
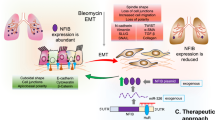
Abstract
Idiopathic pulmonary fibrosis (IPF) is a fatal and devastating lung disease of unknown etiology, described as the result of multiple cycles of epithelial cell injury and fibroblast activation. Despite this impressive increase in understanding, a therapy that reverses this form of fibrosis remains elusive. In our previous study, we found that miR-29b has a therapeutic effect on pulmonary fibrosis. However, its anti-fibrotic mechanism is not yet clear. Recently, our study identified that F-Actin Binding Protein (TRIOBP) is one of the target genes of miR-29b and found that deficiency of TRIOBP increases resistance to lung fibrosis in vivo. TRIOBP knockdown inhibited the proliferation of epithelial cells and attenuated the activation of fibroblasts. In addition, deficiency of Trio Rho Guanine Nucleotide Exchange Factor (TRIO) in epithelial cells and fibroblasts decreases susceptibility to lung fibrosis. TRIOBP interacting with TRIO promoted abnormal epithelial–mesenchymal crosstalk and modulated the nucleocytoplasmic translocation of β-catenin. We concluded that the miR-29b‒TRIOBP–TRIO–β-catenin axis might be a key anti-fibrotic axis in IPF to regulate lung regeneration and fibrosis, which may provide a promising treatment strategy for lung fibrosis.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a fatal and devastating lung disease of unknown etiology [1]. Several risk factors, including aging, smoking, gastroesophageal reflux, and environmental variables, have been proposed associated with IPF [2,3,4]. In IPF conditions, this becomes a progressive and unstoppable process, resulting in severe lung scarring and respiratory failure. Although it has been challenging to pinpoint the pathogenic factors causing IPF, lung epithelial cells are likely to play a major role in IPF [5, 6]. Injuries to lung epithelial cells caused by a variety of insults are thought to be the first step in the pathogenesis of pulmonary fibrosis. This is followed by basement membrane (BM) disruption, epithelial cell death and the epithelial–mesenchymal transition (EMT), infiltration of inflammatory cells, fibroblasts differentiation into active myofibroblasts, extracellular matrix (ECM) overproduction and deposition, and scar formation. Alveolar epithelial type II cells (AECIIs) can serve as progenitor cells that preserve the homeostasis of the epithelium and restore damaged epithelium following injury [7,8,9]. Activated alveolar epithelial cells could release a variety of cytokines and profibrogenic growth factors, such as transforming growth factor β (TGF β), resulting in aberrant epithelial–mesenchymal crosstalk and recruitment of highly synthetic and contractile myofibroblasts, with deposition and remodeling of the ECM.
MicroRNAs (MiRNAs) are deeply involved in regulating processes implicated in IPF such as lung development [10,11,12,13] and maintenance of cellular phenotypes [10, 11, 14,15,16]. Furthermore, IPF lungs exhibit a significantly altered miRNA repertoire [17, 18]. Among miRNAs, the microRNA-29 (miR-29) family is well characterized for their ability to regulate extracellular matrix proteins and has been extensively studied as a potential anti-fibrotic regulator [19]. MiR-29 is decreased in kidney, lung, liver, and myocardial fibrosis and it is important to fibrosis molecules, such as collagen I and III, insulin-like-growth factor-1 (IGF-1), and connective tissue growth factor (CTGF) [17, 20,21,22,23,24,25,26,27]. Consistent with this finding, using a miR-29 mimics developed by miRagen that exhibits preferential lung distribution when administered systemically, which were able to blunt bleomycin (BLM)-induced pulmonary fibrosis in mice [28, 29], we observed that patients with low serum levels of miR-29 experienced significantly shortened survival times. Supplementing miR-29 could be a therapeutic strategy for reversing or mitigating organ fibrosis. With specific relevance to IPF, expression of members of the miR-29 family is decreased in the human IPF lung as well as in animal models of lung fibrosis and their targets are increased. Our previous studies have revealed that miR-29b was downregulated in IPF and BLM-induced lung fibrosis; in addition, treatment with miR-29b mimic in the context of pulmonary fibrosis significant preserves the BLM-induced decrease of miR-29b and reverses pulmonary fibrosis [29]. Several previous studies also reported that miR-29 has been verified to act as a vital regulator in various diseases [30].
Trio Rho Guanine Nucleotide Exchange Factor (TRIO) can encode a large protein that functions as a GDP to GTP exchange factor, which promotes the reorganization of the actin cytoskeleton [31]. Several studies have linked TRIO with intellectual disability [32] and autism spectrum disorders [33,34,35]. One report implicated that the absence of TRIO results in embryonic mortality as well as defective skeletal muscle development [36]. TRIO-mediated RhoA activation is important during early eye development [37] and TRIO-regulated MYH9 activation is essential for craniofacial abnormalities in zebrafish [38]. Recently, studies showed that TRIO regulates cell migration, growth, or invasion, thereby promoting the development of cervical cancer [39], osteosarcoma [40], breast cancer [41], colorectal cancer [42], and liver cancer [43]. TRIO and F-actin-binding protein (TRIOBP) also referred to as Tara, was originally isolated as a cytoskeleton remodeling protein [44]. Recent studies provide clues that TRIOBP variants are associated with other human diseases including cancer and brain diseases. However, TRIOBP and TRIO exact function in IPF as well as the underlying mechanism are still largely unknown. In our research, we demonstrate that TRIOBP was one of target genes of miR-29b. TRIOBP interacts with TRIO and positively regulates TRIO expression. TRIO is upregulated in IPF lung tissues and experimental pulmonary fibrosis. TRIO knockdown significantly reduced the proliferation, migration, and EMT process of AECIIs, as well as inhibits lung fibroblast activation. TRIO knockdown specifically inhibited the nuclear deposition of β-catenin, thereby inhibited some profibrotic genes expression. TRIO and TRIOBP play a stimulative role in aberrant epithelial–mesenchymal crosstalk, which might drive the progress of IPF. Our study not only expands on the understanding of the mechanism for IPF, but also provides a theoretical basis for the research and development of new target for pulmonary fibrosis.
Materials and methods
Lung tissue sampling
Lung tissue samples were obtained from the Henan Provincial Chest Hospital. The IPF samples were surgical remnants of biopsies or lungs explanted from patients with IPF undergoing pulmonary transplant. Control was normal histology tissue obtained from normal disease-free margin of lung cancer resection specimens. IPF was diagnosed based on ATS/ERS/JRS/ALAT Clinical Practice Guidelines [45]. The clinical characteristics of all patients are summarized in Supporting Information Table S1. All studies were approved by the Henan Provincial Chest Hospital Medical Research Ethics Committee (No.2020-03-06). The research conformed to the principles of the Declaration of Helsinki. Oral and written informed consent was obtained from all patients.
Generation of mouse model with lung fibrosis
All animal procedures were approved and performed in accordance with the guidelines of the Institutional Animal Care and Use Committee at the Henan Normal University (IACUC, SMKX-2118BS1018). C57BL/6 mice (5–6 weeks old) were randomly divided into four groups: a saline group, a BLM-induced pulmonary fibrotic group, AAV-shTriobp + saline group, and AAV-shTriobp + BLM group. C57BL/6 mice were allowed to acclimate for 1 week prior to intratracheal administration of either 2.5 × 1011 µg of AAV-shTriobp or AAV-NC (OBiO Technology Corp., Ltd.). After 7 days, pulmonary fibrosis was induced by tracheal infusion of bleomycin (1.5 U/kg), which was carried out under light anesthesia (3–4% isoflurane). ShTriobp: GACGGATTCAAGCCTCAAATA. 14 days after BLM injection, for tissue collection, mice were given a lethal dose of urethane intraperitoneally.
Hydroxyproline assay
Collagen deposition was determined by measuring the total hydroxyproline content in wet lung tissue, which was measured by a hydroxyproline assay kit (Nanjing Jiancheng Bioengineering Institute, AO303-1), according to the manufacturer’s protocol. The concentration of right lung hydroxyproline (µg/mg of wet tissue) was calculated.
Cell culture and treatment
A549, MRC5, 293 T, and MLE-12 cells were obtained from American Type Culture Collection. A549, MRC5, MLE-12, primary mouse lung fibroblast (PMLFs), and 293 T cells were cultured in DME/F-12 or DMEM at 37 °C and 5% CO2. The medium contained 10% fetal bovine serum (FBS), 100 units/mL penicillin, 100 g/mL streptomycin, and 1 mM sodium pyruvate. Media were changed every 3 days.
Mimic miR-29b was synthesized at miRagen Therapeutics, Inc. utilizing standard phosphoramidite solid phase synthesis. Inhibitor miR-29b was obtained from Sangon Biotech (Shanghai) Co., Ltd. The control duplex sequence does not target any known murine or human transcripts by BLAST analysis. The transfection reagent used to mimic miR-29b and inhibitor miR-29b was obtained from Guangzhou RiboBio Co., Ltd.
Isolation of PMLFs
In brief, mice were euthanized with anesthetics and cleaned with 75% ethanol. The lung tissue was collected and washed twice with sterile PBS before being cut into 1 mm3 pieces. The pellet was resuspended in DMEM medium containing 15% FBS and cultured in 10-cm culture dishes at 37 °C after centrifugation (600 g, 5 min). The attached fibroblasts were collected after 4–5 days of incubation for sub-culturing or other assays.
Hematoxylin and eosin (H&E) and Masson’s trichrome staining
H&E and Masson’s trichrome staining were performed according to the instructions of the kit (Solarbio, Beijing, China), and histopathological changes were observed under a microscope.
Immunohistochemistry (IHC)
IHC was performed on TRIOBP (Abcam, ab151320) and TRIO (Affinity, DF2685) expression in IPF and BLM-treated mouse lung. The steps of IHC were as follows: lung sections dewaxed transparently and treated with 3% H2O2 dropwise for 10 min; washed with distilled water and immersed the slides in 0.01% sodium citrate solution, microwave to boiling, 50% goat serum blocked 30 min, overnight incubation with primary antibody. The following day, the horseradish peroxidase-labeled antibody was incubated for 30 min at 37 °C before developing with DAB chromogen. After staining the nuclei with hematoxylin for 5 min, the sections were rinsed with running water for 10 min. Five visual fields were randomly selected for evaluation in each section after being sealed with neutral balsam.
Quantitative real-time PCR
Total RNA was isolated using TRIzol reagent. cDNA was synthesized by M-MLV Reverse Transcriptase (Promega, M1708). RT-qPCR was conducted using SYBR green kit (Yeasen, 11201ES03) according to the manufacturer’s instructions. Each sample and experiments were tested in triplicate. Delta CT values of target gene were normalized to GAPDH or ACTB. The data were evaluated by the 2−ΔΔCt method. RT-qPCR primers used are as described in Table 1.
Western blot analysis and antibodies
Proteins were extracted from cell lysates in lysis buffer and used to quantify protein levels. Proteins were separated on polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, and immunoprobed with specific antibodies. The proteins were detected using a chemiluminescence reagent kit purchased from Thermo Fisher Scientific. The imager station captured the images (Odyssey Software Version 5.2, LI-COR Biosciences). Anti-TRIO (Affinity, DF2685), anti-TRIOBP (Proteintech, 16124-1-AP), anti-β-actin (Affinity, AF7018), and anti-GAPDH (Affinity, AF7021) antibodies were purchased from Affinity Biosciences LTD. Anti-α-SMA antibodies were purchased from Abcam (Abcam, ab124964). Anti-vimentin (Proteintech, 10366-1-AP), anti-E-cadherin (Cell Signaling Technology, 14472S), anti-type I collagen (Proteintech, 14695-1-AP) and anti-fibronectin (Proteintech, 15613-1-AP) antibodies were purchased from Proteintech Group.
Analysis of cell survival and migration
The CCK8 (APExBLO company) and Cell-Light EdU DNA cell proliferation kit (RiboBio, Guangzhou, China) were used to detect the ability of cell proliferation following each manufacturer’s protocol. Cells were plated and grown into a confluent monolayer in six-well plates to test cell migration. After that, scratches were made with a pipette tip. A microscope was used to observe the cell migration process, the following wounding at 0 and 24 or 48 h. Transwell assay was also used to analyze cell migration. Serum-free DME/F12 was used to resuspend the cells, and DME/F12 containing FBS was used in the lower chambers (10%). Calculating the cell numbers from five random fields allowed for the staining, imaging, and analysis of the migrated cells.
Annexin V-FITC/PI flow cytometry
The experiment was carried out with the Apoptosis Detection Kit (Solarbio, CA1020). A549 cells were digested with trypsin without EDTA (Invitrogen), and centrifuged at 1000 rpm for 5 min to be collected. 5 µL of Annexin V-FITC dye was added into the binding buffer (200 µL). Then they were mixed well, and reacted in room temperature for 15 min away from light. Subsequently, 5 µL PI dyes were added. Similarly, they were mixed well and reacted. Flow cytometry was performed within 1 h using FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA).
EdU incorporation assay
An Apollo567 in vitro Imaging Kit was purchased from RiboBio Corporation (Guangzhou, China) and was applied for the EdU incorporation assay. After culturing with EdU (10 μM) for 2 h, the cells were fixed with paraformaldehyde (4%), permeabilized with Triton X-100 (0.2%), and contained with 4′,6-diamidino-2-phenylindole (DAPI, 5 μg/mL) and Apollo fluorescent dyes.
Immunofluorescence assay
Cells fixed with 4% paraformaldehyde were washed three times for 5 min, blocked with 1% goat serum for 1 h at room temperature (RT), incubated with the required antibody, washed three times in PBS, incubated with secondary antibody, washed three times in PBS again, treated with DAPI to dye the nuclei, and finally washed three times in PBS.
Plasmid construction, lentivirus package, and stable-infected cell lines construction
Silencing TRIOBP was achieved by targeting the sequences “GCTGACAGATTCAAGTCTCAA” in the pLKO.1 vector; silencing TRIO was achieved by targeting the sequences “CCACGAAGAATGGATTGAAAT” in the pLKO.1 vector; silencing Triobp was achieved by targeting the sequences “GACGGATTCAAGCCTCAAATA” in the pLKO.1 vector; silencing Trio was achieved by targeting the sequences “TCGACCTATCCGTAGCATTAA” in the pLKO.1 vector. These sequences were acquired from sigmaaldrich.cn. Sub-confluent cultures were overnight infected with concentrated lentivirus particles in the presence of 5 g/mL polybrene. In media containing 2 g/mL puromycin, cells were chosen 24 h after transfection. Details for primers are provided in Table 2.
Isolation of conditioned medium (CM)
The medium was replaced and cells were incubated with fresh medium for 72 h. CM was collected, centrifuged, and immediately used for recipient cells incubation (72 h) or stored at − 20 °C for later use.
ChIP-qPCR assay
ChIP–qPCR was performed as described [39]. A549 cells were cross-linked with 1% formaldehyde for 10 min at RT. Fixation was stopped by 125 mM glycine for 10 min and the samples were washed twice with ice-cold PBS. Cell pellets were then resuspended in 1 mL of cytolysis buffer, mixed and incubated on ice for 10–15 min with occasional inversion every 2 min. Cells were then centrifuged for 5 min at 3500 rpm at 4 °C, the supernatant was discarded, and the remaining nuclear pellet (white) was resuspended in 500 μL of nuclear lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–Cl, pH 8.1, 1 × protease inhibitor cocktail). The average length of the shared chromatin is about 250 bp or less. The soluble chromatin was collected by centrifuging for 10 min at 14,000 rpm, and the supernatant was diluted 1:10 with dilution buffer (150 mM NaCl, 20 mM Tris–HCl, pH 8.1, 2 mM EDTA, 1% Triton X-100 and 1 × protease inhibitor cocktail). Chromatin was incubated at 4 °C overnight with protein A/G beads. Anti-rabbit IgG was used as a negative control. The precipitated complexes were washed twice in low-salt buffer (150 mM NaCl, 20 mM Tris–HCl, pH 8.1, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), twice in high-salt buffer (500 mM NaCl, 20 mM Tris–HCl, pH 8.1, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), twice in LiCl buffer (250 mM LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, pH 8.0, 10 mM Tris–HCl, pH 8.1), and twice in TE buffer (pH 8.0). The thoroughly washed beads were eluted twice with 150 μL of elution buffer (0.1 M NaHCO3, 1% SDS) by vortexing at 70 °C at 1000 rpm for 10 min on a Thermo Mixer C (Eppendorf). The elutes were pooled and heated at 65℃ for overnight to reverse the formaldehyde cross-link. The enriched DNA fragments were then purified with QIAquick Spin column and quantified by Nanodrop. The published primers of VIM and CDH2 were used for β-catenin occupancy analysis with SYBR green master mix (Yeasen, 11201ES03). The following antibodies were used: anti-β-catenin (proteintech, 51067-2-AP) and anti-IgG (Beyotime, A7016). Primers used for ChIP–qPCR are listed in Table 3.
Dual-luciferase reporter analysis
Whether there was a direct target between miR-29b and TRIOBP was confirmed by luciferase reporter gene assay. The potential TRIOBP 3ʹUTR’s targets of miR‐29b were acquired from TargetScan (http://www.targetscan.org/vert_72/). The primers of the predicted target gene TRIOBP were from the Sangon Biotech (Shanghai) Co., Ltd. To obtain the sequence of deletion‐type TRIOBP, we use TRIOBP-3ʹUTR-1 and TRIOBP-3ʹUTR-2 primers to construct two short sequences and then let these two sequences use each as a template for another PCR to acquire the deletion‐type TRIOBP. Primer sequences are listed in Table 4. Sequences of wild‐type TRIOBP and deletion‐type TRIOBP were inserted into the pGL3.0 luciferase reporter vector. Ligase 4 was used to ligate the target gene and the TRIOBP‐wild‐type‐Luc and TRIOBP‐deletion‐type‐Luc plasmid vectors. A total of 20 µL plasmid and 100 µL competent cells containing DH5α were then added into 1.5 mL centrifuge tubes, left on ice for 30 min, water‐bathed at 42℃ for heat shock for 90 s, and immediately chilled on ice for 2 min. After this, 37 ℃ pre‐heated 800 µL Luria–Bertani (LB) liquid medium was added to the tube and centrifuged at 37 ℃ at 220 rpm for 45 min. A total of 150 µL DH5α liquid was tiled on the Amp (+) LB plate, which was reversely placed and incubated overnight at 37 ℃. The 293 T, A549, and MRC5 cells were cultured in 24-well plates and transfected with 400 ng luciferase reporter plasmids together with 50 nM miR-29b mimic/inhibitor/ negative control (NC). The empty was only transferred pGL3.0 vector with no insertion sequence. At 36 h after transfection, the Dual-Luciferase Reporter Assay System purchased from Yeasen Biotechnology (Shanghai) Co., Ltd (11402ES80) was used to detect luciferase activity.
Lung single cell RNA-seq data analysis from human and mouse
The human single-cell data used in this study are sourced from the Human Cell Atlas, specifically focusing on the cell types present in the human lung. The identification of cell identities is based on the research conducted by Sikkema et al. [46]. Mouse single-cell data are obtained from the work previously published by Wang et al. [47]. The human single-cell data are analyzed using Scanpy [48], a powerful Python library designed for single-cell RNA-seq analysis, with UMAP plots being generated to visualize the cellular landscape. Similarly, Seurat [49], a popular R toolkit for single-cell genomics analysis, is employed for the analysis of the mouse single-cell data.
Statistical analysis
Every experiment was repeated at least three times. The results in the control and experimental groups were analyzed by GraphPad software 9.0. The Shapiro–Wilk normality test was used to test normal distribution. The results were analyzed by the Mann–Whitney U test for comparisons between two groups when sample data were not normally distributed, and by unpaired Student’s t test for comparisons between two groups with normal distribution. Data are presented as mean ± SD and were considered statistically significant at P < 0.05.
Results
TRIOBP is a directly target gene of miR-29b
We predicted that TRIOBP was one of the potential targets of miR-29b for its binding to the specific sequence of at the 3ʹ UTR of TRIOBP in Targetscan database (http://www.targetscan.org/vert_72/) (Fig. 1A). Using two luciferase reporter vectors, containing TRIOBP‐wild-type and TRIOBP‐deletion-type to confirm that TRIOBP was the target gene of miR-29b as shown in Fig. 1B, miR-29b reduced TRIOBP‐wild-type luciferase vector activity obviously, and in contrast, we were not able to observe a significant change in TRIOBP‐deletion-type vector activity. Then we transfected the miR-29b mimic into A549 and MRC5 cells, the protein expression of TRIOBP in A549 and MRC5 cells was reduced, expression of α-SMA and vimentin was reduced in A549 cells (Fig. 1C), expression of collagen I, fibronectin, and α-SMA was decreased in MRC5 cells (Fig. 1D). Subsequently, we transfected the inhibitor of miR-29b to A549 and MRC5 cells, the activity of the TRIOBP-wild-type luciferase vector was enhanced (Fig. 1E, F), the expression of TRIOBP and profibrotic markers, such as collagen I and fibronectin, was increased both in A549 and MRC5 cells at mRNA (Figs. S1A-B) and protein levels (Fig. 1G, H). All these data indicated that TRIOBP is one of the directly target genes of miR-29b and promote the profibrotic phenotype.

TRIOBP is one of the direct target genes of miR-29b. A Bioinformatics prediction analysis shows miR-29b could target the region of TRIOBP’s 3ʹUTR. B Luciferase assay shows mimic of miR-29b significantly decreased the luciferase activity of TRIOBP‐wild-type. *p < 0.05 when compared with the plasmid co-transfected with empty vector and TRIOBP‐wild-type; data were evaluated by two-way ANOVA with Šídák’s multiple comparisons test for pairwise comparisons (n = 3). C Western blot of TRIOBP, α-SMA, vimentin, and GAPDH in the NC and mimic miR-29b A549 cells groups. D Western blot of TRIOBP, α-SMA, fibronectin, collagen I, and β-actin in the NC and mimic miR-29b MRC5 cells groups. E The luciferase assay shows inhibitor of miR-29b significantly increased the luciferase activity of TRIOBP‐wild-type in A549 cells. *p < 0.05, data were evaluated by two-way ANOVA with Šídák’s multiple comparisons test for pairwise comparisons (n = 3). F The luciferase assay shows inhibitor of miR-29b significantly increased the luciferase activity of TRIOBP‐wild-type in MRC5 cells. *p < 0.05 when compared with the plasmid co-transfected with empty vector and TRIOBP‐wild-type; data were evaluated by two-way ANOVA with Šídák’s multiple comparisons test for pairwise comparisons (n = 3). G Western blot of TRIOBP, collagen I and β-actin in the NC and mimic miR-29b A549 cells groups. H Western blot of TRIOBP, collagen I, fibronectin and β-actin in the NC and mimic miR-29b MRC5 cells groups
Deficiency of TRIOBP increases resistance to lung fibrosis in vivo
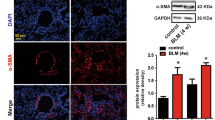
Increased TRIOBP expression was detected in the lung tissue of IPF patients as compared with control samples (Fig. 2A). We also observed upregulation of Triobp expression in BLM-induced mice model of pulmonary fibrosis (Fig. 2B). To evaluate the potential role of TRIOBP in lung injury and fibrosis in vivo, we generated the mice model of lung fibrosis and shRNA adenovirus targeting Triobp treatment mice model (Fig.S2). The X-ray micro-computed tomography (micro-CT) performances showed that opacifications were clearly moderated in sh-Triobp group (Fig. 2C). The lung pathology of BLM-treated mice showed extensive fibrosis with markedly massive collagen deposits, fibrotic lesions, and distorted lung architecture by H&E and Masson Trichrome staining and hydroxyproline assay (Fig. 2D–F), the sh-Triobp infected mice showed reduction collagen deposition and prevention in their weight loss (Fig. 2G), along with reduction in the inflammatory cells counts in bronchoalveolar lavage fluid (BALF) (Fig. 2H), the expression of fibronectin and vimentin was markedly reduced; however, the expression of epithelial marker E-cadherin was increased in the lungs of the sh-Triobp infection mice (Fig. 2I). These data showed that deficiency of TRIOBP decreases susceptibility to lung fibrosis in vivo.

Triobp knockdown blocks the lung fibrosis in BLM-induced mice. A Immunohistochemical analysis indicated that TRIOBP was upregulated in IPF lung tissues. Bar = 50 μM. B Immunohistochemistry indicated that Triobp was upregulated in BLM-treated mice model of pulmonary fibrosis. Bar = 50 μM. C Micro-CT imaging system for small animals (left). Quantification of fibrotic area. Representative micro-CT images of mouse lungs (right). Micro-CT, micro-computed tomography. D Hydroxyproline content in mice. Data were evaluated by two-way ANOVA with Šídák’s multiple comparisons test for pairwise comparisons. Data ****p < 0.0001. E H&E and staining. F Masson staining. G Body weight monitoring revealed that BLM-treated mice lost substantial body mass compared with sham mice, and sh-Triobp effectively blocked this loss compared with the control group. Data were evaluated by two-way ANOVA with Šídák’s multiple comparisons test for pairwise comparisons. H Count of total number of cells in BALF. Data were evaluated by two-way ANOVA with Šídák’s multiple comparisons test for pairwise comparisons. Data ***p < 0.001 and ****p < 0.0001. I Western blot of fibronectin and vimentin, and E-cadherin expression
TRIOBP knockdown inhibited the proliferation, migration, and profibrotic gene expression of epithelial cell
In vivo studies indicate that loss of TRIOBP showed robust anti-fibrotic activity, the abnormally expression of E-cadherin and vimentin in BLM-induced mice lung initiated us to assess the effect of TRIOBP in alveolar epithelium. First, TRIOBP knockdown inhibited vimentin, α-SMA, N-cadherin expression, increased E-cadherin expression in A549 cells at both mRNA and protein levels (Fig. 3A, B). Knockdown of TRIOBP also induced a decreasing of vimentin positive cells (Fig. 3C). Moreover, reduced TRIOBP expression inhibited significantly the epithelial cell proliferation and migration (Fig. 3D–F), while promoted the epithelial cell apoptosis (Fig. 3G). These findings suggested that TRIOBP elicited substantial effects in epithelium cell proliferation, migration, and EMT process, which might participate in lung tissue injury-remodeling process.

Lack of TRIOBP reduced abnormal cell proliferation, migration, EMT and promoted cell apoptosis in epithelial cells. A RT-qPCR of TRIOBP and EMT marker genes (ACTA2, VIM, CDH1, CDH2) expression (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001. B Western blot of TRIOBP, α-SMA, vimentin, E-cadherin, and β-actin in the shNC and sh-TRIOBP A549 cells groups. shNC: short hairpin control (control lentivirus); sh-TRIOBP: short hairpin TRIOBP (TRIOBP lentivirus). C Immunofluorescence images (using Fire LUT) of vimentin (n = 3). The nuclei were counterstained with DAPI. Bar = 20 μm. D The EdU proliferation assay was used to measure A549 cell proliferation (n = 3). Bar = 50 μm. *p < 0.05. E Wound healing assays conducted and photographed at 0 and 48 h, with quantification (n = 3). Bar = 100 μm. F Transwell migration assay of A549 cells at 36 h after TRIOBP knockdown (n = 3). Bar = 100 μm. G Cell apoptosis measured by cytometry, with quantification analysis (n = 3). **p < 0.01. The results were analyzed by the unpaired Student’s t test for comparisons between two groups with normal distribution, data are presented as mean ± SD. Data *p < 0.05, **p < 0.01, and ***p < 0.001
TRIOBP silencing attenuated the activation of fibroblasts in MRC5 cells and PMLFs
Fibroblast-to-myofibroblast differentiation and myofibroblast proliferation and migration are major clinical manifestations of this disease; hence, blocking these processes is a practical treatment strategy [50]. Thus, we investigated the role of TRIOBP in lung fibroblasts using a shRNA to knockdown the TRIOBP in MRC5 cell line. TRIOBP knockdown reduced collagen I, fibronectin, and vimentin expression at both mRNA and protein levels in MRC5 cells (Fig. 4A, B). The shrinking of vimentin positive cells was obviously observed in Fig. 4C. TRIOBP knockdown also reduced the excessive proliferation and migration of MRC5 (Fig. 4D–F). We isolated PMLFs and silenced the expression of Triobp in PMLFs to further validate the function of Triobp in mice. Triobp knockdown inhibited the expression of fibronectin in PMLFs (Fig.S3). These results indicate that TRIOBP silencing attenuated the activation of lung fibroblast.

TRIOBP silencing reduced lung fibroblast activation. A RT-qPCR of TRIOBP and fibrotic marker genes (COL1A1, FN1, VIM) expression (n = 3). *p < 0.05 and **p < 0.01. B Western blot of TRIOBP, α-SMA, vimentin, collagen I, fibronectin, and β-actin in the shNC and sh-TRIOBP MRC5 cells groups. shNC: short hairpin control (control lentivirus); sh-TRIOBP: short hairpin TRIOBP (TRIOBP lentivirus). C Immunofluorescence images (using Fire LUT) of vimentin (n = 3). The nuclei were counterstained with DAPI. Bar = 20 μm. ****p < 0.0001. D The EdU proliferation assay was used to measure MRC5 cell proliferation (n = 3). Bar = 50 μm. E Transwell migration assay of MRC5 cells at 36 h after TRIOBP knockdown (n = 3). Bar = 100 μm. F Wound healing assays conducted and photographed at 0 and 24 h, with quantification (n = 3). Bar = 100 μm. The results in A, C were analyzed by the unpaired Student’s t test for comparisons between two groups with normal distribution, data are presented as mean ± SD. Data *p < 0.05, **p < 0.01, and ****p < 0.0001
Deficiency of TRIO in epithelial cell and fibroblast decreases susceptibility to lung fibrosis
As known, TRIOBP interacts with TRIO, which is controlling actin cytoskeleton organization, cell motility, and cell growth [51]. We found that expression of TRIO was higher in lungs of patients with IPF compared with healthy donors and in BLM-induced lung of mice (Fig. 5A–D). The TRIO and TRIOBP expression and distribution were shown in the UMAP by analyzing the single-cell RNA-seq data from Human Lung Cell Atlas (Fig.S4A) and mouse single-cell data from our previous work (Fig.S4B). In A549 and MRC5 cells, the expression of TRIOBP was positively correlated with the expression of TRIO (Fig.S4C–F). To determine the role of TRIO in epithelial injury and fibrosis, we generated the TRIO knockdown A549 cell and MRC5 cell. TRIO knockdown inhibited cell proliferation and migration significantly (Figs. 5E–H, 6A–C) in A549 and MRC5 cell, resulted in apoptosis in A549 (Fig. 5I).

A549 cells lacking TRIO inhibits abnormal cell proliferation, migration, apoptosis, and differentiation. A Immunohistochemical analysis indicated that TRIO was upregulated in IPF lung tissues. Bar = 20 μM. B Immunohistochemistry indicated that TRIO was upregulated in BLM-treated mouse lung. Bar = 20 μM. C RT-qPCR tested the mRNA level of TRIO in BLM-treated mouse lung. The results were analyzed by the unpaired Student’s t test for comparisons between two groups with normal distribution, data are presented as mean ± SD. Data *p < 0.05. D Western blot tested the protein level of TRIO in BLM-treated mouse lung tissues. E CCK8 assay at 0, 24, 48, 72, 96 h (n = 5). *p < 0.05 and ****p < 0.0001. F The effect of TRIO knockdown on the colony formation of A549 cells (n = 3). shNC: short hairpin control (control lentivirus); sh-TRIO: short hairpin TRIO (TRIO lentivirus). **p < 0.01. G Wound healing assays conducted and photographed at 0 and 72 h, with quantification (n = 3). Bar = 100 μm. H Transwell migration assay of A549 cells at 36 h after TRIO knockdown (n = 3). Bar = 100 μm. ****p < 0.0001. I Cell apoptosis measured by cytometry, with quantification analysis (n = 3). ***p < 0.001. J RT-qPCR of TRIO and EMT marker genes (ACTA2, VIM, CDH1, CDH2) expression (n = 3). *p < 0.05, **p < 0.01 and ****p < 0.0001. K Western blot of TRIO, α-SMA, vimentin, E-cadherin and β-actin in the shNC and sh-TRIO A549 cells groups. L Immunofluorescence images (using Fire LUT) of vimentin (n = 3). The nuclei were counterstained with DAPI. Bar = 20 μm. Data in A were evaluated by two-way ANOVA with Šídák’s multiple comparisons test for pairwise comparisons. The results were analyzed by the unpaired Student’s t test for comparisons between two groups with normal distribution, data are presented as mean ± SD. Data *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001

TRIO silencing decreases the fibroblast proliferation, migration, and activation. A The EdU proliferation assay was used to measure MRC5 cell proliferation (n = 3). Bar = 50 μm. B Transwell migration assay of MRC5 cells at 36 h after TRIO knockdown (n = 3). Bar = 100 μm. C Wound healing assays conducted and photographed at 0 and 24 h (n = 3). Bar = 100 μm. D RT-qPCR of TRIO and fibrotic marker genes (ACTA2, COL1A1, FN1, VIM) expression (n = 3). ***p < 0.001 and ****p < 0.0001. E Immunofluorescence images (using Fire LUT) of vimentin (n = 3). The nuclei were counterstained with DAPI. Bar = 20 μm. ***p < 0.001. F Western blot of TRIO, α-SMA, vimentin, collagen I, fibronectin, and β-actin in the shNC and sh-TRIO MRC5 cells groups. The results were analyzed by the unpaired Student’s t test for comparisons between two groups with normal distribution, data are presented as mean ± SD. Data ***p < 0.001 and ****p < 0.0001
A549 cells lacking TRIO resulted in significant lower transcript level of ACTA2, VIM, CDH2 and lower protein level of α-SMA accompanied by higher transcript level of CDH1 and E-cadherin (Fig. 5J–L). Similarly, Trio knockdown inhibited the expression of vimentin and α-SMA, as well as reduced the proliferation of mouse epithelium MLE-12 cells (Fig.S5). In MRC5 cells, TRIO knockdown decreased the transcript level of ACTA2, VIM, COL1A1, FN1 (Fig. 6D), and the protein expression of α-SMA, vimentin, collagen I, and fibronectin (Fig. 6E, F). Therefore, we concluded that deficiency of TRIO in epithelial cell and fibroblast decreases susceptibility to lung fibrosis.
TRIOBP interacting with TRIO promoted abnormal epithelial–mesenchymal crosstalk
As previously proved that TRIOBP silencing inhibits the proliferation and migration of epithelial cell, TRIO knockdown reduces the proliferation of A549 and MLE-12 cells, while TRIOBP was positively correlated with the expression of TRIO in A549 and MRC5 cells; therefore, we proposed that the TRIOBP might cause abnormal AECIIs injury and repair, and this abnormal activation might cause dysregulated crosstalk between the epithelium and mesenchymal cells as well as an accumulation of myofibroblasts. As shown in Fig. 7A, TRIOBP knockdown of A549 was co-culture with fibroblast to investigate the expression of α-SMA, vimentin, collagen I, and fibronectin in MRC5 cells at both the mRNA and protein levels, all of them were inhibited compared to the controls (Fig. 7B–D), the same results were observed when TRIO knockdown of A549 was co-cultured with fibroblast (Fig. 7E–H). These results support our in vitro findings that TRIOBP promoted abnormal epithelial–mesenchymal crosstalk by regulating TRIO expression.

TRIO and TRIOBP knockdown attenuated the abnormal epithelial–mesenchymal crosstalk. A The model of A549 cells co-cultured with human lung fibroblasts. A549 shNC and sh-TRIOBP cell lines, as well as normal human lung fibroblast (MRC5 cells) were used in this system. The MRC5 cells were used to experiment after cultured for 48 h. B RT-qPCR of co-cultured MRC5 cells’ (A) ACTA2, COL1A1, FN1, and VIM expression. *p < 0.05 and ***p < 0.001. C Western blot of co-cultured MRC5 cells’ (A) α-SMA, collagen I, fibronectin, vimentin, and GAPDH expression (n = 3). D Above A’s co-cultured system, immunofluorescence images (using Fire LUT) of vimentin (n = 3). The nuclei were counterstained with DAPI. Bar = 20 μm. ****p < 0.0001. E The model of A549 cells co-cultured with human lung fibroblasts. A549 shNC and sh-TRIO cell lines, as well as normal human lung fibroblast (MRC5 cells) were used in this system. The MRC5 cells were used to experiment after cultured for 48 h. F RT-qPCR of co-cultured MRC5 cells’ (E) ACTA2, COL1A1, FN1 and VIM expression. *p < 0.05 and **p < 0.01. G Western blot of co-cultured MRC5 cells’ (E) α-SMA, collagen I, fibronectin, vimentin, and GAPDH expression (n = 3). H Above E’s co-cultured system, immunofluorescence images (using Fire LUT) of vimentin (n = 3). The nuclei were counterstained with DAPI. Bar = 20 μm. ***p < 0.001. The results were analyzed by the unpaired Student’s t test for comparisons between two groups with normal distribution, data are presented as mean ± SD. Data *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001
TRIOBP/TRIO modulates the nucleocytoplasmic translocation of β-catenin
Transcription factor β-catenin is involved in alveolar epithelial–mesenchymal transition during pulmonary fibrosis, we hypnotized that TRIOBP interacts with TRIO modulating fibrosis through β-catenin signal pathway. Analysis of the nuclear and cytoplasmic protein levels of β-catenin demonstrated that knockdown of TRIOBP increased the level of β-catenin in the cytoplasmic, while decreased it in the nucleus (Fig. 8A). Immunofluorescence staining also revealed that knockdown of TRIOBP inhibited the localization of β-catenin in nucleus (Fig. 8B). To further examine whether TRIOBP affects the binding of β-catenin as a transcription factor to the promoter regions of VIM and CDH2, ChIP-qPCR results revealed that β-catenin exhibited significant binding to the promoter regions of CDH2 and VIM in the control group; however, TRIOBP knockdown markedly reduced the binding ability of β-catenin to the promoter regions of CDH2 and VIM in A549 cells (Fig. 8C, D). Next, we investigated whether TRIO knockdown also changes the nucleocytoplasmic translocation of β-catenin. Western blot and immunofluorescence staining showed that TRIO silencing also deregulated the disposition of β-catenin in nucleus (Fig. 8E, F). Moreover, similarly, ChIP-qPCR results revealed that TRIO knockdown blocked β-catenin bounds to promoter regions of VIM and CDH2 (Fig. 8G, H). These data suggest that TRIOBP and TRIO are critical for the binding of β-catenin to profibrotic genes and affected epithelial–mesenchymal crosstalk.

TRIO knockdown triggers the nucleocytoplasmic translocation of β-catenin. A Western blot of β-catenin in the nucleus and cytoplasm in the shNC and sh-TRIOBP groups in A549 cells. B Immunofluorescence images (using Fire LUT) of β-catenin. The nuclei were counterstained with DAPI. Bar = 20 μm. C ChIP assay of β-catenin binding to the CDH2 promoter in A549 cells from shNC and sh-TRIOBP group. D ChIP assay of β-catenin binding to the VIM promoter in A549 cells from shNC and sh-TRIOBP group. E Western blot of β-catenin in the nucleus and cytoplasm in the shNC and sh-TRIO groups in A549 cells. F Immunofluorescence staining of β-catenin. The nuclei were counterstained with DAPI. Bar = 20 μm. G ChIP assay of β-catenin binding to the CDH2 promoter in A549 cells from shNC and sh-TRIO group. H ChIP assay of β-catenin binding to the VIM promoter in A549 cells from shNC and sh-TRIO group. Data were evaluated by two-way ANOVA with Šídák’s multiple comparisons test for pairwise comparisons. Data *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001
Taken together, these findings imply that TRIOBP is one of target genes of miR-29b. TRIOBP interacts with TRIO to promote abnormal epithelial–mesenchymal crosstalk and downstream β-catenin signaling in IPF. The miR-29b–TRIOBP–TRIO–β-catenin axis might be a key anti-fibrotic axis and provide a promising treatment strategy for lung fibrosis.
Discussion
Our previous studies demonstrated that miR-29 mimicry inhibited pulmonary fibrosis in BLM-treated mice, rats, and non-human primates [28, 29], but little was known about the mechanism underlined in the models and patients. Here, we showed that miR-29b could bind to TRIOBP which interact with TRIO, the miR-29–TRIOBP–TRIO axis that modulates activation of the β-catenin signaling pathway in IPF. Our findings exhibit that TRIOBP is one of the target genes and discovers a novel mechanism that miR-29b blocks pulmonary fibrosis by regulation of TRIOBP and TRIO.
MiRNAs have been shown to predict IPF mortality and facilitate early diagnosis. miR-185, miR-210, miR-302c, miR-376c, and miR-423-5p were increased in IPF lung tissue of rapid progressors, the reduced miR-29 expression in peripheral blood was associated with increased mortality of patients with IPF [52]. MiRNAs act as negative regulators of gene expression by inhibiting the translation or promoting the degradation of target mRNAs, individual miRNAs often regulate the expression of multiple target genes with related functions, a change in the expression of a single miRNA, or a family sharing the same targets, can influence an entire gene network and thereby modify complex disease phenotypes in principle [53]. In the context of their targets, miRNAs are deeply involved in regulating processes implicated in IPF such as lung differentiation and development, regulation of ECM deposition and TGF β signaling [54]. The miR-29 family is probably the most relevant to fibrosis. The family consists of miR-29a, -29b and -29c, which are expressed as 2 bicistronic clusters (miR-29a/-29b-1 and miR-29b-2/-29c), and are largely homologous in sequence [27]. Among the predicted and proven targets of the miR-29 family are multiple ECM proteins and profibrotic molecules, herein we discovered that TRIOBP is one of the target genes of miR-29b, miR-29b inhibited the expression of TRIOBP at the mRNA and protein levels in A549 and MRC5 cells. Silencing TRIOBP reduced the ECM disposition and inhibited the EMT process in vivo. TRIOBP knockdown inhibited the abnormal activation of AECIIs and fibroblast. The most accepted in pathogenesis of IPF is the epithelial-driven process triggered by AECIIs injury which leads to altered crosstalk with immune cells, fibroblasts, myofibroblast activation, and ECM accumulation [1, 50, 55, 56]. Next, we explored the molecular mechanism of TRIOBP in pulmonary fibrosis. As a binding partner of TRIO [51], TRIOBP positively regulated TRIO at mRNA and protein levels in this study. TRIO knockdown inhibited profibrotic genes expression, while increased the expression of E-cadherin in AECIIs. TRIO knockdown inhibited the fibroblast differentiation to myofibroblast and ECM disposition in vitro. TRIOBP positively regulated TRIO expression and TRIO plays a similar role of TRIOBP in AECIIs and lung fibroblast. Activated alveolar epithelial cells could release a variety of cytokines and profibrogenic growth factors, which resulting in aberrant epithelial–mesenchymal crosstalk and myofibroblast activation, with deposition and remodeling of the ECM. Interestingly, we found that TRIOBP and TRIO knockdown in AECIIs both inhibited the activation of lung fibroblast. Thus, we think that the expression of TRIOBP in AECIIs affects lung fibroblast through regulating TRIO expression. To further explore the mechanism of TRIO regulating epithelial–mesenchymal crosstalk, we found that TRIO silencing decreased transportation of β-catenin to the nucleus and TRIO is critical for the binding of β-catenin to profibrotic genes chromatin in A549 cells. These results pinpoint that TRIO affects the translocation of β-catenin in nuclear, thereby regulating the activation of lung fibroblasts and suggest that miR-29b blocks EMT and lung fibroblast activation in IPF by targeting TRIOBP regulation of TRIO. Further TRIO is critical for the binding of β-catenin to chromatin for profibrotic genes expression and TRIO affected epithelial–mesenchymal crosstalk through regulating AECIIs’ β-catenin transfer to nuclear.
In conclusion, the present work revealed the role and mechanism of the miR-29b–TRIOBP–TRIO–β-catenin axis in lung fibrosis and provided a better understanding for IPF. The results from in vitro and in vivo experiments indicated that increasing with miR-29b or interfering with TRIOBP expression may be effective strategies for the prevention and treatment of lung fibrosis.
Data availability
All data are contained within the manuscript.
Abbreviations
- AECIIs:
-
Alveolar epithelial type II cells
- IPF:
-
Idiopathic pulmonary fibrosis
- TGF-β:
-
Transforming growth factor-β
- CM:
-
Conditioned media
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial–mesenchymal transition
- α-SMA:
-
α-Smooth muscle actin
- PI:
-
Propidium iodide
- TRIO:
-
Trio rho guanine nucleotide exchange factor
- TRIOBP:
-
TRIO And F-Actin Binding Protein
- IGF-1:
-
Insulin-like-growth factor-1
- CTGF:
-
Connective tissue growth factor
- miR-29b:
-
MicroRNA-29b
- ID:
-
Intellectual disability
- ASD:
-
Autism spectrum disorders
- OS:
-
Osteosarcoma
- MiRNAs:
-
MicroRNAs
- IHC:
-
Immunohistochemistry
- FBS:
-
Fetal bovine serum
References
Richeldi L, Collard HR, Jones MG (2017) Idiopathic pulmonary fibrosis. Lancet 389:1941–1952
(2000) American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med 161: 646–664
Xylourgidis N, Min K, Ahangari F, Yu G, Herazo-Maya JD, Karampitsakos T, Aidinis V, Binzenhofer L, Bouros D, Bennett AM, Kaminski N, Tzouvelekis A (2019) Role of dual-specificity protein phosphatase DUSP10/MKP-5 in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 317:L678–L689
Ntatsoulis K, Karampitsakos T, Tsitoura E, Stylianaki EA, Matralis AN, Tzouvelekis A, Antoniou K, Aidinis V (2021) Commonalities between ARDS, pulmonary fibrosis and COVID-19: the potential of autotaxin as a therapeutic target. Front Immunol 12:687397
Dong Y, Geng Y, Li L, Li X, Yan X, Fang Y, Li X, Dong S, Liu X, Li X, Yang X, Zheng X, Xie T, Liang J, Dai H, Liu X, Yin Z, Noble PW, Jiang D, Ning W (2015) Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J Exp Med 212:235–252
Noble PW, Barkauskas CE, Jiang D (2012) Pulmonary fibrosis: patterns and perpetrators. J Clin Invest 122:2756–2762
Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BL (2013) Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123:3025–3036
Desai TJ, Brownfield DG, Krasnow MA (2014) Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507:190–194
Hogan BL, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CC, Niklason L, Calle E, Le A, Randell SH, Rock J, Snitow M, Krummel M, Stripp BR, Vu T, White ES, Whitsett JA, Morrisey EE (2014) Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15:123–138
Dong J, Jiang G, Asmann YW, Tomaszek S, Jen J, Kislinger T, Wigle DA (2010) MicroRNA networks in mouse lung organogenesis. PLoS ONE 5:e10854
Lu Y, Okubo T, Rawlins E, Hogan BL (2008) Epithelial progenitor cells of the embryonic lung and the role of microRNAs in their proliferation. Proc Am Thorac Soc 5:300–304
Bhaskaran M, Wang Y, Zhang H, Weng T, Baviskar P, Guo Y, Gou D, Liu L (2009) MicroRNA-127 modulates fetal lung development. Physiol Genomics 37:268–278
Nimmo RA, Slack FJ (2009) An elegant miRror: microRNAs in stem cells, developmental timing and cancer. Chromosoma 118:405–418
Peter ME (2009) Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle 8:843–852
Warburton D, El-Hashash A, Carraro G, Tiozzo C, Sala F, Rogers O, De Langhe S, Kemp PJ, Riccardi D, Torday J, Bellusci S, Shi W, Lubkin SR, Jesudason E (2010) Lung organogenesis. Curr Top Dev Biol 90:73–158
Tomankova T, Petrek M, Kriegova E (2010) Involvement of microRNAs in physiological and pathological processes in the lung. Respir Res 11:159
Pandit KV, Milosevic J, Kaminski N (2011) MicroRNAs in idiopathic pulmonary fibrosis. Transl Res 157:191–199
Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, Konishi K, Yousem SA, Singh M, Handley D, Richards T, Selman M, Watkins SC, Pardo A, Ben-Yehudah A, Bouros D, Eickelberg O, Ray P, Benos PV, Kaminski N (2010) Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 182:220–229
He Y, Huang C, Lin X, Li J (2013) MicroRNA-29 family, a crucial therapeutic target for fibrosis diseases. Biochimie 95:1355–1359
Liu GX, Li YQ, Huang XR, Wei L, Chen HY, Shi YJ, Heuchel RL, Lan HY (2013) Disruption of Smad7 promotes ANG II-mediated renal inflammation and fibrosis via Sp1-TGF-beta/Smad3-NF.kappaB-dependent mechanisms in mice. PLoS ONE 8:e53573
Fang Y, Yu X, Liu Y, Kriegel AJ, Heng Y, Xu X, Liang M, Ding X (2013) miR-29c is downregulated in renal interstitial fibrosis in humans and rats and restored by HIF-alpha activation. Am J Physiol Renal Physiol 304:F1274-1282
Wang B, Komers R, Carew R, Winbanks CE, Xu B, Herman-Edelstein M, Koh P, Thomas M, Jandeleit-Dahm K, Gregorevic P, Cooper ME, Kantharidis P (2012) Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol 23:252–265
Noetel A, Kwiecinski M, Elfimova N, Huang J, Odenthal M (2012) microRNA are central players in anti- and profibrotic gene regulation during liver fibrosis. Front Physiol 3:49
Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi M, Tacke F, Trautwein C, Luedde T (2011) Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53:209–218
Cushing L, Kuang PP, Qian J, Shao F, Wu J, Little F, Thannickal VJ, Cardoso WV, Lu J (2011) miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol 45:287–294
Li Z, Hassan MQ, Jafferji M, Aqeilan RI, Garzon R, Croce CM, van Wijnen AJ, Stein JL, Stein GS, Lian JB (2009) Biological functions of miR-29b contribute to positive regulation of osteoblast differentiation. J Biol Chem 284:15676–15684
van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN (2008) Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A 105:13027–13032
Chioccioli M, Roy S, Newell R, Pestano L, Dickinson B, Rigby K, Herazo-Maya J, Jenkins G, Ian S, Saini G, Johnson SR, Braybrooke R, Yu G, Sauler M, Ahangari F, Ding S, DeIuliis J, Aurelien N, Montgomery RL, Kaminski N (2022) A lung targeted miR-29 mimic as a therapy for pulmonary fibrosis. EBioMedicine 85:104304
Montgomery RL, Yu G, Latimer PA, Stack C, Robinson K, Dalby CM, Kaminski N, van Rooij E (2014) MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol Med 6:1347–1356
Wang H, Wang B, Zhang A, Hassounah F, Seow Y, Wood M, Ma F, Klein JD, Price SR, Wang XH (2019) Exosome-mediated miR-29 transfer reduces muscle atrophy and kidney fibrosis in mice. Mol Ther 27:571–583
Govek EE, Newey SE, Van Aelst L (2005) The role of the Rho GTPases in neuronal development. Genes Dev 19:1–49
Ba W, Yan Y, Reijnders MR, Schuurs-Hoeijmakers JH, Feenstra I, Bongers EM, Bosch DG, De Leeuw N, Pfundt R, Gilissen C, De Vries PF, Veltman JA, Hoischen A, Mefford HC, Eichler EE, Vissers LE, Nadif Kasri N, De Vries BB (2016) TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum Mol Genet 25:892–902
Pengelly RJ, Greville-Heygate S, Schmidt S, Seaby EG, Jabalameli MR, Mehta SG, Parker MJ, Goudie D, Fagotto-Kaufmann C, Mercer C, Debant A, Ennis S, Baralle D (2016) Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J Med Genet 53:735–742
Sadybekov A, Tian C, Arnesano C, Katritch V, Herring BE (2017) An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat Commun 8:601
Katrancha SM, Wu Y, Zhu M, Eipper BA, Koleske AJ, Mains RE (2017) Neurodevelopmental disease-associated de novo mutations and rare sequence variants affect TRIO GDP/GTP exchange factor activity. Hum Mol Genet 26:4728–4740
O’Brien SP, Seipel K, Medley QG, Bronson R, Segal R, Streuli M (2000) Skeletal muscle deformity and neuronal disorder in Trio exchange factor-deficient mouse embryos. Proc Natl Acad Sci U S A 97:12074–12078
Plageman TF Jr, Chauhan BK, Yang C, Jaudon F, Shang X, Zheng Y, Lou M, Debant A, Hildebrand JD, Lang RA (2011) A Trio-RhoA-Shroom3 pathway is required for apical constriction and epithelial invagination. Development 138:5177–5188
Guo S, Meng L, Liu H, Yuan L, Zhao N, Ni J, Zhang Y, Ben J, Li YP, Ma J (2021) Trio cooperates with Myh9 to regulate neural crest-derived craniofacial development. Theranostics 11:4316–4334
Chen J, Cai Z, Bai M, Yu X, Zhang C, Cao C, Hu X, Wang L, Su R, Wang D, Wang L, Yao Y, Ye R, Hou B, Yu Y, Yu S, Li J, Xue Y (2018) The RNA-binding protein ROD1/PTBP3 cotranscriptionally defines AID-loading sites to mediate antibody class switch in mammalian genomes. Cell Res 28:981–995
Wang J, Yuan L, Xu X, Zhang Z, Ma Y, Hong L, Ma J (2021) Rho-GEF Trio regulates osteosarcoma progression and osteogenic differentiation through Rac1 and RhoA. Cell Death Dis 12:1148
Chapman HA (2011) Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol 73:413–435
Sonoshita M, Itatani Y, Kakizaki F, Sakimura K, Terashima T, Katsuyama Y, Sakai Y, Taketo MM (2015) Promotion of colorectal cancer invasion and metastasis through activation of NOTCH-DAB1-ABL-RHOGEF protein TRIO. Cancer Discov 5:198–211
Wang B, Fang J, Qu L, Cao Z, Zhou J, Deng B (2015) Upregulated TRIO expression correlates with a malignant phenotype in human hepatocellular carcinoma. Tumour Biol 36:6901–6908
Park S, Lee H, Kim M, Park J, Kim SH, Park J (2018) Emerging roles of TRIO and F-actin-binding protein in human diseases. Cell Commun Signal 16:29
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F, Flaherty KR, Wells A, Martinez FJ, Azuma A, Bice TJ, Bouros D, Brown KK, Collard HR, Duggal A, Galvin L, Inoue Y, Jenkins RG, Johkoh T, Kazerooni EA, Kitaichi M, Knight SL, Mansour G, Nicholson AG, Pipavath SNJ, Buendia-Roldan I, Selman M, Travis WD, Walsh S, Wilson KC, American Thoracic Society, E. R. S. J. R. S., and Latin American Thoracic, S (2018) Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 198:e44–e68
Sikkema L, Ramirez-Suastegui C, Strobl DC, Gillett TE, Zappia L, Madissoon E, Markov NS, Zaragosi LE, Ji Y, Ansari M, Arguel MJ, Apperloo L, Banchero M, Becavin C, Berg M, Chichelnitskiy E, Chung MI, Collin A, Gay ACA, Gote-Schniering J, Hooshiar Kashani B, Inecik K, Jain M, Kapellos TS, Kole TM, Leroy S, Mayr CH, Oliver AJ, von Papen M, Peter L, Taylor CJ, Walzthoeni T, Xu C, Bui LT, De Donno C, Dony L, Faiz A, Guo M, Gutierrez AJ, Heumos L, Huang N, Ibarra IL, Jackson ND, Kadur Lakshminarasimha Murthy P, Lotfollahi M, Tabib T, Talavera-Lopez C, Travaglini KJ, Wilbrey-Clark A, Worlock KB, Yoshida M, Lung Biological Network, C, van den Berge M, Bosse Y, Desai TJ, Eickelberg O, Kaminski N, Krasnow MA, Lafyatis R, Nikolic MZ, Powell JE, Rajagopal J, Rojas M, Rozenblatt-Rosen O, Seibold MA, Sheppard D, Shepherd DP, Sin DD, Timens W, Tsankov AM, Whitsett J, Xu Y, Banovich NE, Barbry P, Duong TE, Falk CS, Meyer KB, Kropski JA, Pe’er D, Schiller HB, Tata PR, Schultze JL, Teichmann SA, Misharin AV, Nawijn MC, Luecken MD, Theis FJ (2023) An integrated cell atlas of the lung in health and disease. Nat Med 29:1563–1577
Wang L, Li Z, Wan R, Pan X, Li B, Zhao H, Yang J, Zhao W, Wang S, Wang Q, Yan P, Ma C, Yuan H, Zhao M, Rosas I, Ding C, Sun B, Yu G (2023) Single-cell RNA sequencing provides new insights into therapeutic roles of thyroid hormone in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 69:456–469
Wolf FA, Angerer P, Theis FJ (2018) SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19:15
Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R (2021) Integrated analysis of multimodal single-cell data. Cell 184(3573–3587):e3529
Mora AL, Rojas M, Pardo A, Selman M (2017) Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat Rev Drug Discov 16:755–772
Seipel K, O’Brien SP, Iannotti E, Medley QG, Streuli M (2001) Tara, a novel F-actin binding protein, associates with the Trio guanine nucleotide exchange factor and regulates actin cytoskeletal organization. J Cell Sci 114:389–399
Karampitsakos T, Juan-Guardela BM, Tzouvelekis A, Herazo-Maya JD (2023) Precision medicine advances in idiopathic pulmonary fibrosis. EBioMedicine 95:104766
Schulz MH, Pandit KV, Lino Cardenas CL, Ambalavanan N, Kaminski N, Bar-Joseph Z (2013) Reconstructing dynamic microRNA-regulated interaction networks. Proc Natl Acad Sci U S A 110:15686–15691
Fernandez IE, Eickelberg O (2012) The impact of TGF-beta on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc 9:111–116
Ng B, Dong J, D’Agostino G, Viswanathan S, Widjaja AA, Lim WW, Ko NSJ, Tan J, Chothani SP, Huang B, Xie C, Pua CJ, Chacko AM, Guimaraes-Camboa N, Evans SM, Byrne AJ, Maher TM, Liang J, Jiang D, Noble PW, Schafer S, Cook SA (2019) Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci Transl Med 11:eaaw1237
Bueno M, Calyeca J, Khaliullin T, Miller MP, Alvarez D, Rosas L, Brands J, Baker C, Nasser A, Shulkowski S, Mathien A, Uzoukwu N, Sembrat J, Mays BG, Fiedler K, Hahn SA, Salvatore SR, Schopfer FJ, Rojas M, Sandner P, Straub AC, Mora AL (2023) CYB5R3 in type II alveolar epithelial cells protects against lung fibrosis by suppressing TGF-beta1 signaling. JCI Insight 8
Acknowledgements
This research was financially supported by the Ministry of Science and Technology of the People’s Republic of China, the Science and Technology Department of Henan Province, and the Science and Technology Bureau of Xinxiang City. We are grateful for their generous funding, which has greatly contributed to the successful completion of this study.
Funding
This work was supported by Ministry of Science and Technology, PR China, 2019YFE0119500, State Innovation Base for Pulmonary Fibrosis (111 Project), the Education Ministry of National, 202200382, and Henan Project of Science and Technology, GZS2023008, 232102521025, 231111310400, and Xinxiang Major Project 21ZD002.
Author information
Authors and Affiliations
Contributions
LW and GY: conceptualization; WZ and CX: data generation; LW, WZ, CX, ZL: data analysis; WZ, HL, JY, YL, and CX: investigation; NW, LD, SM, LC, and YW: methodology; LW and WZ: draft; LW and GY: resources; IR: manuscript editing; GY: supervision, funding acquisition, validation, project administration, manuscript editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest concerning the contents of this article.
Ethical approval
The clinical studies were approved by the Henan Provincial Chest Hospital Medical Research Ethics Committee (No.2020-03-06). All animal procedures were approved and performed in accordance with the guidelines of the Institutional Animal Care and Use Committee at the Henan Normal University (IACUC, SMKX-2118BS1018).
Consent for publication
All authors have read and approved its submission to this journal.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
18_2023_5080_MOESM1_ESM.tif
Supplementary file1 Supplemental figure 1 (A) RT-qPCR test showed that inhibitor miR-29b increased the mRNA level of TRIOBP, ACTA2, VIM, COL1A1 and CDH2 in A549 cells. (B) RT-qPCR test showed that inhibitor miR-29b increased the mRNA level of TRIOBP, ACTA2, VIM, COL1A1 and FN1 in MRC5 cells. The results were analyzed by the unpaired Student’s t-test for comparisons between two groups with normal distribution, data are presented as mean±SD. Data *p<0.05, **p<0.01 and ***p<0.001 (TIF 60334 KB)
18_2023_5080_MOESM2_ESM.tif
Supplementary file2 Supplemental figure 2 (A) The model of AVVs and BLM-induced mouse model. (B-C) RT-qPCR (n=5) and western blot (n=3) test the knockdown efficiency of sh-TRIOBP-shRNA. (D) IHC staining showed TRIOBP increased in fibrotic lung. Date were analyzed by the unpaired Student’s t-test for comparisons between two groups with normal distribution, data are presented as mean±SD. Data **p<0.01 and ***p<0.001 (TIF 66293 KB)
18_2023_5080_MOESM3_ESM.tif
Supplementary file3 Supplemental figure 3 Western blot test showed the expression of Triobp, fibronectin and Gapdh in PMLFs (TIF 6615 KB)
18_2023_5080_MOESM4_ESM.tif
Supplementary file4 Supplemental figure 4 (A) Lung single cell RNA-seq data analysis of human, specifically focusing on the cell types present in the human lung. (B) Analysis of mouse single-cell data. (C) RT-qPCR test showed that TRIOBP silencing inhibited the mRNA level of TRIO in A549 cells. (D) Western blot of TRIO and GAPDH expression. TRIOBP knockdown inhibited the expression of TRIO in A549 cells. (E) RT-qPCR test showed that TRIOBP silencing inhibited the mRNA level of TRIO in MRC5 cells. (F) Western blot of TRIO and β-actin expression. TRIOBP knockdown inhibited the expression of TRIO in MRC5 cells. Date were analyzed by the unpaired Student’s t-test for comparisons between two groups with normal distribution, data are presented as mean±SD. Data *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 (TIF 119535 KB)
18_2023_5080_MOESM5_ESM.tif
Supplementary file5 Supplemental figure 5 (A) Western blot of Trio, E-cadherin, vimentin, α-SMA and β-actin expression (n=3). Trio knockdown inhibited the EMT process of MLE-12 cells. (B) The effect of Trio knockdown on the colony formation of MLE-12 cells. shNC: control lentivirus; shTrio: Trio lentivirus (TIF 24640 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Zhao, W., Xia, C. et al. TRIOBP modulates β-catenin signaling by regulation of miR-29b in idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 81, 13 (2024). https://doi.org/10.1007/s00018-023-05080-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-05080-4