
Abstract
Abstract
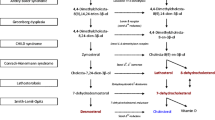
An earlier described patient with combined sphingolipidoses, Farber and Sandhoff disease, had two healthy older brothers and two further sibs, one with Sandhoff disease and one (a fetus) with Farber disease, showing segregation of the respective genes. The prenatal diagnosis in the latter was performed using lipid (sphingomyelin and glucosylceramide) loading tests on the cultured amniotic fluid cells. After 1–3 days of incubation the cells' lipid extract revealed radioactive ceramide to be released and highly accumulated. The deficiency in acid ceramidase was known from the patient with the combined diseases. Confirmation of the prenatal Farber diagnosis was done by similar loading tests on the fetal fibroblasts and by analysis of liver lipids of the less than 18-week-old fetus.
Conclusion
This is the first report on the use of lipid loading tests on intact cultured cells for prenatal diagnosis of Farber disease. The postnatal diagnosis of Farber disease can also be readily made using those tests, as was shown in four further cases.
Similar content being viewed by others


Abbreviations
- CER :
-
acid ceramidase
- cer :
-
ceramide
- HEXB :
-
β-hexosaminidase B
References
Bradová V, Smid F, Ulrich-Bou B, Roggendorf W, Paton BC, Harzer K (1993) Prosaposin deficiency: further characterization of the sphingolipid activator protein-deficient sibs. Multiple glycolipid elevations (including lactosylceramidosis), partial enzyme deficiencies and ultrastructure of the skin in this generalized sphingolipid storage disease. Hum Genet 92: 143–152
Chen WW, Moser AB, Moser HW (1981) Role of lysosomal acid ceramidase in the metabolism of ceramide in human skin fibroblasts. Arch Biochem Biophys 208: 444–455
Conzelmann E, Sandhoff K (1984) Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci 6: 58–71
Fensom AH, Benson PF, Neville BRG, Moser HW, Moser AB, Dulaney JT (1979) Prenatal diagnosis of Farber's disease. Lancet 2: 990–992
Fusch C, Huenges R, Moser HW, Sewell AC, Roggendorf W, Kustermann-Kuhn B, Poulos A, Carey WF, Harzer K (1989) A case of combined Farber and Sandhoff disease. Eur J Pediatr 148: 558–562
Graber D, Salvayre R, Levade T (1994) Accurate differentiation of neuronopathic and nonneuronopathic forms of Niemann-Pick disease by evaluation of the effective residual lysosomal sphingomyelinase activity in intact cells. J Neurochem 63: 1060–1068
Harzer K, Sandhoff K, Schall H, Kollmann F (1971) Enzymatische Untersuchungen im Blut von Überträgern einer Variante der Tay-Sachsschen Erkrankung (Variante 0). Klin Wochenschr 49: 1189–1191
Harzer K, Paton BC, Poulos A, Kustermann-Kuhn B, Roggendorf W, Grisar T, Popp M (1989) Sphingolipid activator protein deficiency in a 16-week-old atypical Gaucher disease patient and his fetal sibling: biochemical signs of combined sphingolipidoses. Eur J Pediatr 149: 31–39
Kudoh T, Wenger DA (1982) Diagnosis of metachromatic leukodystrophy, Krabbe disease, and Farber disease after uptake of fatty acid-labeled cerebroside sulfate into cultured skin fibroblasts. J Clin Invest 70: 89–97
Kustermann-Kuhn B, Harzer K (1983) Prenatal diagnosis of Tay-Sachs disease. Reflectometry of hexosaminidase A, B, and C/S. bands on zymograms. Hum Genet 65: 172–175
Leinekugel P, Michel S, Conzelmann E, Sandhoff K (1992) Quantitative correlation between the residual activity of β-hexosaminisase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet 88: 513–523
Levade T, Tempesta M-C, Salvayre R (1993) The in situ degradation of ceramide, a potential lipid mediator, is not completely impaired in Farber disease. FEBS Letters 329: 306–312
Marquard K, Harzer K, Keimer R, Köhler B, Wörle H, Schöning M, Enders H, Kustermann-Kuhn B (1994) M. Farber (Lipogranulomatose) — eine wenig bekannte Sphingolipidose. Monatsschr Kinderheilkd 142: 753–772
Moser HW, Moser AB, Chen WW, Schram AW (1989) Ceramidase deficiency: Farber lipogranulomatosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 6th edn. McGraw-Hill, New York, pp 1645–1654
Paton BC, Schmid B, Kustermann-Kuhn B, Poulos A, Harzer K (1992) Additional biochemical findings in a patient and fetal sibling with a genetic defect in the sphingolipid activator protein (SAP) precursor, prosaposin. Evidence for a deficiency in SAP-1 and for a normal lysosomal neuraminidase. Biochem J 285: 481–488
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Levade, T., Enders, H., Schliephacke, M. et al. A family with combined Farber and Sandhoff, isolated Sandhoff and isolated fetal Farber disease: postnatal exclusion and prenatal diagnosis of Farber disease using lipid loading tests on intact cultured cells. Eur J Pediatr 154, 643–648 (1995). https://doi.org/10.1007/BF02079069
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF02079069