
Abstract
Homologous recombination (HR) is a pathway to faithfully repair DNA double-strand breaks (DSBs). At the core of this pathway is a DNA recombinase, which, as a nucleoprotein filament on ssDNA, pairs with homologous DNA as a template to repair the damaged site. In eukaryotes Rad51 is the recombinase capable of carrying out essential steps including strand invasion, homology search on the sister chromatid and strand exchange. Importantly, a tightly regulated process involving many protein factors has evolved to ensure proper localisation of this DNA repair machinery and its correct timing within the cell cycle. Dysregulation of any of the proteins involved can result in unchecked DNA damage, leading to uncontrolled cell division and cancer. Indeed, many are tumour suppressors and are key targets in the development of new cancer therapies. Over the past 40 years, our structural and mechanistic understanding of homologous recombination has steadily increased with notable recent advancements due to the advances in single particle cryo electron microscopy. These have resulted in higher resolution structural models of the signalling proteins ATM (ataxia telangiectasia mutated), and ATR (ataxia telangiectasia and Rad3-related protein), along with various structures of Rad51. However, structural information of the other major players involved, such as BRCA1 (breast cancer type 1 susceptibility protein) and BRCA2 (breast cancer type 2 susceptibility protein), has been limited to crystal structures of isolated domains and low-resolution electron microscopy reconstructions of the full-length proteins. Here we summarise the current structural understanding of homologous recombination, focusing on key proteins in recruitment and signalling events as well as the mediators for the Rad51 recombinase.
Similar content being viewed by others

Introduction
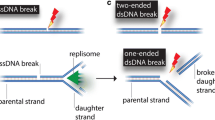
In eukaryotic cells, there are two major processes that act to repair double-strand breaks (DSB): end-joining (EJ) and homologous recombination (HR). Processes that involve end-joining are template independent and can be subdivided further into non-homologous EJ and microhomology-mediated EJ. End-joining (or more accurately ligation) without a template, or sister chromatid, can result in the loss of genetic material or chromosomal rearrangements when a large number of DSBs occur [1]. Homologous recombination, on the other hand, requires a DNA template for the repair process and is therefore considered more accurate as any missing genetic information lost in the DSB or during end processing is recovered. Similarly to end-joining, recombination can also be subdivided into distinct pathways known broadly as Single-Stranded Annealing (SSA), synthesis-dependent strand annealing (SDSA), break-induced replication (BIR) and canonical DSB Repair. In this review we focus on current structural insights into key proteins involved in canonical DSB repair via homologous recombination.
Overview of DNA damage response signal transduction
Eukaryotes have evolved sophisticated and highly organised processes in response to DNA damage. This coordinated effort is known as the DNA damage response (DDR) and operates to sense and signal genotoxic events, which are subsequently resolved by DNA repair machineries or cell death if DNA remains unrepaired. The DDR signal transduction pathway is primarily mediated by Serine/Threonine protein kinases belonging to the phosphatidylinositol 3-kinase-like protein kinase (PIKK) family, which include ATM (Ataxia Telangiectasia Mutated), ATR (ATM-related) and DNA-PK (DNA-dependent protein kinase). ATM and DNA-PK are recruited to a DNA double-strand break [2], with the activation of each kinase directing distinct repair pathways. For instance, DNA-PK regulates proteins involved in DSB end joining (NHEJ), whereas ATM regulates hundreds of substrates that ultimately bring about HR and cell cycle arrest. Whilst ATM responds to DSB, ATR, along with its partner ATRIP (ATR-interacting protein), is activated after its recruitment to replication protein A (RPA)-coated single-stranded DNA (ssDNA), generated at stalled replication forks or as intermediates during the processing of DSB [3] (Fig. 1). ATM and ATR phosphorylate serine/threonine residues of hundreds of substrate proteins at S/T-Q motifs and can elicit a second wave of phosphorylation events via their activation of other kinases such as Chk1 and Chk2, for example. DNA-PK catalytic subunit (DNA-PKcs) also shows preferences for phosphorylating serine/threonines residues at S/T-Q motifs. Additional residues in the vicinity of the S/T-Q motifs have been shown to be determinants of phosphorylation, such as hydrophobic or acidic amino acids promoting phosphorylation, whereas basic residues are negative determinants. Thus, the ATM, ATR and DNA-PK kinases have overlapping targets and can be considered ‘master transducers’ of the DDR [4].

Brief overview of DDR signalling. Canonical double-strand break (DSB) DNA damage response signalling pathway (see text for details)
In the HR pathway, the signalling of a DSB is initiated via the binding of the MRN complex (Mre11, Rad50 and Nbs1) to the broken DNA ends. The MRN complex plays a critical role in recruiting and activating ATM at DSB sites (Fig. 1). Once activated, ATM is able to phosphorylate a variety of proteins involved in the execution of the DDR, for example histone H2AX [5], where its phosphorylated form serves as a platform for further recruitment of additional DDR factors around the break site. DSBs also activate ATM yeast orthology Tel1 kinase signalling [6], with both ATM and ATR being critical for DSB repair and checkpoint activation. ATR signalling is elicited whenever ssDNA is generated, and this is reflected in ATR’s roles in a broad range of DNA damage, particularly those that interfere with DNA replication. Thus ATM and ATR kinases have DNA damage specificities and non-redundant functions despite overlapping substrates [7].
Architecture of the MRN complex: the initiator of the DSB repair
The MRN complex initiates DSB repair by recognising and resecting the free DNA ends at damaged sites. The complex has a multitude of functions: (1) facilitating ATP-dependent endonucleolytic cleavage near the double stand break and short range 3′–5′ resection from the nick site towards the DNA end to generate a 3′ overhang, (2) promoting recruitment of endonuclease ExoI or BLM complex to perform bulk 5′–3′ DNA resection, (3) loading of RPA on ssDNA overhang and activation of ATM (Fig. 1) [8–10]. A recent study suggests that MRN also acts as a processive factor for ExoI during long range resection [10]. In addition to its prominent roles in HR as the primary recruitment and activation factor for ATM, MRN can also stimulate NHEJ in an ATM-independent manner [11]. Furthermore, studies suggest recruitment of Cdk2 phosphorylated CtIP and BRCA1 at G2/S phase to MRN facilitates the removal of Ku70-Ku80 (Ku) cap at DNA end and promotes the dephosphorylation of 53BP1 [12], which in turn simulates HR and inhibit NHEJ [13, 14].
Mre11 and Rad50 are evolutionarily conserved enzymes. Mre11-Rad50 complexes found in archaea and bacteria share the same enzymatic activities and morphologies as their human counterpart, whereas Nbs1 only shows homology in eukaryotes [15]. Early atomic force microscopy (AFM) and electron microscopy studies on human and Saccharomyces cerevisiae Mre11-Rad50 reveal a heterodimeric architecture comprising of two Mre11 and two Rad50 molecules. As a structural maintenance of chromosomes (SMC) family protein, Rad50 has a nucleotide-binding head domain and a long anti-parallel coiled-coil insertion (Fig. 2a). Two Rad50 head domains are brought together by two long, 15–50 nm coiled-coils with Zn2+ hook domain dimerising at the middle hinge [16, 17] (Fig. 2a). Mre11 also dimerises and attaches to the globular domains of Rad50 through a helix-loop-helix (HLH) motif to form a globular head that binds DNA end [18–20]. Mre11 is composed of two α/β domains, an N-terminal Mn2+ bound nuclease domain and a C-terminal capping domain responsible for substrate recognition (Fig. 2b) [21, 22].

Structures of the MRN complex. a Domain structure of human Rad50, along with crystal structure of Pyrococcus furiosus RAD50 ATPase dimer (left, PDB ID: 3QKR) and human RAD50 coiled-coil domain (right, PDB ID: 5GOX) The N-terminal Walker A domain is shown in pink, C-terminal Walker B domain in orange, coiled-coil domain in green and Zn-hook in blue. Zn2+ is indicated as a red sphere. Bound Mre11 helix-loop-helix is shown in violet. b Domain structure of human Mre11 and crystal structure of human Mre11 N-terminal domain (PDB ID: 3T1I). Nuclease domain is shown in cyan, capping domain in red. Mn2+ is indicated as grey spheres. The latching loops that bind Nbs1 are disordered in this structure and are annotated as grey dash line. c Domain structure of human Nbs1 and crystal structure of Schizosaccharomyces pombe Nbs1 (PDB ID: 3HUF) with FHA domain shown in blue, BRCT 1 shown in yellow and BRCT 2 in orange. d Surface representation of ATP-hydrolysis driven RAD50 dimerisation (PDB ID: 5F3 W for the close conformation and 4FBW for the open conformation). e Intramolecular and intermolecular complex forms of MRN
Nbs1 attaches to Mre11 through two binding motifs, a Mre11 binding (MB) motif and a highly conserved FKXXFXK motif, which binds across the Mre11 dimer to the eukaryotic specific insertion loops on both Mre11 molecules [23]. Although the stoichiometry of Nbs1 to Mre11 is 2:2, only one FKXXFXK motif of the two Nbs1 is involved in Mre11 interface, sterically occluding the other Nbs1 [23]. The N-terminal domain of Nbs1 extends away from the globular head of Mre11-Rad50 and consists of a canonical fork head associated domain (FHA) and tandem BRCT repeats that are able to accommodate diverse phosphorylated proteins (Fig. 2c). The crystal structure of S. pombe Nbs1 shows that a 20° rotation of BRCT2 towards BRCT1-BRCT2 interface is triggered by bound CHK-dependent phosphorylated Ctp1, an orthologue of human CtIP [24]. As a result, the N-terminal domain of Nbs1 shifts 10 Å closer towards the C-terminus and possibly, the Mre11-Rad50 core. These structural transitions are likely to influence Mre11 and Rad50 in a regulatory manner [23–26]. However, structural evidence of the interplay between Nbs1 and the core of the MR complex, to facilitate regulation, remains unclear.
Crystal structures of ATP analogue-bound Mre11-Rad50 in complex with double-strand DNA (dsDNA) and recent cryoEM structure of the E. coli Mre11-Rad50 homolog in complex with DNA reveal an ATP-dependent clamp-like mechanism (Fig. 2d) [27–30]. Upon ATP binding, the two Rad50 head domains close up to facilitate the binding of DNA. Upon ATP hydrolyses, the Rad50 dimeric head domains dissociate, exposing the nuclease site of Mre11 to carry out DNA resection [28–32]. However, how the ATP-dependent open-close transition is regulated upon DSB recognition, repair initiation and downstream signalling transduction is still not well understood. A recent solution NMR study characterising fast timescale dynamics on side chain methyl groups of valines and isoleucines in Rad50 hinge helices adjacent to the Mre11-Rad50 interface reveals that ATP hydrolysis and Rad50 dimerisation are coupled through a dynamic allosteric network [27, 33]. Although only cause subtle structural changes, mutations disrupting the allosteric pathway increase the activity of ATP hydrolysis and the propensity for dimerization, altering the correlation of ATP hydrolysis and Mre11 nuclease activity [34].
Long coiled-coil structures are shown to extend from the Mre11-Rad50 core (Fig. 2a). A recent crystallographic study reveals two rod-like human Rad50 coiled-coil dimers in which Zn2+ hook is located at apex (Fig. 2a) [35]. Two different assemblies, intermolecular and intramolecular complexes, were observed as a result of Zn2+ hook dimerization (Fig. 2e) [35–37]. Crystallographic studies on Pyrococcus furiosus Rad50 show that an octamer formed by two MR complexes extends in opposite directions from the Zn2+ hook dimer at the centre of coiled-coils (Fig. 2e, right). This result is supported by a later atomic force microscopy observation of a DNA-induced parallel arrangement of yeast MR complex, which together suggest the coiled-coils could act as a tether to bridge sister chromatids together [36, 38]. The intramolecular complex involving the dimerization of the Zn2+ hook at the apex of the coiled coil has also been proposed (Fig. 2a, e left). This model is consistent with the results that mutations of conserved Zn2+ hook cysteines impaired Zn2+ binding and dimerisation, but not sister chromatid cohesion [27, 37, 39, 40]. The predominant state in solution and their functional significances thus remain unknown.
Despite the significance advance, there is still a lack of structural information of the C-terminal domain of eukaryotic Mre11 which contains a glycine-arginine rich (GAR) domain, a region that a number of disease-associate variants are reported to be located [41, 42]. Furthermore, the structural information of the intact MRN, and how it recruits and regulates other DSB associated proteins, remains unknown.
Structures of the large kinases that signal DDR
ATM, ATR and DNA-PKcs are key regulators of the DDR and perhaps some of the first transducers of damage signals. ATM, ATR and DNA-PKcs share structural similarities and domain organisations with other PIKK family members, such as mTOR (mammalian/mechanistic target of rapamycin) [43]. All PIKK family members share a conserved C-terminal PI3K-like kinase domain sandwiched between a unique region, N-terminal to the kinase, known as FAT (FRAP [FKBP12-rapamycin-associated protein], ATR, TRAPP [transformation/transcription domain-associated protein]) and, on the C-terminal side of the kinase domain, a PIKK regulatory domain (PRD) and FAT C-terminal domain (FATC). This catalytic portion only constitutes less than half of the polypeptide chain with the other comprising long stretches of HEAT (Huntington, elongation factor 3A, protein phosphatase 2A, TOR1) repeats. All PIKKs have between ~ 1400 and 3000 amino acid residues at the N-terminus that are arranged into these HEAT-repeat helical bundles described as a solenoid, that are likely important for recruitment of other proteins [44, 45]. ATR requires an integral partner, ATRIP, another HEAT-repeat containing protein (Fig. 3a, b). Due to the large size and limited sample quantity, these DDR PIKKs have been recalcitrant to traditional structural biology techniques such as X-ray crystallography, with the exception of DNA-PKcs and the C-terminal portion of mTOR [43, 46, 47]. More recently, with the advance of single particle cryoEM, there is a rapid increase in the number of low resolution and now higher resolution structures of ATM [48–52], ATR-ATRIP [53, 54] DNA-PK [44, 55] and mTOR [45, 56], providing significant insights into the precise domain organisation of these kinases. However, there is a clear lack of information on how these kinases are recruited and activated.

Structure of ATM and ATR-ATRIP. a Domain structure of human ATM (HsATM) and the Saccharomyces cerevisiae orthologue Tel1ATM (ScTel1ATM) and b human ATR (HsATR) with its binding partner ATR-interacting protein (HsATRIP) with Saccharomyces cerevisiae Mec1ATR (ScMec1ATR) and the ATRIP orthologue, Ddc2 (ScDdc2). The N-terminal HEAT-repeats are shown in cyan, the FAT (FRAP, ATR, TRAPP) in purple, the kinase domain in yellow, the PRD (PIKK regulatory domain) in orange and the FATC (FAT C-terminal domain) in dark blue. ATRIP/Ddc2, the specific ATR-interacting protein is also shown coloured green. Residue numbers are also labelled above. c The cryo-EM structure of human ATM and d the cryoEM structure of yeast Mec1ATR-Ddc2ATRIP, which is the most complete among the yeast and human structures, both coloured as in the schematic above, showing the dimeric architecture of these kinases along with approximate dimensions. e, f Features important for activity (see text), including the active site loop, are shown in a close up view of the atomic model, 90° rotated with respect to c, d
ATM and ATR Structures
Several higher resolution (~ 4 to 9 Å) cryoEM structures have now been determined for the human ATM [50] and ATR-ATRIP [53], alongside structures of the yeast orthologues Tel1A™ [49] and Mec1ATR-Ddc2ATRIP [54]. Overall, ATM and ATR-ATRIP adopt a similar “butterfly” architecture, and dimeric state with the kinase domain and tetratricopeptide repeat domains (TPR-D) of the FAT region forming a major dimerization interface [50, 53]. Additional dimer interfaces are also present in ATM and ATR contributed by the PIKK regulatory domain (PRD) and specifically for ATR/Mec1 an extensive interface contributed by ATRIP/Ddc2 dimerization along with the N-terminal region of the HEAT-repeats [53, 54] (Fig. 3c, d). DNA-PKcs differ in its quaternary structure as it exists as a monomer [47]. The kinase domain structure is highly conserved between the PIKKs, comprising a classical bi-lobed structure that binds ATP and magnesium ions in its cleft. The conserved activation loop, P-loop and catalytic loop are essential features for catalysis and are ordered in some of the high-resolution PIKK structures, which provides some insight into the regulation of the PIKK activity. Dimerisation of PIKKs has been described as a major mechanism for regulating the proper activity of such enzymes. The activation of these dimeric PIKKs has been proposed to rely on a dimer-to-monomer transition thus relieving the auto-inhibited state, resulting in a more accessible active site. Biochemical evidence for such a phenomenon is most compelling for ATM, with auto-phosphorylation at Ser1981 the switch for its activation [57]. ATR is also suggested to have an equivalent activation mechanism, with Thr1989 auto-phosphorylation the switch [58]. However, in both the ATM and ATR-ATRIP structures, the catalytic pockets of the kinase domain face away from each other. This outward facing arrangement, similar to mTOR, presents the active sites for substrate phosphorylation and does not explain how PIKK auto-inhibition and subsequent auto-phosphorylation occur, which is required for the activation of ATM and ATR [49, 50, 53, 54]. However, all the structures so far are in the absence of recruitment and activator factors, which might be required for a structural transition.
Recruitment and activation of ATM
ATM is recruited to DSBs via its binding to the C-terminus of Nbs1 [2] (Fig. 1). The MRN complex can stimulate the activity of ATM directly in vitro [59], suggesting that the MRN complex is responsible for recruitment and activation of ATM. However, the exact details of how ATM is activated by MRN remain to be uncovered. The ATM cryoEM structures shed some light on its activation with two different structures captured. Two distinct populations of ATM dimer exist in solution and possibly in a dynamic equilibrium; a closely packed ‘inactive’ dimer, and a more open accessible active conformation [50]. In the ATM structure of the closed ‘inactive’ conformation, the FATC, PRD and activation loop form a closely packed structural feature denoted as the FLAP (FATC, LBE, activation loop and PRD) that associates with a helical hairpin from a TPR domain of the FAT region from the other monomer (denoted as the FLAP-binding element [FLAP-BE]) [50]. This intimate arrangement between the monomers reduces accessibility to the active site of either kinase domain due to a short region of the PRD being pushed into the active site pocket and the FLAP-BE occluding substrate binding [50]. In the ‘open’ dimer structure, determined from the same cryoEM dataset, the ATM dimer is asymmetric with one kinase domain rotated 24° with respect to the other, as compared to the “closed” dimer. This conformational difference reduces the extensive dimer interface and alleviates the restriction to the kinase active site by removing the interaction between the FLAP-BE of one monomer with the FLAP of another resulting in the PRD becoming disordered [50]. This equilibrium between a closed and an open ATM dimer would allow regulation from inactive to active by shifting the conformational equilibrium. This model is in agreement with earlier data showing that ATM is activated upon DNA damage by the acetylation of Lys3016 that sit close to one another and to the FLAP-BE in the closed dimer. Lysine acetylation, catalysed by the Tip60 complex [60], could weaken the dimer interface thus favouring an open active population. However, many questions remain, such as how ATM is activated by auto-phosphorylation, how is the equilibrium shifted by MRN and if a dimer to monomer transition occurs or is required for activation.
Recruitment and activation of ATR
ATR has little basal activity and its activation is a multi-step process. In contrast to ATM and DNA-PKcs, which are activated by the recruitment of MRN and Ku70/80 complex to a DSB site, a range of genotoxic stresses elicit ATR signalling due to the fact that ATR, along with its obligatory binding partner ATRIP, is recruited to tracts of ssDNA that are coated with Replication Protein A (RPA) [61] (Fig. 1, see next section). These RPA-coated ssDNA recruitment platforms are typically generated as a result of stalled replication fork via the uncoupling of helicase and polymerase function, or the nucleolytic processing of DNA damage intermediates. It has also been observed that longer tracts of RPA-ssDNA serves as a more efficient platform for the recruitment of ATR-ATRIP [61]. The recruitment ATR-ATRIP (yeast Mec1-Ddc2) to RPA is mediated through interactions of an acidic patch (comprising Asp/Glu) of residues in the N-terminal domain of ATRIP/Ddc2 with a basic cleft of the N-terminal OB fold of the large subunit within RPA, RPA70 (and its yeast orthologue Rfa1) [61, 62]. The crystal structure of the N-terminal domain of Ddc2 (residues 1-109), which consists of the RPA binding domain (RBD) and coiled-coil domain (CCD), in complex with the Rfa1 N-terminal domain shows a mixture of hydrophobic and electrostatic interactions that drive the recruitment process [62]. The structure also shows that Ddc2 dimerises via its elongated CCD domain while each RBD domain interacts with a single Rfa1 N domain, consistent with earlier biochemical data of Ddc2 and ATRIP [3, 62, 63]. This crystal structure of the Ddc2 N-terminus is in stark contrast with the structural model proposed based on the 3.9 Å Mec1-Ddc2 cryoEM reconstruction, where a helical bundle domain, not directly involved in the dimer interface, has been built [54]. The crystal structure of Ddc2 is a domain in isolation whereas the cryoEM structure has limited resolution, thus the exact structures of Ddc2 and Mec1-Ddc2 complex require further investigation to reconcile these differences.
Unlike ATM with MRN, binding of ATR-ATRIP to RPA is insufficient to activate its kinase activity. Optimal ATR activation is achieved by the presence and interaction of additional activator proteins. The replication and DDR protein TopBP1 and its yeast orthologue Dpb11 are well characterised activators, which interact with ATRIP (Ddc2) and the PRD of ATR (Mec1) to stimulate the activation via its ATR-activation domain [64, 65], and can be recruited via its interaction with the damage clamp protein complex 9-1-1 (RAD9-RAD1-HUS1) (Fig. 1). A second ATR activator, ETAA1, was more recently discovered and is directly recruited via its interaction with RPA bound to ssDNA [64–68]. In budding yeast, alongside Dpb11, both Dna2 and Ddc1 (RAD9 in human) of 9-1-1 are Mec1/ATR activators. RPA and the 9-1-1 clamp are sufficient to recruit and activate Mec1 signalling in G1 phase and are required throughout the whole cell cycle. Dpb11 is additionally required in G2 phase whereas both Dpb11 and Dna2 are required in S phase [69]. Mec1 activation in yeast is mediated via two aromatic residues found in a long unstructured region of these activators [70]. Despite a wealth of biochemical and cellular work, the exact mechanism of ATR/Mec1 activation is still poorly understood. The recent cryoEM structures of Mec1-Ddc2 at 3.9 Å and the C-terminal catalytic core of ATR at 3.9 Å may partly explain the requirement of activators [53, 54].
In a similar fashion to ATM, the PRD plays an important role in ATR/Mec1 activation [64]. PRD is proposed to regulate dimerization and active site accessibility in both ATM and ATR/Mec1. The PRD of ATR/Mec1 forms an important dimer-interface of the global structure and this has implications on the conformation of the activation loop, P-loop and catalytic loop of the kinase domain. In the Mec1-Mec1 dimer, the PRD and FATC seem to hold and enclose the activation and catalytic loops of the kinase domain preventing access to the active site to substrates [54]. A key hydrophobic interaction between Met2312 of the PRD and Phe2244 of the conserved DFG motif in the activation loop has been shown to hold the kinase in an inactive state, with activators proposed to disrupt the PRD and alleviate the auto-inhibition [54, 64]. Intriguingly, the PRD is visible in the yeast Mec1 complex structure [54], but not in the human ATR complex, despite being at a similar resolution [53]. The structural insights thus far provide a framework to explain the low basal activity of ATR/Mec1 and the necessary requirement of activators to stimulate its kinase activity and why the PRD represents an important regulatory feature although the precise activation mechanisms requires the studies of activator bound complexes.
RPA-coated ssDNA
The signalling at a DSB by ATM-MRN and further signalling by ATR-ATRIP generate long stretches of single-stranded DNA produced by DNA resection processes (dsDNA unwinding and nucleolytic digestion) mediated by Exo1, DNA2, and BLM-Sgs1 [71]. RPA plays significant roles both co-ordinating this process and simultaneously preserving the integrity of the resultant ssDNA. RPA is a ubiquitous heterotrimeric ssDNA binding protein that is essential to nearly all DNA processing events. Comprised of three protein subunits, RPA70, RPA32 and RPA14 (Rfa1, Rfa2 and Rfa3 in yeast), RPA contains multiple oligonucleotide/oligosaccharide (OB)-folds that interact with both ssDNA and proteins [72] (Fig. 4a). The large subunit, RPA70, contains 4 OB-folds; an N-Terminal Domain, DNA-binding domain-A [DbdA], B [DbdB] and C [DbdC] connected by flexible linkers. RPA32 possesses a single DNA-binding OB-fold [DbdD] with a long flexible N-terminal hyperphosphorylation region and a C-terminal Winged-Helix (WH) domain [73]. The smallest subunit RPA14 has a single OB-fold (DbdE). RPA associates with ssDNA with very high affinity via its core DNA binding domains DbdA, B, C from RPA70, and DbdD, from RPA32. The N-terminal domain of RPA70, as discussed earlier, and the winged-helix (WH) domain of RPA32 mediate the majority of the protein–protein interactions [74, 75]. RPA binds nucleic acids in two conformational states with different affinities for ssDNA; a lower affinity mode (dissociation constant Kd ~ 50 nM) occludes a binding site length of ~ 8 to 10 nucleotides [76–78] and involves DbdA and DbdB of RPA70 [76, 78–80], whereas a higher-affinity mode (Kd in the low nM—pM range) occludes 30-nucleotides and engages all four DNA-binding domains [76, 81]. This switch is coupled to a conformational change that has been captured by X-ray crystallography of U. maydis RPA [76]. The compact structure provides some clues to the coupling of RPA-ssDNA binding and allosteric coupling with protein recruitment, particularly in replication [76] (Fig. 4a). However, RPA is flexible [78], associates with ssDNA in many numbers and is heavily modified by ubiquitylation and phosphorylation during DDR [75]. The way in which multiple RPAs associate on ssDNA and coordinate its vast array of processes remains to be determined.

Known structures of RPA and Rad51. a Structure and domain organisation of RPA. RPA consist of three subunits: RPA70, RPA32, and RPA14. The structure shown is RPA from of U. maydis (PDB ID: 4GNX) showing secondary structure elements of the major OB folds involved in ssDNA binding. b Structure of human Rad51 in the presence of ATP showing secondary structure elements and the right-handed helical formation (PDB ID: 5NWL). Rad51 consists of a N-terminal DNA binding domain (residues 1–84), a small linker domain (residues 85–97), and a C-terminal ATPase domain (residues 98–339). Important residues involved in ATP binding are highlighted and are located at the dimer interface of two Rad51 monomers within the filamentous crystal structure. c CryoEM structure of the presynaptic Rad51 filament in the presence of AMP-PNP bound to ssDNA (EMDB ID: 9566, PDB ID: 5H1B). Due to the intercalation of Arg235 and Val273, the Rad51 filament engages DNA as triplet clusters extending the DNA length by nearly 1.5 times in comparison to B-DNA. This mode of binding may help in the formation of the synaptic filament during the search for homologous regions
A crucial feature of RPA is that, whilst being able to bind nucleic acids with very high affinity, it must also be easily displaced to ‘hand-off’ ssDNA to other enzymes for further downstream processing. Single molecule work has shown that RPA can be displaced and exchanged with other ssDNA-binding proteins when there is sufficient concentration of unbound molecules [82]. This in vitro concentration-dependent exchange, referred to as facilitated exchange [83], is thought to rely on submicroscopic dissociated intermediates, where one of the DNA-binding domains is not bound to ssDNA, exposing a small patch of nucleic acid for other proteins to bind [82, 83]. However, this poses significant issues in vivo when considering inappropriate exchange, and there exists a number of proteins that facilitate the exchange of RPA for other genomic factors, such as Rad51.
The Rad51 recombinase and its interactions with RPA
Structure of Rad51 and nucleoprotein filament formation
The catalyst of DNA strand exchange and ultimately DNA repair is Rad51 recombinase. First discovered in yeast, it was recognised to be an orthologue of bacterial recombinase A (RecA) [84]. Recruited in an ATP-dependent manner to RPA coated single-strand DNA, mediated by BRCA2 in humans [85–87], this 43 kDa protein has been studied intensely since its discovery. Multiple crystal structures, EM reconstructions along with recent high resolution cryoEM structures have given much insight into its function. Rad51 has a two-lobed architecture of a mostly α-helical N-terminal domain of ~ 84 residues joined by a small helical linker to a larger C-terminal ATPase domain of ~ 240 residues [88–90] (Fig. 4b). Studies have described a DNA binding function to the N-terminal domain [91], possibly regulating the recognition of Rad51 for either ssDNA or dsDNA [92, 93]. The C-terminal domain contains a Walker A/B motif that is responsible for ATP binding and hydrolysis [94] (Fig. 4b). In the presence of ATP or ADP, Rad51 can readily form nucleoprotein filaments with DNA but the disassembly is dependent on the hydrolysis of ATP [95, 96]. Structures of the filamentous forms of the protein have shown that the ATP binding site is present at the interface between two Rad51 monomers [89, 90] (Fig. 4b). Residues Lys133 and Thr134 of human Rad51 from the Walker A motif bind to ATP where loop 315-323 from the adjacent monomer contributes hydrophobic interactions with the adenosine moiety. This stabilises interactions between Arg130 with Tyr315 and Glu322 from the adjacent monomer (Fig. 4b) and in turn communicates to residues involved in the binding of DNA such as Arg130 [97, 98].
Crystal and EM structures have also revealed the dynamic flexibility of Rad51 nucleoprotein filaments [89, 90, 99–102] where they vary in their pitch from 76 to 128 Å as compact and open forms respectively. This variation depends on the presence of DNA along with the state of the bound nucleotide. More specifically, filaments tend to be in more extended and dynamic state when ATP is bound versus when ADP is bound which might be an important feature in efficient homology searching [90, 100]. An example of this inherent flexibility is the recent crystal structure of human Rad51 filament in the presence of ATP where two different alternating dimer conformations were apparent which are due to small rotations around the ATP binding site [90]. Similarly the crystal structure of yeast Rad51 showed the presence of two different alternating dimer conformations around the ATP binding site which was due to a slight rotation between the N and C terminal domains [89].
Furthermore, high resolution cryoEM structures of the presynaptic (in the presence of ssDNA which represents the invading strand) and postsynaptic structures (in the presence of dsDNA, which represents the invading strand bound to a complementary strand) in the presence of AMP-PNP have shed light into the mechanism of DNA binding [101]. These structures show that Rad51 interacts with DNA through its phosphate backbone and very little structural changes occur when bound to ssDNA over dsDNA (Fig. 4c). In the presynaptic complex, each Rad51 monomer engages DNA in triplet clusters by the intercalation of Arg235 and Val273 (Fig. 4c). This mode is further stabilised by Arg235 interacting with the phosphate backbone of the complementary strand while having an electrostatic interaction with Asp274 when dsDNA is present. Hence this mode of DNA binding may be important in homology searching [101]. Though this study obtained a low resolution (~ 12 Å) structure of the synaptic complex and tentatively confirmed a second DNA binding site, much is still not known of how Rad51 binds dsDNA during strand invasion [101].
Interactions with RPA and effects on Rad51 filament
During HR, Rad51 must somehow replace RPA on ssDNA (Fig. 1). This is predominately catalysed and stabilised by various mediator proteins that directly interact with both or either of these proteins (see next section) as RPA inhibits the loading of Rad51 on DNA [103]. Nevertheless the presence of RPA is important in stimulating strand exchange by Rad51 [104, 105]. As such it is interesting to note that Rad51 physically interacts with RPA, specifically between its N-terminal domain and the RPA70 subunit between residues 168-327, which contains DbdA (Fig. 4a) [106, 107]. This interaction is insufficient to fully displace RPA [107]. Recent single-molecule imaging experiments showed that the facilitated exchange of RPA allows the exchange with Rad51. However, free RPA in solution can inhibit Rad51 filament nucleation but has less effects on filaments elongation [83]. The dynamic exchange of RPA therefore facilitates Rad51 nucleation, but when sufficient amount of free RPA exists in solution, resulting from Rad51 nucleation/elongation, RPA then inhibits further nucleation. RPA therefore has a role in both facilitating and regulating Rad51 nucleoprotein filament formation.
The Rad51 mediators
The loading of RAD51 on 3′-overhanged ssDNA at DSB and formation of nucleoprotein filaments are highly regulated events, not just by RPA itself as discussed in the previous section, but by a number of dedicated mediators. The main mediator protein to load Rad51 on ssDNA is the BRCA2, a 3,418 residue protein. In cells, BRCA2 is recruited to DSB sites by Partner and Localizer of BRCA2 (PALB2) and BRCA1 and binds to ssDNA through its C-terminal DNA binding domain that contains several OB-domains (Fig. 5a). Only limited structural information of BRCA2 is available due to its large size, low natural abundance in cells and containing a large amount of intrinsically unstructured regions. Three crystal structures of various isolated domains and regions of BRCA2 have been reported: a BRC4 motif in complex with Rad51, a short N-terminal peptide in complex with the WD40 domain of PALB2, and a C-terminal helical domain followed by three OB-folds in complex with ssDNA and/or a small acidic protein DSS1 [88, 108, 109].

Known structures of the main Rad51 mediators BRCA2, BRCA1, and PALB2. a Domain structure of BRCA2. BRC4 motif is colored in orange in the BRC4-Rad51 complex (upper left, PDB ID: 1N0 W). Helical domain in BRCA2 C-terminal Domain (BRCA2DBD) complex with DSS1 and ssDNA (upper right, PDB ID: 1MJE) is colored in sky blue, three OB folds in cyan and tower domain in pink. Magentas coil near helical and OB1 represents DSS1. ssDNA is shown in orange. EM map of full length BRCA2 (bottom, EMDB ID: 2779) is colored in yellow with its orthogonal view. b Domain structure of PALB2 and crystal structure of its WD40 domain (PDB ID: 3EU7) bound to BRCA2 N-terminal motif is colored (ruby). c Domain structure of BRCA1. NMR structure of RING heterodimer of BRCA1 and BARD1, BRCA1 is colored in red and BARD1 in light blue, bound Zn2+ ions are shown in sphere representation (PDB ID: 1JM7). Crystal structure of tandem BRCT repeats (purple and blue, PDB ID: 1T29). EM map of BRCA1-BARD1 is shown in violet (EMDB ID: 8833)
The C-terminal domain is the most conserved region of BRCA2. The crystal structures of ~ 800 amino acid C-terminal domain of mouse BRCA2 (BRCA2DBD) in complex with DSS1 and ssDNA determined more than 15 years ago [109], reveal an α-helical domain followed by three OB domains, forming a linear and elongated structure. The OB domains, both structurally and functionally, are similar to the OB domains in RPA as the BRCA2 OB domains can be replaced by RPA70 [110]. Both OB2 and OB3 have a canonical DNA binding groove accommodating the bound ssDNA. A long insertion in OB2 forms an α-helical coil which extends above and makes up the tower domain. The 70-amino acid DSS1 binds to BRCA2DBD in two sections with a disordered loop in the middle: the N-terminal portion of DSS1 tunnels through the helical domain and crosses the interface of OB1-helical domain whereas the C-terminal portion wraps around OB1 and ends up at OB1-OB2 interface. DSS1 is proposed as a ssDNA mimic that regulates both BRCA2-ssDNA and RPA-ssDNA associations. The latter is supported by biochemical studies demonstrating that DSS1 weakens the affinity of RPA toward ssDNA [111] and that BRCA2DBD-DSS1 complex simulates the displacement of RPA by Rad51 [111].
More recently, negative strain EM reconstructions of full length human BRCA2 and in complex with Rad51 have been reported [112]. In the reconstructions, BRCA2 dimerises to form an elliptical kidney-bean shape with C-terminal domains arching at both vertexes of the ellipse (Fig. 5a). Two sets of BRC repeats with bound Rad51 line up in the middle part. The dimeric BRCA2 is shown to bind to ~ 70nt ssDNA. It promotes RAD51 nucleation, but not filament growth. The structure provides insights into the overall architecture of intact BRCA2 as well as mechanism of Rad51 filament growth although the limited resolution precludes a precise assignment of structural domains [112].
In vivo studies suggest PALB2 recruits BRCA2 to nuclear foci and forms a complex with BRCA1 and BRCA2 [113–115]. However, apart from a C-terminal WD40 motif, a predicted chromatin binding motif and an N-terminal coiled-coil, which interacts with BRCA1, there is little structural information [113]. The structure of the WD40 domain in complex with the N terminal 18 amino-acid-peptide of BRCA2 shows that the BRCA2 peptide is accommodated at the outside of the β-propeller structure of the WD40 domain through polar interactions (Fig. 5b) [108]. The PALB2 binding motif on BRCA2 shows sequence conservation across species, suggesting the importance of the interactions in HR.
BRCA1 is a E3 ubiquitin ligase also involved in a diverse range of biological processes including HR [116–118], cell cycle checkpoint [119, 120] and transcriptional regulation [121, 122]. BRCA1 contains two major functional domains, the RING domain and tandem BRCT repeats at the N- and C-termini respectively (Fig. 5c) [123–125]. The RING domain at the N-terminus contains seven conserved Cys-His-Cys motifs with two bound Zn2+. Biochemical and NMR studies suggest it forms a tight heterodimer with BARD1, required for maintaining BRCA1 stability, E3 ubiquitin ligase activity and interaction with DNA [126–130], although its biological role and substrate in vivo are unclear [131, 132]. The C-terminal BRCT repeats incorporate two BRCT domains containing ~ 90 to 100 amino acids arranged in a linear fashion. The BRCT domains in BRCA1 play regulatory roles through the recognition of phosphorylated peptides with a pSXF motif on effectors such as 53BP1, BACH1, Abraxas, p53 and CtIP [123, 133–138]. The pSXF motif is recognised by a hydrophobic patch with a conserved lysine nearby. Similar features of BRCT domains have been observed in wide array of other DNA repair proteins, which bind to their respective physiological partner [139–141]. Although no high resolution structures of full-length BRCA1 are available, a low resolution negative stain EM structure of the BRCA1-BARD1 complex has been reported, in which it adopts a clamp-like architecture (Fig. 5c) [142].The RING domain and BRCT domain are mapped into the two opposite ends of the map. The presence of additional density near the RING domain is interpreted as ubiquitin in BRCA1 mutants.
Interactions of mediators with Rad51
The main mediator for Rad51 loading on ssDNA is BRCA2 [85]. It interacts with Rad51 via its eight BRC repeats along with a small 36 amino-acid sequence at its C-terminus (Fig. 5a). The first four BRC repeats bind to Rad51 monomers with higher affinity and the binding inhibits Rad51 ATPase activity, thus indirectly enhancing Rad51 binding to ssDNA [143, 144]. The 5th–8th BRC repeats of BRCA2 preferentially bind to and stabilise Rad51 filament [86, 111, 143, 144]. Investigations of the various isolated BRC repeats along with a construct of all eight BRC repeats and the full-length protein have shown that they directly promote the binding of Rad51 onto ssDNA over dsDNA by both preventing ATP hydrolysis of Rad51, thus enhancing Rad51 binding to DNA and through the ssDNA binding of BRCA2 [143–146]. Furthermore, studies show that BRCA2 promotes Rad51 nucleation, thus a mediator role for BRCA2 has been proposed [112]. Besides the BRC repeats, another Rad51 binding site was reported at the C-terminal domain between 3265 and 3330 amino acid of BRCA2 (Fig. 5a) [147]. The interaction is regulated by CDK dependent phosphorylation on S3291. Loss of phosphorylation results in HR defect phenotype, speculating a regulatory mechanism of cell cycle check point on homologous recombination.
Structurally the BRC motif consists of ~ 30 amino acids that bind to the C-terminal ATPase domain of RAD51 (Fig. 5a). A β -hairpin structure is formed at N-terminus, leading to an amphipathic α-helix segment followed by a 310 helix at the C-terminus. The hairpin extends the β -sheet on RAD51, mimicking the monomer–monomer contacts in a RAD51 filament [99]. BRC4 thus inhibits RAD51 oligomerisation. The ATPase active site is also inhibited allosterically through a BRC4 induced conformational closure to preclude ATP binding. The conformational change would also expose the BRC binding sites on adjacent promoters in a Rad51 filament/oligomer (Fig. 5a) [88, 148, 149].
It is also of note that the BRCA2 interacting proteins of PALB2 and the BRCA1-BARD1 complex have also been reported to bind RAD51, however no evidence of BRC repeats are present in their sequences [150–152]. PALB2 has been shown to bind Rad51 at both its N and C termini (residues 1–200 and residues 853–1186). On the other hand, Rad51 has been shown to bind to the centre of BRCA1 (residues 758–1064) and near the N-terminus of BARD1. Both PALB2 and the BRCA1-BARD1 complex have been reported to stabilise Rad51 nucleoprotein filaments during recombination and promote strand exchange [150–152]. As PALB2 is the bridging protein to form a complex between BRCA1 (possibly BARD1) and BRCA2 [113, 153], these findings imply that it is possible that all components of the resulting complex are involved in the loading and stabilisation of Rad51 on ssDNA (Fig. 1). However, further information is required on the exact roles of these proteins and more importantly, on the mechanisms of their actions.
A number of other complexes have also been reported to promote and stabilise the Rad51 filament during homology searching and strand invasion (Fig. 1) [154]. Two such complexes are Rad51 paralogues complexes, BCDX2 (Rad51B-Rad51C-Rad51D-XRCC2) and CX3 (Rad51C-XRCC3), though they have other roles earlier in the process of homologous recombination [155–157]. These paralogues have 20–30% sequence homology with RAD51, share a similar domain structure, [155], interact with Rad51 [158], bind to DNA [159], and are essential for homologous recombination [160]. Another complex involved in maintaining the Rad51 filament is the Shu complex in yeast consisting of Shu1, Shu2, Psy3, and Csm2 [161, 162]. A recent crystal structure of the complex revealed a V-shaped architecture and showed that Shu1, Psy3, and Csm2 are Rad51 paralogues, whereas Shu2 formed a novel fold with a zinc-finger domain [163]. In humans SWS1 and SWSAP1 have been identified as homologues of the Shu complex and are likely to have a similar role [163–165].
Future perspectives
Although structural and biochemical studies have revealed a plethora of information on the various roles of major players within the homologous recombination pathway, it is still unclear how their activities are regulated and coordinated. Due to the large size and difficultly in obtaining these proteins recombinantly and in assembling stable and homogenous functional complexes [85–87], structural insights have been elusive, especially for larger complexes with multiple components. However, with the recent advance in cryoEM [166] and the ability to produce recombinant complexes [167], we envisage that structural insights of these important complexes should be attainable in the near future.
Abbreviations
- ADP:
-
Adenosine diphosphate
- AMP-PNP:
-
Adenylyl-imidodiphosphate
- ATM:
-
Ataxia telangiectasia mutated
- ATP:
-
Adenosine triphosphate
- ATR:
-
Ataxia telangiectasia and Rad3-related protein
- BARD1:
-
BRCA1-associated RING domain protein 1
- BRCA1:
-
Breast cancer type 1 susceptibility protein
- BRCA2:
-
Breast cancer type 2 susceptibility protein
- BRCT:
-
BRCA1 C-terminal domain
- cryoEM:
-
Cryo electron microscopy
- DDR:
-
DNA damage response
- DSB:
-
Double-strand break
- EJ:
-
End joining
- MRE11:
-
Meiotic recombination 11 homolog 1
- MRN:
-
MRE11 RAD50 NBS1
- NBS1:
-
Nijmegen breakage syndrome protein 1
- OB:
-
Oligonucleotide/oligosaccharide-binding
- PALB2:
-
Partner and localiser of BRCA2
- RPA:
-
Replication protein A
References
Chang HHY, Pannunzio NR, Adachi N, Lieber MR (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18(8):495–506
Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434(7033):605–611
Ball HL, Myers JS, Cortez D (2005) ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol Biol Cell 16(5):2372–2381
Blackford AN, Jackson SP (2017) ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell 66(6):801–817
Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276(45):42462–42467
Hailemariam S, Kumar S, Burgers PM (2019) Activation of Tel1ATM kinase requires Rad50 ATPase and long nucleosome-free DNA but no DNA ends. J Bio Chem 294(26):10120–10130
Brown EJ, Baltimore D (2003) Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 17(5):615–628
Cannavo E, Cejka P (2014) Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514(7520):122–125
Mimitou EP, Symington LS (2011) DNA end resection–unraveling the tail. DNA Repair (Amst) 10(3):344–348
Myler LR et al (2017) Single-molecule imaging reveals how Mre11-Rad50-Nbs1 initiates DNA break repair. Mol Cell 67(5):891–898.e4
Rass E et al (2009) Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol 16(8):819–824
Isono M et al (2017) BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep 18(2):520–532
Langerak P, Mejia-Ramirez E, Limbo O, Russell P (2011) Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet 7(9):e1002271
Yun MH, Hiom K (2009) CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 459(7245):460–463
Demogines A et al (2010) Ancient and recent adaptive evolution of primate non-homologous end joining genes. PLoS Genet 6(10):e1001169
Anderson DE, Trujillo KM, Sung P, Erickson HP (2001) Structure of the Rad50 x Mre11 DNA repair complex from Saccharomyces cerevisiae by electron microscopy. J Biol Chem 276(40):37027–37033
de Jager M et al (2001) Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell 8(5):1129–1135
Das D et al (2010) Crystal structure of the first eubacterial Mre11 nuclease reveals novel features that may discriminate substrates during DNA repair. J Mol Biol 397(3):647–663
Park YB, Chae J, Kim YC, Cho Y (2011) Crystal structure of human Mre11: understanding tumorigenic mutations. Structure 19(11):1591–1602
Hopfner KP et al (2001) Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 105(4):473–485
Williams RS et al (2008) Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135(1):97–109
Sung S et al (2014) DNA end recognition by the Mre11 nuclease dimer: insights into resection and repair of damaged DNA. EMBO J 33(20):2422–2435
Schiller CB et al (2012) Structure of Mre11-Nbs1 complex yields insights into ataxia-telangiectasia-like disease mutations and DNA damage signaling. Nat Struct Mol Biol 19(7):693–700
Williams RS et al (2009) Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 139(1):87–99
Lloyd J et al (2009) A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell 139(1):100–111
Rai R et al (2017) NBS1 phosphorylation status dictates repair choice of dysfunctional telomeres. Mol Cell 65(5):801–817.e4
Deshpande RA et al (2014) ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J 33(5):482–500
Lim HS, Kim JS, Park YB, Gwon GH, Cho Y (2011) Crystal structure of the Mre11-Rad50-ATPγS complex: understanding the interplay between Mre11 and Rad50. Genes Dev 25(10):1091–1104
Liu Y et al (2016) ATP-dependent DNA binding, unwinding, and resection by the Mre11/Rad50 complex. EMBO J 35(7):743–758
Käshammer L, Saathoff JH, Lammens K, Gut F, Bartho J, Alt A, Kessler B, Hopfner KP (2019) Mechanism of DNA end sensing and processing by the Mre11-Rad50 complex. Mol Cell 76(3):382–394
Lammens K et al (2011) The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell 145(1):54–66
Seifert FU, Lammens K, Stoehr G, Kessler B, Hopfner K-P (2016) Structural mechanism of ATP-dependent DNA binding and DNA end bridging by eukaryotic Rad50. EMBO J 35(7):759–772
Williams GJ et al (2011) ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat Struct Mol Biol 18(4):423–431
Boswell ZK, Rahman S, Canny MD, Latham MP (2018) A dynamic allosteric pathway underlies Rad50 ABC ATPase function in DNA repair. Sci Rep 8(1):1639
Park YB et al (2017) Eukaryotic Rad50 functions as a rod-shaped dimer. Nat Struct Mol Biol 24(3):248–257
Hopfner K-P et al (2002) The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 418(6897):562–566
Wiltzius JJW, Hohl M, Fleming JC, Petrini JHJ (2005) The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat Struct Mol Biol 12(5):403–407
Moreno-Herrero F et al (2005) Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature 437(7057):440–443
Hohl M et al (2011) The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat Struct Mol Biol 18(10):1124–1131
Hohl M et al (2015) Interdependence of the rad50 hook and globular domain functions. Mol Cell 57(3):479–491
Déry U et al (2008) A glycine-arginine domain in control of the human MRE11 DNA repair protein. Mol Cell Biol 28(9):3058–3069
Boisvert F-M, Hendzel MJ, Masson J-Y, Richard S (2005) Methylation of MRE11 regulates its nuclear compartmentalization. Cell Cycle 4(7):981–989
Yang H et al (2013) mTOR kinase structure, mechanism and regulation. Nature 497(7448):217–223
Sharif H et al (2017) Cryo-EM structure of the DNA-PK holoenzyme. Proc Natl Acad Sci USA 114(28):7367–7372
Aylett CH et al (2016) Architecture of human mTOR complex 1. Science (80-) 351(6268):48–52
Sibanda BL, Chirgadze DY, Blundell TL (2010) Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature 463(7277):118–121
Sibanda BL, Chirgadze DY, Ascher DB, Blundell TL (2017) DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science (80-) 355(6324):520–524
Lau WC et al (2016) Structure of the human dimeric ATM kinase. Cell Cycle 15(8):1117–1124
Wang X et al (2016) Structure of the intact ATM/Tel1 kinase. Nat Commun 7:11655
Baretić D et al (2017) Structures of closed and open conformations of dimeric human ATM. Sci Adv 3(5):e1700933
Llorca O, Rivera-Calzada A, Grantham J, Willison KR (2003) Electron microscopy and 3D reconstructions reveal that human ATM kinase uses an arm-like domain to clamp around double-stranded DNA. Oncogene 22(25):3867–3874
Sawicka M et al (2016) The dimeric architecture of checkpoint kinases mec1atr and tel1 atm reveal a common structural organization. J Biol Chem 291(26):13436–13447
Rao Q et al (2018) Cryo-EM structure of human ATR-ATRIP complex. Cell Res 28(2):143–156
Wang X et al (2017) 3.9 Å structure of the yeast Mec1-Ddc2 complex, a homolog of human ATR-ATRIP. Science (80-) 358(6367):1206–1209
Yin X, Liu M, Tian Y, Wang J, Xu Y (2017) Cryo-EM structure of human DNA-PK holoenzyme. Cell Res 27(11):1341–1350
Yang H et al (2017) Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552(7685):368–373
Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421(6922):499–506
Liu S et al (2011) ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell 43(2):192–202
Lee JH, Paull TT (2004) Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science (80-) 304(5667):93–96
Sun Y, Jiang X, Chen S, Fernandes N, Price BD (2005) A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA 102(37):13182–13187
Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science (80-) 300(5625):1542–1548
Deshpande I et al (2017) Structural basis of Mec1-Ddc2-RPA assembly and activation on single-stranded DNA at sites of damage. Mol Cell 68(2):431–445.e5
Ball HL et al (2007) Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol Cell Biol 27(9):3367–3377
Mordes DA, Glick GG, Zhao R, Cortez D (2008) TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev 22(11):1478–1489
Kumagai A, Lee J, Yoo HY, Dunphy WG (2006) TopBP1 activates the ATR-ATRIP complex. Cell 124(5):943–955
Haahr P et al (2016) Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol 18(11):1196–1207
Bass TE et al (2016) ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol 18(11):1185–1195
Feng S et al (2016) Ewing tumor-associated antigen 1 interacts with replication protein A to promote restart of stalled replication forks. J Biol Chem 291(42):21956–21962
Majka J, Niedziela-Majka A, Burgers PM (2006) The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol Cell 24(6):891–901
Wanrooij PH, Tannous E, Kumar S, Navadgi-Patil VM, Burgers PM (2016) Probing the Mec1ATR checkpoint activation mechanism with small peptides. J Biol Chem 291(1):393–401
Nimonkar AV et al (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25(4):350–362
Kim C, Paulus BF, Wold MS (1994) Interactions of human replication protein A with oligonucleotides. Biochemistry 33(47):14197–14206
Fanning E, Klimovich V, Nager AR (2006) A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res 34(15):4126–4137
Feldkamp MD, Mason AC, Eichman BF, Chazin WJ (2014) Structural analysis of replication protein A recruitment of the DNA damage response protein SMARCAL1. Biochemistry 53(18):3052–3061
Maréchal A, Zou L (2015) RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res 25(1):9–23
Fan J, Pavletich NP (2012) Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev 26(20):2337–2347
Bochkareva E, Korolev S, Lees-Miller SP, Bochkarev A (2002) Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. EMBO J 21(7):1855–1863
Brosey CA et al (2013) A new structural framework for integrating replication protein A into DNA processing machinery. Nucleic Acids Res 41(4):2313–2327
Brosey CA et al (2015) Functional dynamics in replication protein A DNA binding and protein recruitment domains. Structure 23(6):1028–1038
Arunkumar AI, Stauffer ME, Bochkareva E, Bochkarev A, Chazin WJ (2003) Independent and coordinated functions of replication protein A tandem high affinity single-stranded DNA binding domains. J Biol Chem 278(42):41077–41082
Blackwell LJ, Borowiec JA (1994) Human replication protein A binds single-stranded DNA in two distinct complexes. Mol Cell Biol 14(6):3993–4001
Gibb B et al (2014) Concentration-dependent exchange of replication protein A on single-stranded DNA revealed by single-molecule imaging. PLoS One 9(2):e87922
Ma CJ, Gibb B, Kwon Y, Sung P, Greene EC (2017) Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res 45(2):749–761
Aboussekhra A, Chanet R, Adjiri A, Fabre F (1992) Semidominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to procaryotic RecA proteins. Mol Cell Biol 12(7):3224–3234
Jensen RB, Carreira A, Kowalczykowski SC (2010) Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467(7316):678–683
Liu J, Doty T, Gibson B, Heyer W-D (2010) Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol 17(10):1260–1262
Thorslund T et al (2010) The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat Struct Mol Biol 17(10):1263–1265
Pellegrini L et al (2002) Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 420(6913):287–293
Conway AB et al (2004) Crystal structure of a Rad51 filament. Nat Struct Mol Biol 11(8):791–796
Brouwer I et al (2018) Two distinct conformational states define the interaction of human RAD51-ATP with single-stranded DNA. EMBO J 37(7):e98162. https://doi.org/10.15252/embj.201798162
Aihara H, Ito Y, Kurumizaka H, Yokoyama S, Shibata T (1999) The N-terminal domain of the human Rad51 protein binds DNA: structure and a DNA binding surface as revealed by NMR. J Mol Biol 290(2):495–504
Galkin VE et al (2006) The Rad51/RadA N-terminal domain activates nucleoprotein filament ATPase activity. Structure 14(6):983–992
Subramanyam S, Jones WT, Spies M, Spies MA (2013) Contributions of the RAD51 N-terminal domain to BRCA2-RAD51 interaction. Nucleic Acids Res 41(19):9020–9032
Tombline G, Fishel R (2002) Biochemical characterization of the human RAD51 protein. I. ATP hydrolysis. J Biol Chem 277(17):14417–14425
van Mameren J et al (2009) Counting RAD51 proteins disassembling from nucleoprotein filaments under tension. Nature 457(7230):745–748
Hilario J, Amitani I, Baskin RJ, Kowalczykowski SC (2009) Direct imaging of human Rad51 nucleoprotein dynamics on individual DNA molecules. Proc Natl Acad Sci USA 106(2):361–368
Tombline G, Shim K-S, Fishel R (2002) Biochemical characterization of the human RAD51 protein. II. Adenosine nucleotide binding and competition. J Biol Chem 277(17):14426–14433
Cloud V, Chan Y-L, Grubb J, Budke B, Bishop DK (2012) Rad51 is an accessory factor for Dmc1-mediated joint molecule formation during meiosis. Science (80-) 337(6099):1222–1225
Short JM et al (2016) High-resolution structure of the presynaptic RAD51 filament on single-stranded DNA by electron cryo-microscopy. Nucleic Acids Res 44(19):9017–9030
Špírek M et al (2018) Human RAD51 rapidly forms intrinsically dynamic nucleoprotein filaments modulated by nucleotide binding state. Nucleic Acids Res 46(8):3967–3980
Xu J et al (2017) Cryo-EM structures of human RAD51 recombinase filaments during catalysis of DNA-strand exchange. Nat Struct Mol Biol 24(1):40–46
Yu X, Jacobs SA, West SC, Ogawa T, Egelman EH (2001) Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc Natl Acad Sci USA 98(15):8419–8424
Shinohara A, Ogawa T (1998) Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature 391(6665):404–407
Gupta RC, Golub EI, Wold MS, Radding CM (1998) Polarity of DNA strand exchange promoted by recombination proteins of the RecA family. Proc Natl Acad Sci USA 95(17):9843–9848
Baumann P, West SC (1997) The human Rad51 protein: polarity of strand transfer and stimulation by hRP-A. EMBO J 16(17):5198–5206
Golub EI, Gupta RC, Haaf T, Wold MS, Radding CM (1998) Interaction of human rad51 recombination protein with single-stranded DNA binding protein, RPA. Nucleic Acids Res 26(23):5388–5393
Stauffer ME, Chazin WJ (2004) Physical interaction between replication protein A and Rad51 promotes exchange on single-stranded DNA. J Biol Chem 279(24):25638–25645
Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH (2009) Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep 10(9):990–996
Yang H et al (2002) BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science (80-) 297(5588):1837–1848
Jacobs DM et al (1999) Human replication protein A: global fold of the N-terminal RPA-70 domain reveals a basic cleft and flexible C-terminal linker. J Biomol NMR 14(4):321–331
Zhao W et al (2015) Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Mol Cell 59(2):176–187
Shahid T et al (2014) Structure and mechanism of action of the BRCA2 breast cancer tumor suppressor. Nat Struct Mol Biol 21(11):962–968
Sy SMH, Huen MSY, Chen J (2009) PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA 106(17):7155–7160
Zhang F et al (2009) PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol 19(6):524–529
Xia B et al (2006) Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 22(6):719–729
Lou Z, Chini CCS, Minter-Dykhouse K, Chen J (2003) Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J Biol Chem 278(16):13599–13602
Venkitaraman AR (2001) Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J Cell Sci 114(Pt 20):3591–3598
Wu J, Lu L-Y, Yu X (2010) The role of BRCA1 in DNA damage response. Protein Cell 1(2):117–123
Zou J, Rezvani K, Wang H, Lee KS, Zhang D (2013) BRCA1 downregulates the kinase activity of Polo-like kinase 1 in response to replication stress. Cell Cycle 12(14):2255–2265
Wang B (2014) Analyzing cell cycle checkpoints in response to ionizing radiation in mammalian cells. Methods Mol Biol 1170:313–320
Lee JS, Collins KM, Brown AL, Lee CH, Chung JH (2000) hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 404(6774):201–204
Savage KI, Harkin DP (2015) BRCA1, a “complex” protein involved in the maintenance of genomic stability. FEBS J 282(4):630–646
Yu X, Chini CCS, He M, Mer G, Chen J (2003) The BRCT domain is a phospho-protein binding domain. Science (80-) 302(5645):639–642
Au WWY, Henderson BR (2005) The BRCA1 RING and BRCT domains cooperate in targeting BRCA1 to ionizing radiation-induced nuclear foci. J Biol Chem 280(8):6993–7001
Wu LC et al (1996) Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet 14(4):430–440
Mallery DL, Vandenberg CJ, Hiom K (2002) Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J 21(24):6755–6762
Christensen DE, Brzovic PS, Klevit RE (2007) E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat Struct Mol Biol 14(10):941–948
Stewart MD et al (2017) Tuning BRCA1 and BARD1 activity to investigate RING ubiquitin ligase mechanisms. Protein Sci 26(3):475–483
Paull TT, Cortez D, Bowers B, Elledge SJ, Gellert M (2001) Direct DNA binding by Brca1. Proc Natl Acad Sci USA 98(11):6086–6091
Simons AM et al (2006) BRCA1 DNA-binding activity is stimulated by BARD1. Cancer Res 66(4):2012–2018
Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE (2001) Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol 8(10):833–837
Xia Y, Pao GM, Chen H-W, Verma IM, Hunter T (2003) Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J Biol Chem 278(7):5255–5263
Wu Q et al (2016) Structure of BRCA1-BRCT/abraxas complex reveals phosphorylation-dependent BRCT dimerization at DNA damage sites. Mol Cell 61(3):434–448
Botuyan MVE et al (2004) Structural basis of BACH1 phosphopeptide recognition by BRCA1 tandem BRCT domains. Structure 12(7):1137–1146
Williams RS, Green R, Glover JN (2001) Crystal structure of the BRCT repeat region from the breast cancer-associated protein BRCA1. Nat Struct Biol 8(10):838–842
Liu X, Ladias JAA (2013) Structural basis for the BRCA1 BRCT interaction with the proteins ATRIP and BAAT1. Biochemistry 52(43):7618–7627
Clapperton JA et al (2004) Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat Struct Mol Biol 11(6):512–518
Shiozaki EN, Gu L, Yan N, Shi Y (2004) Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide. Mol Cell 14(3):405–412
Zappulla DC, Maharaj ASR, Connelly JJ, Jockusch RA, Sternglanz R (2006) Rtt107/Esc4 binds silent chromatin and DNA repair proteins using different BRCT motifs. BMC Mol Biol 7:40
Thornton K et al (1999) Purification, characterization, and crystallization of the distal BRCT domain of the human XRCC1 DNA repair protein. Protein Expr Purif 16(2):236–242
Rodriguez M, Yu X, Chen J, Songyang Z (2003) Phosphopeptide binding specificities of BRCA1 COOH-terminal (BRCT) domains. J Biol Chem 278(52):52914–52918
Liang Y et al (2017) Structural analysis of BRCA1 reveals modification hotspot. Sci Adv 3(9):e1701386
Carreira A, Kowalczykowski SC (2011) Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc Natl Acad Sci USA 108(26):10448–10453
Carreira A et al (2009) The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 136(6):1032–1043
Shivji MKK et al (2009) The BRC repeats of human BRCA2 differentially regulate RAD51 binding on single- versus double-stranded DNA to stimulate strand exchange. Proc Natl Acad Sci USA 106(32):13254–13259
Chatterjee G, Jimenez-Sainz J, Presti T, Nguyen T, Jensen RB (2016) Distinct binding of BRCA2 BRC repeats to RAD51 generates differential DNA damage sensitivity. Nucleic Acids Res 44(11):5256–5270
Esashi F et al (2005) CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 434(7033):598–604
Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL (1997) RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem 272(51):31941–31944
Bignell G, Micklem G, Stratton MR, Ashworth A, Wooster R (1997) The BRC repeats are conserved in mammalian BRCA2 proteins. Hum Mol Genet 6(1):53–58
Zhao W et al (2017) BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 550(7676):360–365
Buisson R et al (2010) Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol 17(10):1247–1254
Dray E et al (2010) Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat Struct Mol Biol 17(10):1255–1259
Zhang F, Fan Q, Ren K, Andreassen PR (2009) PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res 7(7):1110–1118
Maloisel L, Fabre F, Gangloff S (2008) DNA polymerase delta is preferentially recruited during homologous recombination to promote heteroduplex DNA extension. Mol Cell Biol 28(4):1373–1382
Masson JY et al (2001) Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev 15(24):3296–3307
Kurumizaka H et al (2002) Homologous pairing and ring and filament structure formation activities of the human Xrcc2*Rad51D complex. J Biol Chem 277(16):14315–14320
Kurumizaka H et al (2001) Homologous-pairing activity of the human DNA-repair proteins Xrcc3.Rad51C. Proc Natl Acad Sci USA 98(10):5538–5543
Schild D, Lio YC, Collins DW, Tsomondo T, Chen DJ (2000) Evidence for simultaneous protein interactions between human Rad51 paralogs. J Biol Chem 275(22):16443–16449
Yokoyama H et al (2004) Preferential binding to branched DNA strands and strand-annealing activity of the human Rad51B, Rad51C, Rad51D and Xrcc2 protein complex. Nucleic Acids Res 32(8):2556–2565
Chun J, Buechelmaier ES, Powell SN (2013) Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol Cell Biol 33(2):387–395
Ball LG, Zhang K, Cobb JA, Boone C, Xiao W (2009) The yeast Shu complex couples error-free post-replication repair to homologous recombination. Mol Microbiol 73(1):89–102
Gaines WA et al (2015) Promotion of presynaptic filament assembly by the ensemble of S. cerevisiae Rad51 paralogues with Rad52. Nat Commun 6:7834
Zhang S et al (2017) Structural basis for the functional role of the Shu complex in homologous recombination. Nucleic Acids Res 45(22):13068–13079
Martino J, Bernstein KA (2016) The Shu complex is a conserved regulator of homologous recombination. FEMS Yeast Res 16(6)
Liu T, Wan L, Wu Y, Chen J, Huang J (2011) hSWS1·SWSAP1 is an evolutionarily conserved complex required for efficient homologous recombination repair. J Biol Chem 286(48):41758–41766
Sawicka M, Aramayo R, Ayala R, Glyde R, Zhang X (2017) Single-particle electron microscopy analysis of DNA repair complexes. Meth Enzymol 592:159–186
Rees DM, Willhoft O, Lin C-L, Bythell-Douglas R, Wigley DB (2017) Production and assay of recombinant multisubunit chromatin remodeling complexes. Methods Enzymol 592:27–47
Acknowledgements
We apologize to those whose work we have not cited directly due to the conciseness of this review. We are grateful for the support by a Royal Society Newton International Fellowship to Y.S., a Wolfson Research Merit Award to XZ, Wellcome Trust Investigator Award (Grant Number 098412/Z/12/Z) and Medical Research Council grants to XZ.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sun, Y., McCorvie, T.J., Yates, L.A. et al. Structural basis of homologous recombination. Cell. Mol. Life Sci. 77, 3–18 (2020). https://doi.org/10.1007/s00018-019-03365-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03365-1