
Abstract
The aging process is associated with chronic low-grade inflammation in both humans and rodents, commonly called inflammaging. At the same time, there is a gradual decline in the functional capacity of adaptive and innate immune systems, i.e., immunosenescence, a process not only linked to the aging process, but also encountered in several pathological conditions involving chronic inflammation. The hallmarks of immunosenescence include a decline in the numbers of naïve CD4+ and CD8+ T cells, an imbalance in the T cell subsets, and a decrease in T cell receptor (TCR) repertoire and signaling. Correspondingly, there is a decline in B cell lymphopoiesis and a reduction in antibody production. The age-related changes are not as profound in innate immunity as they are in adaptive immunity. However, there are distinct functional deficiencies in dendritic cells, natural killer cells, and monocytes/macrophages with aging. Interestingly, the immunosuppression induced by myeloid-derived suppressor cells (MDSC) in diverse inflammatory conditions also targets mainly the T and B cell compartments, i.e., inducing very similar alterations to those present in immunosenescence. Here, we will compare the immune profiles induced by immunosenescence and the MDSC-driven immunosuppression. Given that the appearance of MDSCs significantly increases with aging and MDSCs are the enhancers of other immunosuppressive cells, e.g., regulatory T cells (Tregs) and B cells (Bregs), it seems likely that MDSCs might remodel the immune system, thus preventing excessive inflammation with aging. We propose that MDSCs are potent inducers of immunosenescence.
Similar content being viewed by others

Introduction
The aging process in humans is associated with a gradual decline in the functional capacity of adaptive and innate immune systems [1,2,3,4,5]. This age-related immune deficiency has been called immunosenescence. Clinically, immunosenescence reduces vaccination efficiency and impairs anticancer immunity, thus increasing the susceptibility to infections and the prevalence of cancers with aging. Immune deficiencies, similar to those encountered in immunosenescence, also appear in many diseases involving chronic inflammation, e.g., sepsis and autoimmune diseases. It is known that chronic low-grade inflammation, called inflammaging, down-regulates the immune responses of both the adaptive and innate immune system in humans and mice [3]. Not only does aging modulate the phenotypes and functions of immune cells, but it also affects their development and maturation in the bone marrow and spleen. Currently, it is still unclear whether immunosenescence is a significant defense mechanism against age-related chronic inflammation or a detrimental consequence of the chronic low-level inflammatory condition associated with aging. However, it is known that the aging process induces an active remodeling of the immune system rather than causing irreversible cellular senescence such as that occurring in non-immune cells [6, 7].
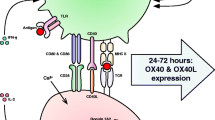
There is convincing evidence that chronic inflammation induces immunosuppression which inhibits both adaptive and innate immunity in different human disorders [8, 9]. Inflammatory factors promote the recruitment of immunosuppressive cells into inflamed tissues, where they suppress persistent inflammation and restore homeostasis in inflamed tissues. Myeloid-derived suppressor cells (MDSC) are specialized immunosuppressors which can control the functions of other immune cells, thus preventing excessive inflammatory responses [10, 11] (Fig. 1). For instance, Bunt et al. [12] demonstrated that chronic inflammation increased the accumulation of MDSCs into mouse mammary carcinoma; this induced immunosuppression, allowing the tumor cells to undergo immune escape. The inhibition of inflammation prevented the recruitment of MDSCs into tumors, a process that prevented the immunosuppression and subsequently blocked tumor growth. It is not only cancer-related inflammation which recruits MDSCs since these cells accumulate in many inflamed, non-neoplastic tissues and consequently suppress the functions of T cells and myeloid cells [13]. We will compare the immune profiles of immunosenescence and MDSC-driven immunosuppression. This comparison clearly highlights how immunosenescence might be driven by MDSCs which modulate the immunosuppressive network to the form encountered in both inflammaging and many inflammatory diseases. Bueno et al. [14] have also speculated that MDSCs could be involved in the generation of immunosenescence.

A schematic representation of MDSC-driven immunosenescence. The age-related cellular stress and senescence induce a condition termed inflammaging, which is associated with increased myelopoiesis. A mild inflammatory profile stimulates the production of MDSCs and other immunosuppressive cells, e.g., Tregs, Bregs, and Mregs. The cooperation between the components of this immunosuppressive network creates an immune-suppressive microenvironment, which after the remodeling of the immune system generates immunosenescence
Immunosenescence
There is a substantial literature indicating that a distinct functional decline occurs in the human immune system with aging, although all age-related alterations are not ubiquitous since many of these modifications seem to be context dependent [1, 3, 5, 15]. Briefly, an involution of the thymus and a marked decline in the numbers of naïve CD4+ and CD8+ T cells are the common biomarkers of immunosenescence in both humans and mice. Correspondingly, there is an expansion of memory CD4+ and CD8+ T cells, which might be a reflection of a persistent antigen load, e.g., induced by cytomegalovirus (CMV) infections with aging in humans [16, 17]. In addition, there are distinct deficiencies in the B cell compartment with aging which disturb the maintenance of humoral immunity, e.g., the decline in antibody production [18, 19]. Overall, the effects of aging are more profound on adaptive immunity than on innate immunity, although there exist also some functional deficiencies in dendritic cells [20], monocytes/macrophages [21], and natural killer cells [22] (Fig. 2). Interestingly, age-related immunosenescence seems to be an evolutionarily conserved phenomenon, e.g., in insects, birds, and mammals [23,24,25]. We will examine more thoroughly the specific changes in immunosenescence and compare their characteristics to those induced by MDSCs.

The comparison of immune cell phenotypes induced by MDSC-driven immunosuppression (upper panel) and age-related immunosenescence (lower panel)
Currently, there is still a debate about whether immunosenescence evokes inflammaging or whether it is inflammaging which reprograms the immune system [7, 15]. Given that inflammation is a consequence rather than the original perpetrator of the aging process, it seems reasonable to argue that chronic inflammation could adapt the immune system to cope with the aging microenvironment. The inhibition of T cells and some components of innate immunity might protect tissues from excessive injuries in conditions where persistent insults cannot be removed and the resolution of inflammation is impaired. Moreover, immunosenescence is associated not only with the aging process, but is also present in different pathological conditions involving chronic inflammation. For instance, inflammation has a crucial role in tumorigenesis where there is a significant increase in the biomarkers of immunosenescence, e.g., in breast cancer [26], multiple myeloma [27], glioma [28], and lung cancer [29]. In addition, it has been recognized that immunosenescence has a crucial role in the pathology of rheumatoid arthritis [30] and cardiovascular diseases [31]. It is known that the impaired resolution of acute inflammation induces chronic inflammation involving immune suppression, which provokes detrimental effects in host tissues [8]. It seems that persistent inflammatory conditions cause an adaptive response in the immune system by inducing a state of immunosuppression, similar to immunosenescence, not only in the aging process but also in inflammatory diseases.
MDSC-induced immunosuppression
MDSCs are a heterogeneous group of immunosuppressive myeloid cells which develop from common myeloid progenitor cells during the myelopoietic process [10, 32, 33]. Generally, we can separate these cells into human/mouse monocytic and granulocytic MDSC subsets which possess distinct phenotypes in their cell-surface markers as well as displaying some differences in their context-dependent immunosuppressive functions [33,34,35]. MDSCs are the major immune-suppressive cells in the body and thus they are involved in the host defense against inflammatory insults induced by either endogenous damages or by environmental insults, such as viral and bacterial infections [10, 36, 37]. Inflammatory factors, e.g., colony-stimulating factors (CSF), chemokines, and some cytokines can provoke emergency myelopoiesis in the bone marrow by stimulating the expansion and release of MDSCs [32, 38]. Chronic inflammation can also stimulate extramedullary myelopoiesis and trigger the generation of MDSCs, e.g., in the spleen and peripheral lymphoid organs. Several chemokines, e.g., CCL2, CXCL2, and IL-8, are able to induce the recruitment of MDSCs into inflamed tissues where they inhibit acute inflammatory responses allowing the resolution of inflammation [36, 37, 39, 40]. In inflamed tissues, many cytokines, e.g., IL-1β, IL-6, and TNF-α, as well as many inflammatory alarmins, such as HMGB1, S100 factors, and PGE2, can activate the immunosuppressive armament of MDSCs [36, 41,42,43]. The JAK-STAT and the NF-κB signaling pathways are the two major mechanisms involved in inducing the immunosuppressive potential of MDSCs [44]. Moreover, hypoxia/HIF-1α is a potent enhancer of MDSC-mediated immunosuppression [45].
MDSCs possess a powerful array of immune-suppressive mechanisms [10, 11, 46]. For instance, MDSCs secrete IL-10 and TGF-β cytokines which are potent anti-inflammatory and immunosuppressive factors. IL-10 and TGF-β are the major regulators of many of the functions performed by myeloid and lymphoid cells [47, 48]. For instance, IL-10 inhibits NF-κB signaling, a crucial inducer of pro-inflammatory reactions as well as stimulating STAT3 signaling, a key factor in the activation of MDSCs and immunosuppressive regulatory T cells (Tregs) [49,50,51]. Moreover, IL-10 can suppress the antigen presentation of dendritic cells and macrophages, and evoke macrophage M2 polarization [52, 53]. Similarly, TGF-β cytokines are potent human immunoregulators since TGF-β signaling can (1) convert naïve CD4+ cells into Tregs [54], (2) prevent T cell proliferation and the differentiation of Th1 and Th2 cells [47], (3) inhibit B cell responsiveness [55], (4) suppress human dendritic cell function [56], and (5) promote alternative M2 polarization in human macrophages [57]. In addition, TGF-β exerts degenerative bystander effects in non-immune cells, e.g., it can increase tissue fibrosis [58] and trigger cellular senescence in human fibroblasts [59]. However, MDSCs are not the only immune cells which secrete IL-10 and TGF-β cytokines, since other immunosuppressive cells also utilize the same mechanism to maintain immune suppression.
MDSCs alter the tissue microenvironment by actively generating reactive oxygen species (ROS) [33, 34, 60]. Corzo et al. [60] revealed that the STAT3 transcription factor, the major regulator of MDSC activation, induced the expression of NADPH oxidase (NOX2) which stimulated the production of ROS compounds in different mouse and human tumor models. MDSCs also generate nitric oxide (NO) by inducing the expression of inducible nitric oxide synthase (iNOS) [61]. Nagaraj et al. [62] demonstrated that peroxynitrite (ONOO−) nitrated the tyrosine residues in the T cell receptor (TCR) in mouse CD8+ T cells, thus preventing the antigen-specific stimulation of CD8+ T cells. It seems that the production of ROS is dependent on the insult and the subtype of MDSCs activated, e.g., a bacterial insult induced ROS generation in mouse granulocytic MDSCs, whereas monocytic MDSCs produced NO [34]. MDSCs are very resistant to ROS compounds; this is thought to be the reason why these cells can survive in inflammatory milieu in conditions of oxidative stress [63, 64]. Beury et al. [63] demonstrated that the increased expression of nuclear factor E2-related factor 2 (Nrf2), a powerful survival factor of cells in oxidative stress, enhanced the survival of MDSCs in tumors by decreasing the level of intracellular ROS and the rate of apoptosis in infiltrated MDSCs. Recently, Ohl et al. [64] revealed that the constitutive activation of Nrf2 in mouse myeloid cells increased the proliferation capacity of MDSCs, which induced the expansion of MDSCs and developed splenomegaly attributable to the accumulation of MDSCs into the spleen. They also reported that the increased expression of Nrf2 affected the metabolism of MDSCs by enhancing the expression of several genes involved in glycolytic energy metabolism. Oxidant species produced by MDSCs not only suppress the activity of immune cells, but also exert robust unspecific responses which affect both immune and non-immune cells, enhancing immunosenescence in inflamed tissues [65, 66]. In addition to ROS generation, the activation of MDSCs induces the expression of arginase 1 (ARG1) and indoleamine 2,3-dioxygenase (IDO) which catabolize arginine and tryptophan amino acids, respectively [67, 68]. A consequence of this enzymatic activation is that there is a shortage of these amino acids which inhibits protein synthesis, thus preventing the proliferation of T cells and other pro-inflammatory cells in inflamed tissues.
MDSCs can also induce immunosuppression of T cells via the cellular contacts mediated by immune checkpoint proteins [69, 70]. There is an abundant literature on the inhibitory checkpoint receptors and their inhibitors, since these membrane receptors are promising targets in cancer therapy. Recently, it was revealed that activated MDSCs expressed programmed death-ligand 1 (PD-L1) receptor protein which can bind to the PD-1 receptor of T cells and thus suppress their function [71, 72]. Many other human immune cells, e.g., B cells, dendritic cells, monocytes, and mast cells, express the proteins of the immunosuppressive PD-1/PD-L1 system [73]. Lei et al. [74] demonstrated that MDSCs acted through the PD-1/PD-L1 pathway to impair the ability of murine alveolar macrophages to respond to a pneumonia infection. Tregs and Bregs also utilize the PD-1/PD-L1 system to induce T cell immunosuppression [75, 76]. The PD-1/PD-L1 system is a potent source of immunosuppression in infections and tumors, but its role in age-related immunosenescence needs to be clarified.
Immunosuppressive cooperation between MDSCs, regulatory T and B cells, and macrophages
The immunosuppressive armament not only contains MDSCs, but also regulatory T cells (Treg), regulatory B cells (Breg), and regulatory macrophages (Mreg) which are also called M2c macrophages [77,78,79] (Fig. 1). There is a significant cross talk between these cell populations in an attempt to induce and maintain an immunosuppressive microenvironment in conditions of chronic inflammation, e.g., present in tumors and many inflammatory diseases. In general, IL-10 and TGF-β cytokines have a crucial role in this kind of communication within this immunosuppressive network. For instance, MDSCs can induce the differentiation of Tregs as well as enhance the expansion of Treg and Breg populations [80,81,82,83]. Consequently, activated Tregs can inhibit the functions of T cells, e.g., they suppress the proliferation of naïve/effector T cells [84]. Correspondingly, the stimulation of Bregs can (1) trigger the production of anti-inflammatory IL-10 and TGF-β cytokines, (2) inhibit the immune reactions mediated by Th1 cells, and (3) prevent autoimmune diseases [85]. In addition, one distinct subset of Bregs can convert resting CD4+ T cells into Tregs in mouse tumors [86]. Tregs and MDSCs can also establish a positive feedback loop, since Tregs stimulate the expansion and immunosuppressive activities of MDSCs [87]. On the other hand, Sinha et al. [88] reported that the activated T cells were able to induce the apoptosis of MDSCs in mice. MDSCs are known to express the death receptor Fas, whereas activated T cells secrete the Fas ligand (FasL). Sinha et al. [88] demonstrated that the Fas–FasL system controlled the numbers of MDSCs in circulation in the context of cancer and metastasis.
Myeloid-derived cells including MDSCs, monocytes, macrophages/microglia, dendritic cells, and natural killer (NK) cells reveal plastic phenotypes. For instance, macrophages can become polarized toward proinflammatory M1 and anti-inflammatory M2 properties in a context-dependent manner [89]. The polarization of macrophages is not fixed in vivo, but there is remarkable plasticity in the properties of the M1 and M2 subpopulations. Microenvironmental conditions control the polarization of macrophages in tissues. In inflammatory conditions, circulating monocytes will be recruited into inflamed tissues, where they differentiate into the M1-type of macrophages [90]. Human monocytic MDSCs can also be converted into inflammatory macrophages [91]. In particular, the exposure of proinflammatory TNF-α enhanced the maturation of human MDSCs into macrophages. Correspondingly, the interaction of MDSCs and macrophages potentiated the immune-suppressive capacities of both cell populations, e.g., in the tumor microenvironment [78, 92]. This bidirectional cross talk, both cell contact dependent and contact independent, robustly increased IL-10 production which consequently activated immunosuppressive Tregs and stimulated Th2 responses, whereas antigen presentation decreased, impairing the cytotoxicity of CD8+ and NK cells [78, 93]. In tumors, infiltrating monocytic MDSCs can be differentiated into the immunosuppressive M2 macrophages, commonly called tumor-associated macrophages (TAM) [94]. TAMs possess specific immune properties, probably educated by cancer cells, and thus they constitute the type M2d macrophage phenotype. It is the cooperation between Tregs and macrophages which also augments the immunosuppression in inflamed tissues. For instance, Tiemessen et al. [95] observed that human Tregs were able to polarize circulating monocytes/macrophages into the immunosuppressive M2 subtype. More recently, Riquelme et al. [96] demonstrated that the human Mregs were able to convert CD4+ T cells into FoxP3+ expressing, IL-10-secreting Tregs, which then suppressed T cell immunity and inhibited dendritic cell maturation. In addition, Lu et al. [97] reported that mouse Mregs induced Treg differentiation and increased the release of Tregs in the local lymph node drainage in mice with nephrosis. In conclusion, it seems that there is extensive cooperation between MDSCs and other immunosuppressive cell populations in the regulation of inflammatory conditions.
Increased myelopoiesis and expansion of MDSCs with aging
The inflammaging process is associated with significant changes in the hematopoietic system affecting the generation of myeloid and lymphoid cells in both humans and mice [98,99,100]. As the individual grows old, there is an increase in the rate of myelopoiesis, whereas lymphopoiesis clearly decreases in the bone marrow. This age-related imbalance in the immune system is caused by the myeloid-biased dominance of the hematopoietic stem cell (HSC) clones as compared to the progenitor clones of B and T lymphocytes. Moreover, a sizeable involution of the thymus with aging reduces lymphopoiesis, since thymus is an important lymphoid tissue in which T cells mature [101]. It is known that inflammatory mediators, e.g., CSFs, TNF-α, and interferons, originating from inflamed tissues can control myelopoiesis during the aging process [102, 103]. Inflammatory changes also appear with aging in the bone marrow which might also enhance aberrant myelopoiesis [104]. Currently, it is not known if it is the activation of MDSCs that controls the functions of HSCs and the progenitors of myeloid and lymphoid lineages in the bone marrow. However, there is substantial evidence that factors secreted by MDSCs, e.g., TGF-β, and IL-10, are potent regulators of HSCs [105, 106]. In particular, it has been claimed that aged HSCs were remarkably sensitive to TGF-β signaling. which might enhance myelopoietic differentiation [107]. Consequently, an age-related increase in myelopoiesis can enhance the production of myeloid cells, including MDSCs, and thus maintain inflammaging.
There is convincing evidence that the aging process increases the frequencies of circulating MDSCs, in both humans [108, 109] and mice [110]. Verschoor et al. [108] revealed that the levels of the CD11b+ CD15+-positive, granulocytic MDSCs were increased in the blood of community-dwelling seniors (61–76 years) and especially in frail elderly people (67–99 years). Recently, Alves et al. [109] demonstrated that the percentage of MDSCs was significantly higher in the blood of old people (80–100 years) than in their younger counterparts (20–30 years). Especially, the percentage of granulocytic MDSCs was robustly upregulated, while that of monocytic MDSCs was unaffected. It still needs to be clarified whether an increased myelopoiesis with aging in humans could increase the generation of MDSCs in the bone marrow, thus provoking the upregulation of MDSCs in the blood. However, there are several studies in mice revealing that the numbers of MDSCs are clearly increased with aging in the bone marrow, spleen, and peripheral lymph nodes [110,111,112,113,114]. The MDSCs isolated from the spleen of aged mice potently suppressed the antigen-induced T cell proliferation and T cell-dependent antibody production as well as inhibited the tumor cytotoxicity of T cells [110, 111]. The increased accumulation of MDSCs with aging was also linked to an enhanced growth of tumors in old mice. Flores et al. [114] reported that there were greater numbers of MDSCs in the bone marrow of two progeroid mouse species, i.e., Ercc1 and BubR1 mutants, than in wild-type mice. Currently, it is difficult to confirm whether the inflammaging process increases the level of MDSCs in peripheral tissues attributable to technical problems and the plasticity of MDSC phenotype.
Not only does the MDSC population of the immunosuppressive network expands with aging, but also the numbers of Tregs (CD25+ FOXP3+) increase in both elderly humans and mice [115,116,117,118,119]. This increase in the number of Tregs was significant in the spleen and lymph nodes, but also present in the skin. There were also age-related changes in the subtypes of Tregs, i.e., the number of naturally occurring thymus-derived Tregs (tTregs) increased with aging, whereas that of inducible Tregs (iTregs) seemed to decline in old mice [120]. Chougnet et al. [121] demonstrated that the aged Treg population was more resistant to apoptosis; this phenomenon was attributable to the reduced expression of pro-apoptotic Bim protein which might enhance the survival of aging Tregs. However, the Tregs from old mice were functionally active, i.e., they were able to prevent the activation of immune responses of effector T cells. Garg et al. [118] demonstrated that the Tregs from aged mice were more potent in inhibiting the proliferation of effector T cells than those isolated from young mice. Aged Tregs also secreted an increased level of the immunosuppressive IL-10 cytokine. Moreover, Garg et al. [118] presented evidences that the age-related increase in the expression of FOXP3+, the master regulator of Tregs, was induced by a hypomethylation of the enhancer sequences of FoxP3 gene. Given that the interactions between MDSCs, Tregs, Bregs, and Mregs maintain the immunosuppressive milieu of tissues (Fig. 1), it is apparent that the age-related functions of Bregs and Mregs need to be clarified. There is an abundant literature on macrophage polarization with aging and in the repair process of tissue injuries [122, 123]. It seems that the responses are remarkably context dependent, probably attributable to the plasticity of macrophages and the complex regulation of the M1/M2 polarization process. Macrophage polarization can also fluctuate during the repair process [124]. However, Jackaman et al. [125] demonstrated that the numbers of anti-inflammatory M2 macrophages were robustly increased in the bone marrow, spleen, and lymph nodes of old mice as compared to their younger counterparts. Wang et al. [126] reported that the aging process in muscles was associated with an increase in the level of M2a macrophages, thus causing fibrosis in muscles. It is likely that the cooperation of tissue-resident macrophages with MDSCs and Tregs might switch these cells toward the immunosuppressive M2 phenotype during the aging process. For instance, MDSCs and Tregs secrete IL-10 and TGF-β, which polarize macrophages into the Mreg phenotype.
Comparison of immune profiles of immunosenescence and MDSC-driven immunosuppression
Given that MDSCs are potent inducers of immunosuppression of adaptive immunity and a significant expansion of MDSCs and Tregs accompanies aging, this could induce and maintain a chronic state of immunosenescence. The MDSC-induced immunosuppression would represent the remodeling mechanism of immunosenescence. The remodeling of immune system might be crucial for the survival of tissues in conditions of chronic inflammation, e.g., in many pathological conditions and even in low-grade inflammaging. It is likely that MDSCs affect immune cells in a direct manner, but some responses detected in in vivo experiments can also be mediated via their interaction with other immunosuppressive cells, e.g., Tregs and Mregs (Fig. 1). Next, we will examine in more detail the similarities in the immune profiles generated by immunosenescence and the MDSC-induced immunosuppression in adaptive and innate immune systems.
Adaptive immunity
T cells
There is an abundant literature indicating that immunosenescence is associated with a progressive decline in the numbers of naïve (CD45RA+) CD4+ and CD8+ T cells, whereas the numbers of the memory type (CD45RO+) of CD4+ and CD8+ T cells gradually increase with aging [7, 127,128,129] (Fig. 2). This hallmark of immunosenescence has been commonly observed in both humans and mice. The age-related loss of CD4+ and CD8+ T cells is attributable to a decline in the clonal expansion of T cell clones in the bone marrow and thymus, as well as to a decrease in their proliferation after maturation. In addition, there are aging-associated changes in the homeostasis of T helper (Th) cells and Tregs. The numbers of Tregs significantly increase with aging and, at the same time, there seems to occur reshaping in the numbers of Th populations. The production of Th1 and Th2 cytokines declines in elderly humans [130] and aging seems to be accompanied by a shift from the Th1 to the Th2 response, although results are more inconsistent in humans [131]. Interestingly, the presence of Th17 cells increases with aging in both humans and mice [132, 133]. Th17 cells are plastic cells which possess proinflammatory properties, e.g., they are involved in autoinflammatory diseases, but they can also exert antifungal and immunosuppressive effects, e.g., they can inhibit T cell-mediated immunity [134]. In addition to the changes in the subsets of T cells, there are significant age-related alterations in the functional capacities of both CD4+ and CD8+ T cells [135, 136]. In general, the responsiveness to distinct external insults decreases with aging in T cells, e.g., there are significant declines in both the proliferation and cytotoxicity of CD8+ T cells [137]. This loss of sensitivity is associated with clear age-related changes in the signaling responses of T cells which affect the differentiation of T cells and the immune outcomes of T cells in host defense [7, 138, 139].
TCRs and their co-receptors, e.g., CD28+, have an important role in the recognition of antigens and the activation of CD4+ and CD8+ T cells (Fig. 2). Recent high-throughput TCR sequencing studies have revealed that the structural TCR repertoire of naïve CD4+ and CD8+ T cell populations in humans dispersed with aging, indicating the non-uniform clonal proliferation of naïve T cells [140]. Moreover, Qi et al. [141] demonstrated that the diversity of the human TCR repertoire declined with aging in both naïve CD4+ and CD8+ T cells, although the clonal sizes of distinct phenotypes expanded with aging. Age-related changes were more modest in the memory CD4+ and CD8+ T cell populations. Several earlier studies have demonstrated that the aging process reduced the functional responses of TCRs [7], e.g., the formation of immune synapses with antigen presenting cells [142]. Aged human T cells also lose the expression and signaling of CD28 receptors which are crucial co-stimulators of TCR activation [143, 144].
T cells are the major target of MDSC-induced immune tolerance in tumors and several inflammatory disorders. MDSCs possess effective mechanisms to suppress the function of T cells and thus provide an immune escape not only for cancer cells, but also for organ allografts in transplantation medicine [145]. For instance, Nagaraj et al. [62] demonstrated that MDSCs were able to nitrate TCR proteins, inducing the dissociation of the TCR complex, which prevented the recognition of foreign antigens and thus suppressed the activation of T cells. Nitration might also affect the dispersion of the TCR repertoire with aging. Recently, Feng et al. [146] revealed that MDSC nitrated the lymphocyte-specific protein tyrosine kinase (LCK) and thus prevented the activation of TCR signaling. The inhibition of TCR signaling with nitration might induce T cell immunosuppression, not only in cancers and inflammatory conditions, but also in immunosenescence. In addition, MDSCs can suppress the functions of T cells by contacting them via the PD-1/PD-L1 checkpoint proteins. For instance, the activation of MDSCs clearly stimulated the expression of PD-L1 in human MDSCs [72]. Lu et al. [71] demonstrated that tumor-infiltrated MDSCs robustly expressed PD-L1 protein in human patients. In addition, Tregs induced the expression of PD-L1 (also called B7-H1) in MDSCs in mouse melanoma [147]. Although there is no direct evidence for the existence of these PD-L1-positive MDSCs in aged tissues, the microenvironment of inflammaging tissues contains different inflammatory factors which are activators of MDSCs [148]. Interestingly, Shimada et al. [149] reported that the expression of PD-1 was clearly increased with aging in mouse memory CD4+ T cells. Given that the PD-1/PD-L1 checkpoint system has an important role in the MDSC-induced immunosuppression, it seems likely that MDSCs could also exploit this mechanism to evoke T cell anergy and immunosenescence in inflammaging.
TGF-β is the major cytokine controlling the functions of the immunosuppressive network in a reciprocal manner. TGF-β is secreted by MDSCs, Tregs, and Bregs and has a crucial role in the proliferation and differentiation of T cells [150,151,152]. For instance, TGF-β inhibits the differentiation of Th1 and Th2 cells, whereas it enhances the differentiation of Tregs and Th17 cells. Interestingly, Th17 cells are very plastic cells which can possess pro-inflammatory properties, e.g., in autoinflammatory diseases, but they also exert immunosuppressive properties inhibiting T cell-based immunity. In addition, TGF-β was reported to reduce the cytotoxicity of CD8+ T cells, and the prevention of TGF-β signaling improved anti-tumor immunity [151]. Moreover, TGF-β enhanced the quiescence of hematopoietic stem cells, especially the development of lymphoid lineages declined with aging [153]. The TGF-β signaling maintains immune tolerance to both self and foreign antigens by controlling the differentiation and functions of effector T cells and Tregs. One could speculate that the increased presence of MDSCs and Tregs with aging might enhance the production of TGF-β and consequently augment the immunosenescence of T cells. The activation of MDSCs stimulates the expression of ARG1 and IDO which induces the metabolic catabolism of l-arginine and tryptophan. Some other myeloid cells can also express ARG1 and IDO proteins. In cancer studies, there is clear evidence that the induction of ARG1 and IDO in MDSCs stimulates T cell tolerance which enhances tumorigenesis [68, 70]. While it is known that aging affects arginine and tryptophan metabolism, it needs to be clarified whether immune effects are mediated by the depletion of these amino acids or through the production of their metabolites, i.e., NO from l-arginine and kynurenine metabolites from tryptophan [154, 155]. In conclusion, it seems that MDSCs induce the immunosuppressive profile of T cells which closely resembles the characteristics of the immunosenescence encountered in T cells.
B cells
B lymphocytes are generated in the bone marrow (BM) and subsequently immature B cells migrate into the spleen to undergo distinctive maturation and activation phases, e.g., the negative selection by self-antigens. In the aged human and mouse BM, there exists a myeloid-biased dominance of hematopoietic stem cell clones which downregulates the generation of lymphopoietic progenitors [100, 156]. The decline in the B cell progenitor clones also affects the development of the mature B cell compartment, another factor which enhances immunosenescence. The age-related increase in inflammatory changes in the BM, e.g., the increased presence of IL-1β and S100A9 factors, impairs B cell lymphopoiesis [19, 157]. Kennedy and Knight [158] demonstrated that MDSCs inhibited B lymphopoiesis through soluble factors in mouse BM cultures. Recently, they reported that inflammasomes might be involved, since exposure to the inflammasome inhibitor, glibenclamide, prevented the decline in B lymphopoiesis in the BM cultures [157]. Interestingly, they observed that the activation of inflammasomes promoted the development of MDSCs in the BM cultures. Flores et al. [114] revealed that the numbers of MDSCs were robustly increased in mouse BM with aging. The failure of B cell generation and the reductions in the numbers of these cells in the inflammatory BM may be caused by the TGF-β produced by MDSCs. It is known that TGF-β is a potent inhibitor of B cell proliferation and activation [47]. It can also enhance the apoptosis of immature and resting B cells. The numbers of human mature naïve B cells and the antigen-experienced memory (CD27+) B cells significantly decline with aging, whereas at the same time, the percentage of human late/exhausted memory B cells (CD27−) increase in the circulation [159, 160] (Fig. 2). Moreover, the diversity of the human B cell repertoire decreases with aging; this may reduce the responsiveness to infections and vaccination as well as increase the production of autoreactive antibodies [19, 156, 160, 161].
In addition to the inhibition of B cell development, MDSCs can also suppress the functions of mature human B cells, e.g., reduce their proliferation, homing, and antibody production [162,163,164]. MDSCs can inhibit the functions of B cells by secreting soluble factors, e.g., NO/ROS, PGE2, and TGF-β. Given that MDSCs can induce the expansion of Bregs and ameliorate autoimmunity [82], it seems that there exists a reciprocal regulation between MDSCs and Bregs, since the tumor-derived Bregs can educate both the monocytic and granulocytic MDSCs of mice and humans, by stimulating their immunosuppressive properties [165]. Moreover, Bregs can convert CD4+ T cells into Tregs [86] which are potent inhibitors of B cell functions. Therefore, it should be clarified whether immunosenescence affects the proliferation and functions of Bregs. However, it is known that certain inflammatory conditions increase the proliferation and immunosuppressive activities of Bregs [79]. It seems that chronic inflammation is the main cause of the functional impairments in the B cell compartment which accompany aging and thus it is likely that MDSCs are involved in the immunosenescence of B cells.
Innate immunity
Currently, there is a debate about the role of innate immunity in the maintenance of immunosenescence. Clearly, age-related changes have been reported in the cells of innate immunity, but many observations are inconsistent, probably due to the fact that alterations are context dependent. The controversies may well be attributed to the high plasticity of myeloid-derived cells i.e., the cells of myeloid lineage can mature/convert into diverse myeloid subsets. For instance, MDSCs can differentiate into macrophages in inflammatory microenvironments (see above). Moreover, both a context-dependent polarization and a modulation of cell subsets are common characteristics of myeloid cells. The age-related changes in innate immunity have been described in detail elsewhere [166, 167]. We will focus on the modifications which have confirmed the potential associations with the MDSC-induced regulation.
Dendritic cells
One major function of dendritic cells (DCs) is the antigen processing and its presentation to T and B lymphocytes. Thus, DCs have a crucial role in the function of the adaptive immune system. It seems that the numbers of circulating DC subsets do not significantly change with aging, although there are conflicting results between studies. However, it appears that there is a decline in the numbers of Langerhans cells with aging in both human and mouse skin [168, 169]. It seems that aging affects several functions of DCs, although the results are not always consistent [20, 170]. There are both human and mouse studies indicating that the migration of DCs is impaired and their capacity to phagocytose antigens, process them, and subsequently present them to T cells decreases with aging [20, 170,171,172] (Fig. 2). However, contradictory observations have been reported. Moreover, Panda et al. [173] demonstrated that there was a significant age-related decrease in the amounts of cytokines induced by the activation of TLRs in human myeloid and plasmacytoid DCs. This may impair the priming of T cells and furthermore inhibit the polarization of Th cells. Currently, the mechanisms of cross talk between DCs and T cells need to be clarified, although a role for checkpoint inhibitors has been proposed [174]. In cancer studies, there is clear evidence that MDSCs inhibit the functions of DCs [78]. For instance, Greifenberg et al. [175] reported that the activation of MDSCs with LPS and IFN-γ prevented the differentiation of DCs in mouse BM cultures. Poschke et al. [176] demonstrated that the DCs which had been generated in a co-culture with human MDSCs displayed a reduced antigen uptake and impaired cytokine production. TGF-β1 is a potent inhibitor of the maturation and function of human and mouse DCs [56, 177]. However, there are differences between DC populations, since TGF-β1 was required for the development and maintenance of Langerhans cells in mice [178]. It seems reasonable to propose that MDSCs can inhibit the functions of DCs and, in this way, contribute to the suppression of T and B cells in cancers and inflammatory conditions.
Natural killer cells
Natural killer (NK) cells have an important role in innate immunity, although they are lymphocytes originating from lymphoid progenitors in the bone marrow. NK cells are cytotoxic cells and undertake similar functions as cytotoxic T cells in the host defense against cancer and viral infections [179]. There are diverse subsets of NK cells which have distinct functions mediated via their surface receptors; this is important not only in cytotoxicity, but also in the cross talk with other immune cells, e.g., DCs and T cells [180, 181]. It is known that aging affects the diversity of NK cell subsets, with this being reflected in the altered surface receptor phenotypes and expression levels, both in humans and mice [22, 182] (Fig. 2). These changes, which can be already detected in the bone marrow, lead to a decline in the cytotoxic capability of NK cells. In addition, the ability of human NK cells to produce cytokines and chemokines significantly decreases with aging. The impaired cytotoxicity of the NK cell population exposes elderly people to tumorigenesis and viral and bacterial infections. There is convincing evidence that MDSCs can inhibit the secretion of cytokines and reduce the cytotoxic properties of NK cells. For instance, Hoechst et al. [183] demonstrated that human MDSCs isolated from hepatocellular carcinoma (HCC) robustly inhibited the cytotoxicity of NK cells from HCC patients. The suppression of NK cells was independent of ARG1 and iNOS expression, but highly dependent on cell contacts which were mediated through the NKp30 receptors present on the NK cells. Infection studies with mice have revealed that granulocytic MDSCs inhibited both the proliferation and the activity of NK cells in response to adenovirus and vaccinia virus infections [184, 185]. There seems to be different mechanisms mediating the inhibition of NK cells evoked by MDSCs. For instance, MDSCs can inhibit the activity of NK cells by secreting ROS [184] and NO [186] or suppress the functions of NK cells via the membrane-bound TGF-β signaling [187]. It is known that TGF-β is a potent inhibitor of the development and differentiation of human NK subsets [188]. These observations indicate that MDSCs could induce the kinds of changes observed in the functions of NK cells with aging.
Monocytes and macrophages
There are many cell types participating in the innate immune system e.g., monocytes, macrophages, and granulocytes; neutrophils make up the largest group of granulocytes. There are less consistent results on the effects of aging on the functions of these cell types, since their functions tend to fluctuate with respect to the phase and intensity of inflammation. For instance, monocytes can differentiate into tissue macrophages in conditions of acute inflammation and macrophages can display M1/M2 polarization. However, there are several studies indicating that there are impairments in the properties of macrophages with aging, e.g., lowering of chemotaxis, antigen presentation, and phagocytosis [21, 123] (Fig. 2). The Toll-like receptor (TLR) signaling and its responses are impaired with aging in both humans and mice [189, 190]. It seems that the age-related alterations in macrophages are dependent on the tissue microenvironment and the disease-associated pathology, especially with respect to macrophage polarization [123]. For instance, tumors contain specific immunosuppressive TAMs which have both overlapping and distinguishing properties as compared to M2 macrophages [191]. Currently, it is not known whether there are specific age-associated M2 macrophages. MDSCs and macrophages have an important role in the resolution of inflammatory reactions [37, 40, 192]. Since MDSCs secrete both IL-10 and TGF-β, they can suppress proinflammatory functions and trigger the resolution phase, e.g., by inducing the M2 polarization of macrophages. Given that inflammaging evokes both inflammatory and anti-inflammatory responses, it seems that MDSCs suppress adaptive immunity and control innate immunity in a context-dependent manner.
Immunosenescence and MDSC-driven immunosuppression in inflammatory disorders
The hallmarks of immune system senescence not only appear with the aging process, but also evidences of premature immunosenescence are present in chronic inflammatory diseases. Sepsis has turned out to be an important model for elucidating the interactions between inflammation-induced immunosuppression and immunosenescence [193, 194]. Sepsis stimulates emergency myelopoiesis which induces the expansion of MDSCs [36, 194]. Consequently, these MDSCs induce a profound immunosuppression which is comparable to that present in cancer or age-related immunosenescence. However, it seems that MDSCs have a complex, phase-dependent role in the pathology of sepsis, causing tissue repair or its destruction. Brudecki et al. [195] utilized the mouse polymicrobial sepsis model to demonstrate that during the early phase, MDSCs secreted NO and pro-inflammatory cytokines, whereas in the later chronic phase, MDSCs expressed ARG1, IL-10, and TGF-β proteins. This indicates that within the course of sepsis, the properties of MDSCs shift from a proinflammatory phenotype to one with a strongly immunosuppressive character. Autocrine and paracrine immune factors induce the generation of immunosuppressive MDSCs in conjunction with Tregs and M2 macrophages, which in cooperation facilitate the resolution of infection. Moreover, the activation of ARG1 produces ornithine and polyamines which enhance the repair process. It has been reported that MDSCs induce immune suppression and also augment repair processes after a spinal cord injury [196] and acute kidney injury [197].
Autoimmune diseases display the hallmarks of premature immunosenescence [198]. Currently, there is convincing evidence that MDSCs have a crucial role in several autoimmune diseases, e.g., multiple sclerosis, rheumatoid arthritis, psoriasis, and autoimmune encephalomyelitis [46, 199,200,201]. In fact, many different autoimmune diseases are associated with an increase in the numbers of MDSCs in the spleen and lymph nodes, as well as in the tissues suffering autoimmune pathology and, moreover, these alterations correlate with the extent of the damage. Iacobaeus et al. [202] demonstrated that there appeared to be clear changes in the numbers of MDSCs between the relapse and remission phases in multiple sclerosis patients. The numbers of both monocytic and granulocytic MDSCs significantly increased during the relapse phase as compared to the stable phase. Experiments conducted in mice have revealed that MDSCs have a protective role against multiple sclerosis [199, 203, 204]. Similar observations have been found in autoimmune arthritis [205, 206]. Fujii et al. [205] demonstrated that collagen-induced arthritis (CIA) in mice robustly increased the numbers of MDSCs in the spleen. Splenic MDSCs effectively suppressed the proliferation of CD4+ T cells and inhibited their differentiation into Th17 cells, the major inducers of arthritic inflammation. They also revealed that the adoptive transfer of MDSCs alleviated the severity of CIA. However, there are contrasting observations indicating that MDSCs might promote the polarization of Th17 and thus augment mouse arthritis and encephalomyelitis during long-term exposures [207, 208]. Moreover, Wang et al. [209] observed that changes could occur in the type of MDSCs and their immunosuppressive properties during the course of disease which might impair efficient immunosuppression. The studies on sepsis and autoimmune diseases have clearly indicated that MDSCs induce immunosuppression (i.e., immunosenescence) which reduces the level of inflammation and the severity of injuries in inflamed tissues. It still needs to be clarified whether the MDSC-induced immunosenescence is also a feasible remodeling mechanism against inflammaging.
Immunosenescence: cellular senescence of immune cells or inflammation-induced remodeling of the immune system?
The primary cause of immunosenescence is still uncertain, although the age-related senescence of the immune system was discovered more than four decades ago. Immunosenescence seems to have a multifaceted origin, since the aging process affects the development and maturation processes of immune cells, e.g., via thymic involution, as well as their functions in peripheral, mildly inflamed tissues (Sects. 4 and 5). Given that the proliferation of T and B cells declines with aging, this implies that immune cells could undergo cellular senescence, in the same way as non-immune cells. Several research groups have investigated the replicative senescence of T cells, both in in vivo and in vitro conditions. There is evidence that changes in surface markers of T cells, e.g., lack of CD28 expression, might cause an attrition of telomeres [210, 211]. However, it seems that the markers of cellular senescence are not identical in fibroblasts and immune cells, although, for instance, CD8+ T and memory B cells can express the senescence-associated secretory phenotype (SASP), a common cellular marker of non-immune senescence [212, 213]. Recently, Ong et al. [214] identified a non-classical monocyte subset in elderly people which displayed a pro-inflammatory SASP phenotype as well as many other hallmarks of cellular senescence. Vicente et al. [215] have reviewed the role of cellular senescence in the control of cell fate and functions of many immune cells. In this respect, we need to take into consideration the difference between quiescence and senescence, since many immune cells, e.g., naïve T cells, are in a quiescent state displaying cell cycle arrest and hyporesponsiveness although they are not senescent [216]. For instance, Tregs can induce and maintain the quiescence of memory CD8+ T cells [217]. In view of the continuous production of immune cells, it seems likely that immune cells are not truly irreversibly senescent but rather exhausted, exhibiting reduced functional capabilities [218].
As long ago as 1978, Roder et al. [219] made the interesting observation that mouse immunological senescence was associated with an increased activity of suppressor cells, especially in the spleen and bone marrow. They reported that suppressor cells secreted soluble mediators, which affected the characteristics of T cells and macrophages. Remarkably, the antibody responses of immune cells could be restored by specifically activated T cells and LPS, which indicated that immunosenescence was not caused by the lack of competent immune cells. After this seminal observation, a network of immunosuppressive cells has been identified. It seems that TGF-β, IL-10, and NO, secreted by MDSCs, are the major soluble mediators maintaining the functions of this age-related immunosuppressive network. There is an abundant literature indicating that TGF-β signaling suppresses the functions of CD4+ [220] and CD8+ [221] T cells as well as DCs [222] and NK cells [223]. In particular, TGF-β inhibits the signaling pathways of CD28 and mTOR kinase. IL-10 also inhibits the CD28-mediated signaling in T cells by activating SHP-1 tyrosine phosphatase-1 [224]. TGF-β also has an important role in the functions of HSCs, e.g., TGF-β signaling promotes the myeloid differentiation of distinct mouse HSC subtypes, thus stimulating myelopoiesis with aging [225]. Flavell et al. [151] have reviewed the immune-suppressive effects of TGF-β on cells in both the innate and adaptive immune systems. Interestingly, many TGF-β-induced responses are the same as those observed in immunosenescence. IL-10, a cytokine produced by MDSCs, Tregs, and Th2 cells, also possesses different immunosuppressive functions and maintains the homeostasis of host tissues [48]. It seems that the MDSC-driven immunosuppressive network is able to generate the phenotypes in the cells of adaptive and innate immunity which are comparable to those appearing in immunosenescence (Fig. 2), although direct evidence on the causal role of MDSCs needs to be clarified.
Outlines for future studies
Currently, the causal role of MDSCs and other immunosuppressive cells in the generation of immunosenescence needs to be clarified, although there is a clear similarity between the immune cell phenotypes induced by either MDSCs or the aging process involving a low-grade inflammation. The studies on cancer therapies have revealed surprisingly many chemotherapeutic and immunotherapeutic treatments which suppress the functions of MDSCs [226,227,228]. There are different therapeutic strategies which target e.g., (1) the maturation process of MDSCs, (2) the trafficking of MDSCs into tumors, (3) the expansion and activation of MDSCs, and (4) the depletion of MDSCs. For instance, distinct compounds, e.g., all-trans retinoic acid (ATRA) and β-glucan, can induce the maturation of MDSCs into the cells of innate immunity [229, 230]. In addition, the inhibitors of signaling pathways, e.g., the inhibitors of STAT3 and COX-2/PGE2, can reduce the activation of MDSCs [228]. Several phytochemicals are also able to inhibit the function of MDSC and thus can alleviate immunosuppression in tumors and inflammatory diseases [231]. However, chemotherapeutic compounds do not specifically target MDSCs and thus there is intense search for the specific antigens of MDSCs which could be targeted in immunotherapies. Recently, Dominguez et al. [232] reported promising results that the agonistic TRAIL-R2 antibody selectively eliminated MDSCs without affecting other immune cells. It is important to understand whether the inhibition of MDSCs function in aged mammals could reverse immunosenescence and thus provide insight into the origin of immunosenescence. As far as we know, this approach has not been utilized in studies attempting to find ways to rejuvenate the immune system of elderly people or primates [233, 234]. Inhibiting the functions of MDSCs and other immunosuppressive regulators could provide the target to reverse the process of immunosenescence (i.e., induce rejuvenation), using the same approach which improves the immune surveillance of tumors and infections. This might also clarify the observations that the aging process increases the risk for cancers and chronic infections. There are studies on the combination therapies indicating that blocking the function of MDSCs, e.g., by ATRA and entinostat, improved immunotherapies in cancers and antibiotic treatments in infections [235,236,237].
There are many studies on tumors where the phenotypes of MDSCs and other immune cells have been identified, whereas in immunosenescence the phenotypes of immune cells in non-immune tissues have not been characterized. Especially, an interesting question is whether there is an accumulation of MDSCs and other immunosuppressive cells in aging tissues in association with an increased level of markers of chronic low-grade inflammation. It is known that the presence of MDSCs remarkably increases with aging in the bone marrow, spleen, and lymph nodes, but no studies exist on peripheral tissues. The great plasticity of MDSCs might cause problems, since MDSCs are disposed to mature toward M2 macrophages in inflamed tissues (see above). Technical problems might also appear with non-immune tissues, since cell sorting techniques are required for the analysis of MDSCs. However, the presence of MDSCs has been verified in studies on age-related diseases in different tissues. This approach will exclude the possibility that the age-related increase in the level of MDSCs in the blood and immune organs could be caused by age-related pathologies, such as tumors. The appearance of other cooperative partners of immunosuppression, i.e., Tregs, Mregs, and Bregs, should also be analyzed at the tissue level, since their immunosuppressive armament not only affects immune cells, but also induces harmful bystander effects on neighboring host tissue [148].
Conclusions
The role of immunosenescence in the aging process still needs to be clarified. There is an extensive literature related to the age-related decline in the function of immune system, but it has proved difficult to determine whether the overall effects are beneficial or detrimental. Given that the perpetrator inducing the age-related mild inflammatory profile cannot be eliminated, immunosenescence seems to be an important remodeling mechanism attempting to maintain tissue homeostasis as the individual grows old. Although it has been known for four decades that the aging process is associated with a suppression of immune system, the mechanism behind this phenomenon has remained elusive. Currently, it is known that there exists a network of immunosuppressive cells which exploit a wide spectrum of mechanisms to inhibit the excessive functions of the immune system. There is abundant evidence indicating that MDSCs are potent immunosuppressive cells in diverse inflammatory conditions, especially in tumor-associated inflammation. MDSCs also co-operate with other immunosuppressive cells, e.g., Tregs, Bregs, and Mregs, to suppress immune functions in inflammatory conditions. Interestingly, the numbers of MDSCs increase with aging which supports the proposal that they have a crucial role in the coordination of immunosenescence.
References
Solana R, Pawelec G (1998) Molecular and cellular basis of immunosenescence. Mech Ageing Dev 102:115–129
Goronzy JJ, Weyand CM (2013) Understanding immunosenescence to improve responses to vaccines. Nat Immunol 14:428–436
Frasca D, Blomberg BB (2016) Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 17:7–19
Nikolich-Zugich J (2018) The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol 19:10–19
Pawelec G (2018) Age and immunity: what is “immunosenescence?”. Exp Gerontol 105:4–9
Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G (2000) Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 908:244–254
Vallejo AN (2007) Immune remodeling: lessons from repertoire alterations during chronological aging and in immune-mediated disease. Trends Mol Med 13:94–102
Kanterman J, Sade-Feldman M, Baniyash M (2012) New insights into chronic inflammation-induced immunosuppression. Semin Cancer Biol 22:307–318
Wang D, DuBois RN (2015) Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 36:1085–1093
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174
Motallebnezhad M, Jadidi-Niaragh F, Qamsari ES, Bagheri S, Gharibi T, Yousefi M (2016) The immunobiology of myeloid-derived suppressor cells in cancer. Tumour Biol 37:1387–1406
Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S (2007) Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 67:10019–10026
Sendo S, Saegusa J, Morinobu A (2018) Myeloid-derived suppressor cells in non-neoplastic inflamed organs. Inflamm Regen 38:19
Bueno V, Sant’Anna OA, Lord JM (2014) Ageing and myeloid-derived suppressor cells: possible involvement in immunosenescence and age-related disease. Age (Dordr) 36:9729
Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, Witkowski JM, Franceschi C (2018) Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol 8:1960
Spyridopoulos I, Martin-Ruiz C, Hilkens C, Yadegarfar ME, Isaacs J, Jagger C, Kirkwood T, von Zglinicki T (2016) CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: results from the Newcastle 85+ study. Aging Cell 15:389–392
Weltevrede M, Eilers R, de Melker HE, van Baarle D (2016) Cytomegalovirus persistence and T-cell immunosenescence in people aged fifty and older: a systematic review. Exp Gerontol 77:87–95
Colonna-Romano G, Bulati M, Aquino A, Vitello S, Lio D, Candore G, Caruso C (2008) B cell immunosenescence in the elderly and in centenarians. Rejuvenation Res 11:433–439
Bulati M, Caruso C, Colonna-Romano G (2017) From lymphopoiesis to plasma cells differentiation, the age-related modifications of B cell compartment are influenced by “inflamm-ageing”. Ageing Res Rev 36:125–136
Agrawal A, Gupta S (2011) Impact of aging on dendritic cell functions in humans. Ageing Res Rev 10:336–345
Linehan E, Fitzgerald DC (2015) Ageing and the immune system: focus on macrophages. Eur J Microbiol Immunol (Bp) 5:14–24
Hazeldine J, Lord JM (2013) The impact of ageing on natural killer cell function and potential consequences for health in older adults. Ageing Res Rev 12:1069–1078
Lavoie ET (2005) Avian immunosenescence. Age (Dordr) 27:281–285
Müller L, Fülop T, Pawelec G (2013) Immunosenescence in vertebrates and invertebrates. Immun Ageing 10:12
Min KJ, Tatar M (2018) Unraveling the molecular mechanism of immunosenescence in Drosophila. Int J Mol Sci 19:2472
Trintinaglia L, Bandinelli LP, Grassi-Oliveira R, Petersen LE, Anzolin M, Correa BL, Schuch JB, Bauer ME (2018) Features of immunosenescence in women newly diagnosed with breast cancer. Front Immunol 9:1651
Zelle-Rieser C, Thangavadivel S, Biedermann R, Brunner A, Stoitzner P, Willenbacher E, Greil R, Jöhrer K (2016) T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol 9:116
Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE (2018) T-cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res 24:3792–3802
Saavedra D, Garcia B, Lorenzo-Luaces P, Gonzalez A, Popa X, Fuentes KP, Mazorra Z, Crombet T, Neninger E, Lage A (2016) Biomarkers related to immunosenescence: relationships with therapy and survival in lung cancer patients. Cancer Immunol Immunother 65:37–45
Chalan P, van den Berg A, Kroesen BJ, Brouwer L, Boots A (2015) Rheumatoid arthritis, immunosenescence and the hallmarks of aging. Curr Aging Sci 8:131–146
Yu HT, Park S, Shin EC, Lee WW (2016) T cell senescence and cardiovascular diseases. Clin Exp Med 16:257–263
Millrud CR, Bergenfelz C, Leandersson K (2017) On the origin of myeloid-derived suppressor cells. Oncotarget 8:3649–3665
Veglia F, Perego M, Gabrilovich D (2018) Myeloid-derived suppressor cells coming of age. Nat Immunol 19:108–119
Dietlin TA, Hofman FM, Lund BT, Gilmore W, Stohlman SA, van der Veen RC (2007) Mycobacteria-induced Gr-1+ subsets from distinct myeloid lineages have opposite effects on T cell expansion. J Leukoc Biol 81:1205–1212
Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, Ochoa AC, Fletcher M, Velasco C, Wilk A, Reiss K, Rodriguez PC (2014) Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer 134:2853–2864
Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, Heyworth PG, Efron PA, Moldawer LL (2011) A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med 17:281–292
Fullerton JN, O’Brien AJ, Gilroy DW (2013) Pathways mediating resolution of inflammation: when enough is too much. J Pathol 231:8–20
Loftus TJ, Mohr AM, Moldawer LL (2018) Dysregulated myelopoiesis and hematopoietic function following acute physiologic insult. Curr Opin Hematol 25:37–43
Saiwai H, Kumamaru H, Ohkawa Y, Kubota K, Kobayakawa K, Yamada H, Yokomizo T, Iwamoto Y, Okada S (2013) Ly6C+ Ly6G− myeloid-derived suppressor cells play a critical role in the resolution of acute inflammation and the subsequent tissue repair process after spinal cord injury. J Neurochem 125:74–88
Arocena AR, Onofrio LI, Pellegrini AV, Carrera Silva AE, Paroli A, Cano RC, Aoki MP, Gea S (2014) Myeloid-derived suppressor cells are key players in the resolution of inflammation during a model of acute infection. Eur J Immunol 44:184–194
Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, Tracey KJ, Ostrand-Rosenberg S (2014) HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res 74:5723–5733
Zhao Y, Wu T, Shao S, Shi B, Zhao Y (2015) Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology 5:e1004983
Ostrand-Rosenberg S, Fenselau C (2018) Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol 200:422–431
Condamine T, Gabrilovich DI (2011) Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol 32:19–25
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI (2010) HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 207:2439–2453
Kwak Y, Kim HE, Park SG (2015) Insights into myeloid-derived suppressor cells in inflammatory diseases. Arch Immunol Ther Exp (Warsz) 63:269–285
Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA (2006) Transforming growth factor-β regulation of immune responses. Annu Rev Immunol 24:99–146
Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG (2011) Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol 29:71–109
Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ (2004) Molecular mechanisms of interleukin-10-mediated inhibition of NF-κB activity: a role for p50. Clin Exp Immunol 135:64–73
Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, Jack RS, Wunderlich FT, Brüning JC, Müller W, Rudensky AY (2011) Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34:566–578
Schmetterer KG, Pickl WF (2017) The IL-10/STAT3 axis: contributions to immune tolerance by thymus and peripherally derived regulatory T-cells. Eur J Immunol 47:1256–1265
Mittal SK, Roche PA (2015) Suppression of antigen presentation by IL-10. Curr Opin Immunol 34:22–27
Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML (2017) IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol 112:33
Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM (2003) Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med 198:1875–1886
Roes J, Choi BK, Cazac BB (2003) Redirection of B cell responsiveness by transforming growth factor β receptor. Proc Natl Acad Sci USA 100:7241–7246
Fogel-Petrovic M, Long JA, Misso NL, Foster PS, Bhoola KD, Thompson PJ (2007) Physiological concentrations of transforming growth factor β1 selectively inhibit human dendritic cell function. Int Immunopharmacol 7:1924–1933
Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, Wang H, Fang R, Bu X, Cai S, Du J (2016) TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 7:52294–52306
Meng XM, Nikolic-Paterson DJ, Lan HY (2016) TGF-β: the master regulator of fibrosis. Nat Rev Nephrol 12:325–338
Frippiat C, Chen QM, Zdanov S, Magalhaes JP, Remacle J, Toussaint O (2001) Subcytotoxic H2O2 stress triggers a release of transforming growth factor-β1, which induces biomarkers of cellular senescence of human diploid fibroblasts. J Biol Chem 276:2531–2537
Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI (2009) Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol 182:5693–5701
Redd PS, Ibrahim ML, Klement JD, Sharman SK, Paschall AV, Yang D, Nayak-Kapoor A, Liu K (2017) SETD1B activates iNOS expression in myeloid-derived suppressor cells. Cancer Res 77:2834–2843
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI (2007) Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 13:828–835
Beury DW, Carter KA, Nelson C, Sinha P, Hanson E, Nyandjo M, Fitzgerald PJ, Majeed A, Wali N, Ostrand-Rosenberg S (2016) Myeloid-derived suppressor cell survival and function are regulated by the transcription factor Nrf2. J Immunol 196:3470–3478
Ohl K, Fragoulis A, Klemm P, Baumeister J, Klock W, Verjans E, Böll S, Möllmann J, Lehrke M, Costa I, Denecke B, Schippers A, Roth J, Wagner N, Wruck C, Tenbrock K (2016) Nrf2 is a central regulator of metabolic reprogramming of myeloid-derived suppressor cells in steady state and sepsis. Front Immunol 9:1552
Cannizzo ES, Clement CC, Sahu R, Follo C, Santambrogio L (2011) Oxidative stress, inflamm-aging and immunosenescence. J Proteomics 74:2313–2323
Bauer ME, De la Fuente M (2016) The role of oxidative and inflammatory stress and persistent viral infections in immunosenescence. Mech Ageing Dev 158:27–37
Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P (2003) L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol 24:302–306
Yu J, Du W, Yan F, Wang Y, Li H, Cao S, Yu W, Shen C, Liu J, Ren X (2013) Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol 190:3783–3797
Fuse H, Tomihara K, Heshiki W, Yamazaki M, Akyu-Takei R, Tachinami H, Furukawa KI, Sakurai K, Rouwan M, Noguchi M (2016) Enhanced expression of PD-L1 in oral squamous cell carcinoma-derived CD11b+Gr-1+ cells and its contribution to immunosuppressive activity. Oral Oncol 59:20–29
Chen J, Ye Y, Liu P, Yu W, Wei F, Li H, Yu J (2017) Suppression of T cells by myeloid-derived suppressor cells in cancer. Hum Immunol 78:113–119
Lu C, Redd PS, Lee JR, Savage N, Liu K (2016) The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 5:e1247135
Ballbach M, Dannert A, Singh A, Siegmund DM, Handgretinger R, Piali L, Rieber N, Hartl D (2017) Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol Lett 192:1–6
Keir ME, Butte MJ, Freeman GJ, Sharpe AH (2008) PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26:677–704
Lei GS, Zhang C, Lee CH (2015) Myeloid-derived suppressor cells impair alveolar macrophages through PD-1 receptor ligation during Pneumocystis pneumonia. Infect Immun 83:572–582
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 206:3015–3029
Khan AR, Hams E, Floudas A, Sparwasser T, Weaver CT, Fallon PG (2015) PD-L1hi B cells are critical regulators of humoral immunity. Nat Commun 6:5997
Josefowicz SZ, Lu LF, Rudensky AY (2012) Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30:531–564
Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK (2012) Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol 22:275–281
Rosser EC, Mauri C (2015) Regulatory B cells: origin, phenotype, and function. Immunity 42:607–612
Serafini P, Mgebroff S, Noonan K, Borrello I (2008) Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res 68:5439–5449
Luan Y, Mosheir E, Menon MC, Wilson D, Woytovich C, Ochando J, Murphy B (2013) Monocytic myeloid-derived suppressor cells accumulate in renal transplant patients and mediate CD4+ Foxp3+ Treg expansion. Am J Transplant 13:3123–3131
Park MJ, Lee SH, Kim EK, Lee EJ, Park SH, Kwok SK, Cho ML (2016) Myeloid-derived suppressor cells induce the expansion of regulatory B cells and ameliorate autoimmunity in the Sanroque mouse model of systemic lupus erythematosus. Arthritis Rheumatol 68:2717–2727
Park MJ, Lee SH, Kim EK, Lee EJ, Baek JA, Park SH, Kwok SK, Cho ML (2018) Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci Rep 8:3753
Ye J, Huang X, Hsueh EC, Zhang Q, Ma C, Zhang Y, Varvares MA, Hoft DF, Peng G (2012) Human regulatory T cells induce T-lymphocyte senescence. Blood 120:2021–2031
van de Veen W, Stanic B, Wirz OF, Jansen K, Globinska A, Akdis M (2016) Role of regulatory B cells in immune tolerance to allergens and beyond. J Allergy Clin Immunol 138:654–665
Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, Malchinkhuu E, Wersto RP, Biragyn A (2011) Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4+ T cells to T-regulatory cells. Cancer Res 71:3505–3515
Lee CR, Kwak Y, Yang T, Han JH, Park SH, Ye MB, Lee W, Sim KY, Kang JA, Kim YC, Mazmanian SK, Park SG (2016) Myeloid-derived suppressor cells are controlled by regulatory T cells via TGF-β during murine colitis. Cell Rep 17:3219–3232
Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S (2011) Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood 117:5381–5390
Murray PJ (2017) Macrophage polarization. Annu Rev Physiol 79:541–566
Italiani P, Boraschi D (2014) From monocytes to M1/M2 macrophages: phenotypical vs functional differentiation. Front Immunol 5:514
Bayik D, Tross D, Klinman DM (2018) Factors influencing the differentiation of human monocytic myeloid-derived suppressor cells into inflammatory macrophages. Front Immunol 9:608
Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, Ostrand-Rosenberg S (2014) Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukoc Biol 96:1109–1118
Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S (2007) Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 179:977–983
Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, Hockstein N, Guarino M, Masters G, Penman E, Denstman F, Xu X, Altieri DC, Du H, Yan C, Gabrilovich DI (2016) CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity 44:303–315
Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS (2007) CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A 104:19446–19451
Riquelme P, Haarer J, Kammler A, Walter L, Tomiuk S, Ahrens N, Wege AK, Goecze I, Zecher D, Banas B, Spang R, Fändrich F, Lutz MB, Sawitzki B, Schlitt HJ, Ochando J, Geissler EK, Hutchinson JA (2018) TIGIT+ iTregs elicited by human regulatory macrophages control T cell immunity. Nat Commun 9:2858
Lu J, Cao Q, Zheng D, Sun Y, Wang C, Yu X, Wang Y, Lee VW, Zheng G, Tan TK, Wang X, Alexander SI, Harris DC, Wang Y (2013) Discrete functions of M2a and M2c macrophage subsets determine their relative efficacy in treating chronic kidney disease. Kidney Int 84:745–755
Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, Rossi DJ (2010) Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci USA 107:5465–5470
Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, Weissman IL (2011) Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci USA 108:20012–20017
Elias HK, Bryder D, Park CY (2017) Molecular mechanisms underlying lineage bias in aging hematopoiesis. Semin Hematol 54:4–11
Dixit VD (2012) Impact of immune-metabolic interactions on age-related thymic demise and T cell senescence. Semin Immunol 24:321–330
Baldridge MT, King KY, Goodell MA (2011) Inflammatory signals regulate hematopoietic stem cells. Trends Immunol 32:57–65
King KY, Goodell MA (2011) Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol 11:685–692
Kovtonyuk LV, Fritsch K, Feng X, Manz MG, Takizawa H (2016) Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front Immunol 7:502
Kang YJ, Yang SJ, Park G, Cho B, Min CK, Kim TY, Lee JS, Oh IH (2007) A novel function of interleukin-10 promoting self-renewal of hematopoietic stem cells. Stem Cells 25:1814–1822
Hinge A, Filippi MD (2016) Deconstructing the complexity of TGFβ signaling in hematopoietic stem cells: quiescence and beyond. Curr Stem Cell Rep 2:388–397
Quere R, Saint-Paul L, Carmignac V, Martin RZ, Chretien ML, Largeot A, Hammann A, Pais de Barros JP, Bastie JN, Delva L (2014) Tif1γ regulates the TGF-β1 receptor and promotes physiological aging of hematopoietic stem cells. Proc Natl Acad Sci USA 111:10592–10597
Verschoor CP, Johnstone J, Millar J, Dorrington MG, Habibagahi M, Lelic A, Loeb M, Bramson JL, Bowdish DM (2013) Blood CD33+HLA-DR- myeloid-derived suppressor cells are increased with age and a history of cancer. J Leukoc Biol 93:633–637
Alves AS, Ishimura ME, Duarte YA, Bueno V (2018) Parameters of the immune system and vitamin D levels in old individuals. Front Immunol 9:1122
Enioutina EY, Bareyan D, Daynes RA (2011) A role for immature myeloid cells in immune senescence. J Immunol 186:697–707
Grizzle WE, Xu X, Zhang S, Stockard CR, Liu C, Yu S, Wang J, Mountz JD, Zhang HG (2007) Age-related increase of tumor susceptibility is associated with myeloid-derived suppressor cell mediated suppression of T cell cytotoxicity in recombinant inbred BXD12 mice. Mech Ageing Dev 128:672–680
Heithoff DM, Enioutina EY, Bareyan D, Daynes RA, Mahan MJ (2008) Conditions that diminish myeloid-derived suppressor cell activities stimulate cross-protective immunity. Infect Immun 76:5191–5199
Chen S, Liu H, Su N, Zhang G, Wang L (2015) Myeloid-derived suppressor cells promote age-related increase of lung cancer growth via B7-H1. Exp Gerontol 61:84–91
Flores RR, Clauson CL, Cho J, Lee BC, McGowan SJ, Baker DJ, Niedernhofer LJ, Robbins PD (2017) Expansion of myeloid-derived suppressor cells with aging in the bone marrow of mice through a NF-κB-dependent mechanism. Aging Cell 16:480–487
Sharma S, Dominguez AL, Lustgarten J (2006) High accumulation of T regulatory cells prevents the activation of immune responses in aged animals. J Immunol 177:8348–8355
Lages CS, Suffia I, Velilla PA, Huang B, Warshaw G, Hildeman DA, Belkaid Y, Chougnet C (2008) Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J Immunol 181:1835–1848
Raynor J, Lages CS, Shehata H, Hildeman DA, Chougnet CA (2012) Homeostasis and function of regulatory T cells in aging. Curr Opin Immunol 24:482–487
Garg SK, Delaney C, Toubai T, Ghosh A, Reddy P, Banerjee R, Yung R (2014) Aging is associated with increased regulatory T-cell function. Aging Cell 13:441–448
Agius E, Lacy KE, Vukmanovic-Stejic M, Jagger AL, Papageorgiou AP, Hall S, Reed JR, Curnow SJ, Fuentes-Duculan J, Buckley CD, Salmon M, Taams LS, Krueger J, Greenwood J, Klein N, Rustin MH, Akbar AN (2009) Decreased TNF-α synthesis by macrophages restricts cutaneous immunosurveillance by memory CD4+ T cells during aging. J Exp Med 206:1929–1940
Jagger A, Shimojima Y, Goronzy JJ, Weyand CM (2014) Regulatory T cells and the immune aging process: a mini-review. Gerontology 60:130–137
Chougnet CA, Tripathi P, Lages CS, Raynor J, Sholl A, Fink P, Plas DR, Hildeman DA (2011) A major role for Bim in regulatory T cell homeostasis. J Immunol 186:156–163
Hamidzadeh K, Christensen SM, Dalby E, Chandrasekaran P, Mosser DM (2017) Macrophages and the recovery from acute and chronic inflammation. Annu Rev Physiol 79:567–592
Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P, Nelson DJ (2017) Aging and cancer: the role of macrophages and neutrophils. Ageing Res Rev 36:105–116
Gensel JC, Zhang B (2015) Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res 1619:1–11
Jackaman C, Radley-Crabb HG, Soffe Z, Shavlakadze T, Grounds MD, Nelson DJ (2013) Targeting macrophages rescues age-related immune deficiencies in C57BL/6J geriatric mice. Aging Cell 12:345–357
Wang Y, Wehling-Henricks M, Samengo G, Tidball JG (2015) Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell 14:678–688
Pawelec G, Remarque E, Barnett Y, Solana R (1998) T cells and aging. Front Biosci 3:d59–d99
Arnold CR, Wolf J, Brunner S, Herndler-Brandstetter D, Grubeck-Loebenstein B (2011) Gain and loss of T cell subsets in old age—age-related reshaping of the T cell repertoire. J Clin Immunol 31:137–146
Le Page A, Dupuis G, Larbi A, Witkowski JM, Fulop T (2018) Signal transduction changes in CD4+ and CD8+ T cell subpopulations with aging. Exp Gerontol 105:128–139
Karanfilov CI, Liu B, Fox CC, Lakshmanan RR, Whisler RL (1999) Age-related defects in Th1 and Th2 cytokine production by human T cells can be dissociated from altered frequencies of CD45RA+ and CD45RO+ T cell subsets. Mech Ageing Dev 109:97–112
Gardner EM, Murasko DM (2002) Age-related changes in Type 1 and Type 2 cytokine production in humans. Biogerontology 3:271–290
Ouyang X, Yang Z, Zhang R, Arnaboldi P, Lu G, Li Q, Wang W, Zhang B, Cui M, Zhang H, Liang-Chen J, Qin L, Zheng F, Huang B, Xiong H (2011) Potentiation of Th17 cytokines in aging process contributes to the development of colitis. Cell Immunol 266:208–217
Lim MA, Lee J, Park JS, Jhun JY, Moon YM, Cho ML, Kim HY (2014) Increased Th17 differentiation in aged mice is significantly associated with high IL-1β level and low IL-2 expression. Exp Gerontol 49:55–62
Martin F, Apetoh L, Ghiringhelli F (2012) Controversies on the role of Th17 in cancer: a TGF-β-dependent immunosuppressive activity? Trends Mol Med 18:742–749
Haynes L, Maue AC (2009) Effects of aging on T cell function. Curr Opin Immunol 21:414–417
Jergovic M, Smithey MJ, Nikolich-Zugich J (2018) Intrinsic and extrinsic contributors to defective CD8+ T cell responses with aging. Exp Gerontol 105:140–145
Quinn KM, Fox A, Harland KL, Russ BE, Li J, Nguyen THO, Loh L, Olshanksy M, Naeem H, Tsyganov K, Wiede F, Webster R, Blyth C, Sng XYX, Tiganis T, Powell D, Doherty PC, Turner SJ, Kedzierska K, La Gruta NL (2018) Age-related decline in primary CD8+ T cell responses is associated with the development of senescence in virtual memory CD8+ T cells. Cell Rep 23:3512–3524
Pawelec G, Hirokawa K, Fulop T (2001) Altered T cell signalling in ageing. Mech Ageing Dev 122:1613–1637
Goronzy JJ, Li G, Yu M, Weyand CM (2012) Signaling pathways in aged T cells—a reflection of T cell differentiation, cell senescence and host environment. Semin Immunol 24:365–372
Egorov ES, Kasatskaya SA, Zubov VN, Izraelson M, Nakonechnaya TO, Staroverov DB, Angius A, Cucca F, Mamedov IZ, Rosati E, Franke A, Shugay M, Pogorelyy MV, Chudakov DM, Britanova OV (2018) The changing landscape of naive T cell receptor repertoire with human aging. Front Immunol 9:1618
Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY, Olshen RA, Weyand CM, Boyd SD, Goronzy JJ (2014) Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci USA 111:13139–13144
Garcia GG, Miller RA (2002) Age-dependent defects in TCR-triggered cytoskeletal rearrangement in CD4+ T cells. J Immunol 169:5021–5027
Weng NP, Akbar AN, Goronzy J (2009) CD28− T cells: their role in the age-associated decline of immune function. Trends Immunol 30:306–312
Broux B, Markovic-Plese S, Stinissen P, Hellings N (2012) Pathogenic features of CD4+ CD28– T cells in immune disorders. Trends Mol Med 18:446–453
Dilek N, Poirier N, Usal C, Martinet B, Blancho G, Vanhove B (2012) Control of transplant tolerance and intragraft regulatory T cell localization by myeloid-derived suppressor cells and CCL5. J Immunol 188:4209–4216
Feng S, Cheng X, Zhang L, Lu X, Chaudhary S, Teng R, Frederickson C, Champion MM, Zhao R, Cheng L, Gong Y, Deng H, Lu X (2018) Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc Natl Acad Sci USA 115:10094–10099
Fujimura T, Ring S, Umansky V, Mahnke K, Enk AH (2012) Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J Invest Dermatol 132:1239–1246
Salminen A, Kaarniranta K, Kauppinen A (2018) The role of myeloid-derived suppressor cells (MDSC) in the inflammaging process. Ageing Res Rev 48:1–10
Shimada Y, Hayashi M, Nagasaka Y, Ohno-Iwashita Y, Inomata M (2009) Age-associated up-regulation of a negative co-stimulatory receptor PD-1 in mouse CD4+ T cells. Exp Gerontol 44:517–522
Li MO, Flavell RA (2008) TGF-β: a master of all T cell trades. Cell 134:392–404
Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P (2010) The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol 10:554–567
Travis MA, Sheppard D (2014) TGF-β activation and function in immunity. Annu Rev Immunol 32:51–82
Blank U, Karlsson S (2015) TGF-β signaling in the control of hematopoietic stem cells. Blood 125:3542–3550
Knott AB, Bossy-Wetzel E (2010) Impact of nitric oxide on metabolism in health and age-related disease. Diabetes Obes Metab 12(Suppl 2):126–133
van der Goot AT, Nollen EA (2013) Tryptophan metabolism: entering the field of aging and age-related pathologies. Trends Mol Med 19:336–344
Cancro MP, Hao Y, Scholz JL, Riley RL, Frasca D, Dunn-Walters DK, Blomberg BB (2009) B cells and aging: molecules and mechanisms. Trends Immunol 30:313–318
Kennedy DE, Knight KL (2017) Inflammatory changes in bone marrow microenvironment associated with declining B lymphopoiesis. J Immunol 198:3471–3479
Kennedy DE, Knight KL (2015) Inhibition of B lymphopoiesis by adipocytes and IL-1-producing myeloid-derived suppressor cells. J Immunol 195:2666–2674
Colonna-Romano G, Bulati M, Aquino A, Pellicano M, Vitello S, Lio D, Candore G, Caruso C (2009) A double-negative (IgD−CD27−) B cell population is increased in the peripheral blood of elderly people. Mech Ageing Dev 130:681–690
Frasca D, Diaz A, Romero M, Landin AM, Blomberg BB (2011) Age effects on B cells and humoral immunity in humans. Ageing Res Rev 10:330–335
Dunn-Walters DK (2016) The ageing human B cell repertoire: a failure of selection? Clin Exp Immunol 183:50–56
Rastad JL, Green WR (2016) Myeloid-derived suppressor cells in murine AIDS inhibit B-cell responses in part via soluble mediators including reactive oxygen and nitrogen species, and TGF-β. Virology 499:9–22
Lelis FJN, Jaufmann J, Singh A, Fromm K, Teschner AC, Pöschel S, Schäfer I, Beer-Hammer S, Rieber N, Hartl D (2017) Myeloid-derived suppressor cells modulate B-cell responses. Immunol Lett 188:108–115
Özkan B, Lim H, Park SG (2018) Immunomodulatory function of myeloid-derived suppressor cells during B cell-mediated immune responses. Int J Mol Sci 19:1468
Bodogai M, Moritoh K, Lee-Chang C, Hollander CM, Sherman-Baust CA, Wersto RP, Araki Y, Miyoshi I, Yang L, Trinchieri G, Biragyn A (2015) Immunosuppressive and prometastatic functions of myeloid-derived suppressive cells rely upon education from tumor-associated B cells. Cancer Res 75:3456–3465
Solana R, Tarazona R, Gayoso I, Lesur O, Dupuis G, Fulop T (2012) Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol 24:331–341
Shaw AC, Goldstein DR, Montgomery RR (2013) Age-dependent dysregulation of innate immunity. Nat Rev Immunol 13:875–887
Bhushan M, Cumberbatch M, Dearman RJ, Andrew SM, Kimber I, Griffiths CE (2002) Tumour necrosis factor-α-induced migration of human Langerhans cells: the influence of ageing. Br J Dermatol 146:32–40
Xu YP, Qi RQ, Chen W, Shi Y, Cui ZZ, Gao XH, Chen HD, Zhou L, Mi QS (2012) Aging affects epidermal Langerhans cell development and function and alters their miRNA gene expression profile. Aging (Albany NY) 4:742–754
Shurin MR, Shurin GV, Chatta GS (2007) Aging and the dendritic cell system: implications for cancer. Crit Rev Oncol Hematol 64:90–105
Grolleau-Julius A, Garg MR, Mo R, Stoolman LL, Yung RL (2006) Effect of aging on bone marrow-derived murine CD11c+ CD4−CD8α− dendritic cell function. J Gerontol A Biol Sci Med Sci 61:1039–1047
Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, Janssen EM (2015) Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol 195:2624–2632
Panda A, Qian F, Mohanty S, van Duin D, Newman FK, Zhang L, Chen S, Towle V, Belshe RB, Fikrig E, Allore HG, Montgomery RR, Shaw AC (2010) Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J Immunol 184:2518–2527
Gardner JK, Mamotte CDS, Jackaman C, Nelson DJ (2017) Modulation of dendritic cell and T cell cross-talk during aging: the potential role of checkpoint inhibitory molecules. Ageing Res Rev 38:40–51
Greifenberg V, Ribechini E, Rössner S, Lutz MB (2009) Myeloid-derived suppressor cell activation by combined LPS and IFN-γ treatment impairs DC development. Eur J Immunol 39:2865–2876
Poschke I, Mao Y, Adamson L, Salazar-Onfray F, Masucci G, Kiessling R (2012) Myeloid-derived suppressor cells impair the quality of dendritic cell vaccines. Cancer Immunol Immunother 61:827–838
Strobl H, Knapp W (1999) TGF-β1 regulation of dendritic cells. Microbes Infect 1:1283–1290
Borkowski TA, Letterio JJ, Mackall CL, Saitoh A, Wang XJ, Roop DR, Gress RE, Udey MC (1997) A role for TGFβ1 in Langerhans cell biology. Further characterization of the epidermal Langerhans cell defect in TGFβ1 null mice. J Clin Invest 100:575–581
Farag SS, Caligiuri MA (2006) Human natural killer cell development and biology. Blood Rev 20:123–137
Fu B, Tian Z, Wei H (2014) Subsets of human natural killer cells and their regulatory effects. Immunology 141:483–489
Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA (2017) The broad spectrum of human natural killer cell diversity. Immunity 47:820–833
Gayoso I, Sanchez-Correa B, Campos C, Alonso C, Pera A, Casado JG, Morgado S, Tarazona R, Solana R (2011) Immunosenescence of human natural killer cells. J Innate Immun 3:337–343
Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, Lehner F, Manns MP, Greten TF, Korangy F (2009) Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 50:799–807
Fortin C, Huang X, Yang Y (2012) NK cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J Immunol 189:1843–1849
Zhu J, Huang X, Yang Y (2012) Myeloid-derived suppressor cells regulate natural killer cell response to adenovirus-mediated gene transfer. J Virol 86:13689–13696
Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, Kendra K, Campbell A, Gautam S, Abood D, Landi I, Hsu V, Duggan M, Wesolowski R, Old M, Howard JH, Yu L, Stasik N, Olencki T, Muthusamy N, Tridandapani S, Byrd JC, Caligiuri M, Carson WE (2018) Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor-mediated natural killer cell function. Clin Cancer Res 24:1891–1904
Li H, Han Y, Guo Q, Zhang M, Cao X (2009) Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-β1. J Immunol 182:240–249
Allan DS, Rybalov B, Awong G, Zuniga-Pflücker JC, Kopcow HD, Carlyle JR, Strominger JL (2010) TGF-β affects development and differentiation of human natural killer cell subsets. Eur J Immunol 40:2289–2295
Dunston CR, Griffiths HR (2010) The effect of ageing on macrophage Toll-like receptor-mediated responses in the fight against pathogens. Clin Exp Immunol 161:407–416
Shaw AC, Panda A, Joshi SR, Qian F, Allore HG, Montgomery RR (2011) Dysregulation of human Toll-like receptor function in aging. Ageing Res Rev 10:346–353
Guttman O, Lewis EC (2016) M2-like macrophages and tumor-associated macrophages: overlapping and distinguishing properties en route to a safe therapeutic potential. Integr Cancer Sci Therap 3:554–561
Ortega-Gomez A, Perretti M, Soehnlein O (2013) Resolution of inflammation: an integrated view. EMBO Mol Med 5:661–674
Martin S, Perez A, Aldecoa C (2017) Sepsis and immunosenescence in the elderly patient: a review. Front Med (Lausanne) 4:20
Venet F, Monneret G (2018) Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol 14:121–137
Brudecki L, Ferguson DA, McCall CE, El Gazzar M (2012) Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun 80:2026–2034
Wang L, Yu WB, Tao LY, Xu Q (2016) Myeloid-derived suppressor cells mediate immune suppression in spinal cord injury. J Neuroimmunol 290:96–102
Zhang C, Wang S, Li J, Zhang W, Zheng L, Yang C, Zhu T, Rong R (2017) The mTOR signal regulates myeloid-derived suppressor cells differentiation and immunosuppressive function in acute kidney injury. Cell Death Dis 8:e2695
Thewissen M, Linsen L, Somers V, Geusens P, Raus J, Stinissen P (2005) Premature immunosenescence in rheumatoid arthritis and multiple sclerosis patients. Ann N Y Acad Sci 1051:255–262
Ioannou M, Alissafi T, Lazaridis I, Deraos G, Matsoukas J, Gravanis A, Mastorodemos V, Plaitakis A, Sharpe A, Boumpas D, Verginis P (2012) Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J Immunol 188:1136–1146
Melero-Jerez C, Ortega MC, Moline-Velazquez V, Clemente D (2016) Myeloid derived suppressor cells in inflammatory conditions of the central nervous system. Biochim Biophys Acta 1862:368–380
Cao LY, Chung JS, Teshima T, Feigenbaum L, Cruz PD Jr, Jacobe HT, Chong BF, Ariizumi K (2016) Myeloid-derived suppressor cells in psoriasis are an expanded population exhibiting diverse T-cell-suppressor mechanisms. J Invest Dermatol 136:1801–1810
Iacobaeus E, Douagi I, Jitschin R, Marcusson-Ståhl M, Andren AT, Gavin C, Lefsihane K, Davies LC, Mougiakakos D, Kadri N, Le Blanc K (2018) Phenotypic and functional alterations of myeloid-derived suppressor cells during the disease course of multiple sclerosis. Immunol Cell Biol 96:820–830
Moline-Velazquez V, Cuervo H, Vila-Del Sol V, Ortega MC, Clemente D, de Castro F (2011) Myeloid-derived suppressor cells limit the inflammation by promoting T lymphocyte apoptosis in the spinal cord of a murine model of multiple sclerosis. Brain Pathol 21:678–691
Wegner A, Verhagen J, Wraith DC (2017) Myeloid-derived suppressor cells mediate tolerance induction in autoimmune disease. Immunology 151:26–42
Fujii W, Ashihara E, Hirai H, Nagahara H, Kajitani N, Fujioka K, Murakami K, Seno T, Yamamoto A, Ishino H, Kohno M, Maekawa T, Kawahito Y (2013) Myeloid-derived suppressor cells play crucial roles in the regulation of mouse collagen-induced arthritis. J Immunol 191:1073–1081
Zhang L, Zhang Z, Zhang H, Wu M, Wang Y (2014) Myeloid-derived suppressor cells protect mouse models from autoimmune arthritis via controlling inflammatory response. Inflammation 37:670–677
Yi H, Guo C, Yu X, Zuo D, Wang XY (2012) Mouse CD11b+Gr-1+ myeloid cells can promote Th17 cell differentiation and experimental autoimmune encephalomyelitis. J Immunol 189:4295–4304
Guo C, Hu F, Yi H, Feng Z, Li C, Shi L, Li Y, Liu H, Yu X, Wang H, Li J, Li Z, Wang XY (2016) Myeloid-derived suppressor cells have a proinflammatory role in the pathogenesis of autoimmune arthritis. Ann Rheum Dis 75:278–285
Wang W, Jiao Z, Duan T, Liu M, Zhu B, Zhang Y, Xu Q, Wang R, Xiong Y, Xu H, Lu L (2015) Functional characterization of myeloid-derived suppressor cell subpopulations during the development of experimental arthritis. Eur J Immunol 45:464–473
Effros RB, Dagarag M, Spaulding C, Man J (2005) The role of CD8+ T-cell replicative senescence in human aging. Immunol Rev 205:147–157
Chou JP, Effros RB (2013) T cell replicative senescence in human aging. Curr Pharm Des 19:1680–1698
Frasca D, Diaz A, Romero M, Blomberg BB (2017) Human peripheral late/exhausted memory B cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways. Exp Gerontol 87(Pt A):113–120
Callender LA, Carroll EC, Bober EA, Henson SM (2018) Divergent mechanisms of metabolic dysfunction drive fibroblast and T-cell senescence. Ageing Res Rev 47:24–30
Ong SM, Hadadi E, Dang TM, Yeap WH, Tan CT, Ng TP, Larbi A, Wong SC (2018) The pro-inflammatory phenotype of the human non-classical monocyte subset is attributed to senescence. Cell Death Dis 9:266
Vicente R, Mausset-Bonnefont AL, Jorgensen C, Louis-Plence P, Brondello JM (2016) Cellular senescence impact on immune cell fate and function. Aging Cell 15:400–406
Chapman NM, Chi H (2018) Hallmarks of T-cell exit from quiescence. Cancer Immunol Res 6:502–508
Kalia V, Penny LA, Yuzefpolskiy Y, Baumann FM, Sarkar S (2015) Quiescence of memory CD8+ T cells is mediated by regulatory T cells through inhibitory receptor CTLA-4. Immunity 42:1116–1129
Larbi A, Fulop T (2014) From “truly naïve” to “exhausted senescent” T cells: when markers predict functionality. Cytometry A 85:25–35
Roder JC, Duwe AK, Bell DA, Singhal SK (1978) Immunological senescence. I. The role of suppressor cells. Immunology 35:837–847
Delisle JS, Giroux M, Boucher G, Landry JR, Hardy MP, Lemieux S, Jones RG, Wilhelm BT, Perreault C (2013) The TGF-β-Smad3 pathway inhibits CD28-dependent cell growth and proliferation of CD4 T cells. Genes Immun 14:115–126
Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI (2009) Cell-intrinsic transforming growth factor-β signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity 31:145–157
Esebanmen GE, Langridge WHR (2017) The role of TGF-β signaling in dendritic cell tolerance. Immunol Res 65:987–994
Zaiatz-Bittencourt V, Finlay DK, Gardiner CM (2018) Canonical TGF-β signaling pathway represses human NK cell metabolism. J Immunol 200:3934–3941
Taylor A, Akdis M, Joss A, Akkoc T, Wenig R, Colonna M, Daigle I, Flory E, Blaser K, Akdis CA (2007) IL-10 inhibits CD28 and ICOS costimulations of T cells via src homology 2 domain-containing protein tyrosine phosphatase 1. J Allergy Clin Immunol 120:76–83
Challen GA, Boles NC, Chambers SM, Goodell MA (2010) Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-β1. Cell Stem Cell 6:265–278
Wesolowski R, Markowitz J, Carson WE 3rd (2013) Myeloid derived suppressor cells—a new therapeutic target in the treatment of cancer. J Immunother Cancer 1:10
Draghiciu O, Lubbers J, Nijman HW, Daemen T (2015) Myeloid derived suppressor cells—an overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology 4:e954829
Shipp C, Speigl L, Janssen N, Martens A, Pawelec G (2016) A clinical and biological perspective of human myeloid-derived suppressor cells in cancer. Cell Mol Life Sci 73:4043–4061
Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI (2007) Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res 67:11021–11028
Tian J, Ma J, Ma K, Guo H, Baidoo SE, Zhang Y, Yan J, Lu L, Xu H, Wang S (2013) β-Glucan enhances antitumor immune responses by regulating differentiation and function of monocytic myeloid-derived suppressor cells. Eur J Immunol 43:1220–1230
Salminen A, Kaarniranta K, Kauppinen A (2018) Phytochemicals inhibit the immunosuppressive functions of myeloid-derived suppressor cells (MDSC): impact on cancer and age-related chronic inflammatory disorders. Int Immunopharmacol 61:231–240
Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, Forero A, Bendell J, Witt R, Hockstein N, Kumar P, Gabrilovich DI (2017) Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin Cancer Res 23:2942–2950
Holland AM, van den Brink MR (2009) Rejuvenation of the aging T cell compartment. Curr Opin Immunol 21:454–459
Meyer C, Kerns A, Haberthur K, Messaoudi I (2012) Improving immunity in the elderly: current and future lessons from nonhuman primate models. Age (Dordr) 34:1157–1168
Lei GS, Zhang C, Shao S, Jung HW, Durant PJ, Lee CH (2013) All-trans retinoic acid in combination with primaquine clears pneumocystis infection. PLoS One 8:e53479
Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, Chintala S, Ordentlich P, Kao C, Elzey B, Gabrilovich D, Pili R (2017) Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res 23:5187–5201
Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, Lewis KD, McCarter MD (2018) Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with ipilimumab. Int Immunopharmacol 63:282–291
Acknowledgements
Open access funding provided by University of Eastern Finland (UEF) including Kuopio University Hospital. This study was financially supported by grants from the Academy of Finland (AK297267, AK307341, and KK296840), the Kuopio University Hospital VTR grant (KK5503743), the Emil Aaltonen Foundation, the Sigrid Juselius Foundation, the Päivikki and Sakari Sohlberg Foundation, the Finnish Cultural Foundation, and the Finnish Eye Foundation. The authors thank Dr. Ewen MacDonald for checking the language of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors state that there are no personal or institutional conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Salminen, A., Kaarniranta, K. & Kauppinen, A. Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell. Mol. Life Sci. 76, 1901–1918 (2019). https://doi.org/10.1007/s00018-019-03048-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03048-x