoh M (2001) Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2:285–293
Lili LN, Farkas AE, Gerner-Smidt C, Overgaard CE, Moreno CS, Parkos CA, Capaldo CT, Nusrat A (2016) Claudin-based barrier differentiation in the colonic epithelial crypt niche involves Hopx/Klf4 and Tcf7l2/Hnf4-alpha cascades. Tissue Barriers 4:e1214038
Tang VW (2006) Proteomic and bioinformatic analysis of epithelial tight junction reveals an unexpected cluster of synaptic molecules. Biol Direct 1:37
Fredriksson K, Van Itallie CM, Aponte A, Gucek M, Tietgens AJ, Anderson JM (2015) Proteomic analysis of proteins surrounding occludin and claudin-4 reveals their proximity to signaling and trafficking networks. PLoS One 10:e0117074
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S (1993) Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 123:1777–1788
Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S (2005) Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol 171:939–945
Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A et al (1998) Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142:117–127
Fanning AS, Van Itallie CM, Anderson JM (2012) Zonula occludens-1 and -2 regulate apical cell structure and the zonula adherens cytoskeleton in polarized epithelia. Mol Biol Cell 23:577–590
Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S (1999) Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol 147:1351–1363
Ivanov AI (2008) Actin motors that drive formation and disassembly of epithelial apical junctions. Front Biosci 13:6662–6681
Van Itallie CM, Tietgens AJ, Anderson JM (2016) Visualizing the dynamic coupling of claudin strands to the actin cytoskeleton through ZO-1. Mol Biol Cell DOI. doi:10.1091/mbc.E16-10-0698
Cunningham KE, Turner JR (2012) Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann N Y Acad Sci 1258:34–42
Quiros M, Nusrat A (2014) RhoGTPases, actomyosin signaling and regulation of the epithelial apical junctional complex. Semin Cell Dev Biol 36:194–203
Weber CR (2012) Dynamic properties of the tight junction barrier. Ann N Y Acad Sci 1257:77–84
Shen L, Weber CR, Raleigh DR, Yu D, Turner JR (2011) Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol 73:283–309
Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, Colegio OR, Anderson JM (2008) The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci 121:298–305
Guttman JA, Finlay BB (2009) Tight junctions as targets of infectious agents. Biochim Biophys Acta 1788:832–841
Hering NA, Fromm M, Schulzke JD (2012) Determinants of colonic barrier function in inflammatory bowel disease and potential therapeutics. J Physiol 590:1035–1044
Meddings J (2008) What role does intestinal permeability have in IBD pathogenesis? Inflamm Bowel Dis 14(Suppl 2):S138–S139
Ahn SH, Shah YM, Inoue J, Morimura K, Kim I, Yim S, Lambert G, Kurotani R, Nagashima K, Gonzalez FJ et al (2008) Hepatocyte nuclear factor 4alpha in the intestinal epithelial cells protects against inflammatory bowel disease. Inflamm Bowel Dis 14:908–920
Farkas AE, Hilgarth RS, Capaldo CT, Gerner-Smidt C, Powell DR, Vertino PM, Koval M, Parkos CA, Nusrat A (2015) HNF4alpha regulates claudin-7 protein expression during intestinal epithelial differentiation. Am J Pathol 185:2206–2218
Krug SM, Amasheh S, Richter JF, Milatz S, Gunzel D, Westphal JK, Huber O, Schulzke JD, Fromm M (2009) Tricellulin forms a barrier to macromolecules in tricellular tight junctions without affecting ion permeability. Mol Biol Cell 20:3713–3724
Schulzke JD, Gunzel D, John LJ, Fromm M (2012) Perspectives on tight junction research. Ann N Y Acad Sci 1257:1–19
Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Burgel N, Fromm M et al (2005) Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 129:550–564
Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD (2007) Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 56:61–72
Ahmad R, Sorrell MF, Batra SK, Dhawan P, Singh AB (2017) Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal Immunol 10:307–317
Garcia-Hernandez V, Quiros M, Nusrat A (2017) Intestinal epithelial claudins: expression and regulation in homeostasis and inflammation. Ann N Y Acad Sci DOI. doi:10.1111/nyas.13360
Tanaka H, Takechi M, Kiyonari H, Shioi G, Tamura A, Tsukita S (2015) Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut 64:1529–1538
Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, Cao J, Chen YH (2012) Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology 142:305–315
van Sommeren S, Visschedijk MC, Festen EA, de Jong DJ, Ponsioen CY, Wijmenga C, Weersma RK (2011) HNF4alpha and CDH1 are associated with ulcerative colitis in a Dutch cohort. Inflamm Bowel Dis 17:1714–1718
Cattin AL, Le Beyec J, Barreau F, Saint-Just S, Houllier A, Gonzalez FJ, Robine S, Pincon-Raymond M, Cardot P, Lacasa M et al (2009) Hepatocyte nuclear factor 4alpha, a key factor for homeostasis, cell architecture, and barrier function of the adult intestinal epithelium. Mol Cell Biol 29:6294–6308
Darsigny M, Babeu JP, Dupuis AA, Furth EE, Seidman EG, Levy E, Verdu EF, Gendron FP, Boudreau F (2009) Loss of hepatocyte-nuclear-factor-4alpha affects colonic ion transport and causes chronic inflammation resembling inflammatory bowel disease in mice. PLoS One 4:e7609
Cornick S, Tawiah A, Chadee K (2015) Roles and regulation of the mucus barrier in the gut. Tissue Barriers 3:e982426
Lu P, Burger-van Paassen N, van der Sluis M, Witte-Bouma J, Kerckaert JP, van Goudoever JB, Van Seuningen I, Renes IB (2011) Colonic gene expression patterns of mucin Muc2 knockout mice reveal various phases in colitis development. Inflamm Bowel Dis 17:2047–2057
Acknowledgements
Support for this work was provided by the Broad Medical Research Program Award at the Crohn’s and Colitis Foundation of America (to CTC), as well as a by NIH (2R01DK074731-04A1 to DK).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Capaldo, C.T., Powell, D.N. & Kalman, D. Layered defense: how mucus and tight junctions seal the intestinal barrier. J Mol Med 95, 927–934 (2017). https://doi.org/10.1007/s00109-017-1557-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-017-1557-x