
Abstract
Methods and techniques are described for automated measurements of greenhouse gases (GHGs) in both the laboratory and the field. Robotic systems are currently available to measure the entire range of gases evolved from soils including dinitrogen (N2). These systems usually work on an exchange of the atmospheric N2 with helium (He) so that N2 fluxes can be determined. Laboratory systems are often used in microbiology to determine kinetic response reactions via the dynamics of all gaseous N species such as nitric oxide (NO), nitrous oxide (N2O), and N2. Latest He incubation techniques also take plants into account, in order to study the effect of plant–soil interactions on GHGs and N2 production. The advantage of automated in-field techniques is that GHG emission rates can be determined at a high temporal resolution. This allows, for instance, to determine diurnal response reactions (e.g. with temperature) and GHG dynamics over longer time periods.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
3.1 Automated Laboratory Techniques
This section focuses on automated high-resolution determinations of anaerobic atmospheric trace gas (H2, NO, N2O, CO2, CH4) production kinetics, and nitrate (NO3−)-nitrite (NO2−) analysis systems designed for ex situ incubations. Typically, such systems combine a temperature-controlled sampling unit holding several septum vials (with or without stirring or shaking) under a fully programmable robotic arm of an autosampler, which is used to repeatedly pierce the septa of the incubation vessels to subsample the headspace for concentration measurements by gas chromatography and/or other dedicated gas analysers. Processes are studied in different matrices, ranging from repacked or suspended soils (Horn et al. 2006; Palmer et al. 2012, 2015, 2016; Zhu et al. 2013; Schmidt et al. 2016), plant material (Keppler et al. 2006), and water samples (McCrackin and Elser 2010) to extracted bacterial cells (Dörsch et al. 2012; Brenzinger et al. 2015) and cultures of isolated (Lycus et al. 2017) or type strain organisms (Bergaust et al. 2012; Horn et al. 2005) including knock-out mutants. Automated incubation setups are a refinement of classical incubation techniques still used to determine process rates of “respiration” (e.g. by soda-lime trapping of evolved CO2) or “denitrification enzyme activity” (DEA; N2O accumulation in the presence of acetylene). While automated batch incubations allow for estimating process potentials and apparent Michaelis–Menten parameters (Vmax, Km) under quasi-steady-state conditions in short-term assays with ample substrates, high-resolution incubations can also be used to study “gas kinetics” of selected processes with finite amounts of substrates (Palmer et al. 2012). Gaseous intermediates (e.g. in denitrification) evolve transiently before gaseous end products accumulate to amounts given by the added substrate. In this way, high-resolution gas kinetics can provide insights into the metabolic functioning under changing substrate conditions. Typically, incubations are initiated under oxic or anoxic conditions by flushing the headspace of the incubation bottles with various gas mixtures. The transition from oxic to anoxic metabolism can be studied by letting the sample consume a finite amount of O2 while monitoring the evolution of gaseous intermediates (NO, N2O, H2), and final products (N2, CH4, H2S) involved in anoxic metabolism (Schlüter et al. 2019). Notably, replacing the bottle atmosphere with He or a mixture of He and O2 allows for quantifying N2, the final product of denitrification which cannot be detected at ambient N2 background levels (but see section on Raman spectroscopy Chap. 2, Sect. 2.10). Using appropriate inhibitors (Yoshinari and Knowles 1976; Bedard and Knowles 1989; Taylor et al. 2015), selected processes can be studied.
High-resolution gas kinetics are also the method of choice to study the induction of enzymatic processes involved in GHG turnover, which can be studied by sequential analysis of gene expression levels or other genetic markers (Vollack and Zumft 2001; Bergaust et al. 2010; Liu et al. 2014; Brenzinger et al. 2015). In this way, the eco-physiology of specific microbial groups or organisms can be studied in great detail and advance our understanding of regulatory aspects involved in GHG turnover (Bakken et al. 2012; Braker and Conrad 2011; Hink et al. 2017).
3.1.1 Technical Challenges
-
Headspace pressure: Subsampling closed incubation bottles repeatedly implies a decrease in headspace pressure. The incubation setup should be able to replace the removed sample volume automatically by an inert gas (e.g. He, Ar) to maintain bottle pressure at ~1 atm. This can be achieved by reversing the sampling pump (if used) or by replacing the volume with an automatically operated syringe. Alternatively, bottles with elevated headspace pressure can be used (Palmer et al. 2010). In both cases, the resulting dilution has to be corrected when calculating rates between sampling increments (Molstad et al. 2007).
-
Sample loss: Irrespective of how the sampled volume is replaced, high-resolution gas kinetics with finite amounts of substrates suffer from sampling loss, i.e. part of the substrates and evolving intermediates are lost during analysis. This has to be taken into account when calculating internal mass balances between substrates and gaseous products.
-
Leakage: When analysing accumulation or depletion of gaseous products (N2) or substrates (O2) which are abundant in the atmosphere, there will be leakage into the bottles or the sampling line. Incubation vessels, gas sampling access, sample transport, and admission to the measurement system must be designed and tested carefully to minimize the unavoidable leakage of O2 and N2, e.g. caused by repeatedly pierced septa. This can be avoided by programming the autosampler so that the point of piercing varies within a predefined radius (Molstad et al. 2007). Most of the leakage happens when the gas sample is entering the analysis unit. If a peristaltic pump is transporting sample to dedicated sampling loops on multi-port valves, the lining in the pump is made of flexible Marprene or Tygon, both of which allow some diffusion of O2 and N2 into the sampling stream. If using an automated syringe, there will always be some dead volume filled with ambient air. In any case, the sampling system should be backflushed continuously between sampling to keep the lines free of O2 and N2. Leakage rates should be determined in each analytical batch using He-filled dry bottles as part of the sampling sequence.
-
Gas analytics: Analysing multi-gas kinetics from singular samples requires complicated column and detector setups, possibly combining different types of instruments (GC, chemoluminescence, tunable diode laser (TDL), etc.). A particular challenge is analysers which require a high flow-through of analyte (chemoluminescence, TDL). If the gaseous compound of interest is low in ambient air, air can be used to flush the analyser while the sample is spiked into the air stream using a multi-port sampling loop. In case it is undesirable to measure the component on top of atmospheric background, the sample has to be mixed with a span gas prior to analysis (cf. Picarro small sampling unit). In both cases, the compound experiences a considerable dilution, which has to be taken into account when considering detection limits.
-
Detection ranges and precision: Multi-gas kinetics cover a large dynamic spectrum ranging from the ppb range (NO, N2O) to the ppm range (CH4, CO2) to the vol.% range (O2, N2). Common detectors used in gas chromatography-based multi-gas detection (TCD, ECD, PDHID, FID) may cover most of the dynamic range expected to occur in closed bottle incubations, partly overlapping in their response to single compounds. For instance, ECD detectors have unmatched sensitivity for low N2O concentrations but suffer from severe non-linearity in higher ppm ranges. On the other hand, TCD detectors are insensitive for N2O but respond linearly over a wide concentration range. Hence, by combining ECD and TCD detectors, N2O concentration can be analysed over a wide dynamic range. A possibility to cope with gas concentrations exceeding the dynamic range of an analyser is to use splits which divert a defined fraction of a compound to later injection as implemented, for instance, in case of NO detection by chemoluminescence (Molstad et al. 2007). It must also be noted that gas concentrations undergo strong changes during batch incubations as gases accumulate or deplete. This can create separation problems in gas chromatography if two adjacent peaks change concentrations in opposite directions. For example, CO2 may accumulate to the vol.% concentrations, while N2O is consumed (in denitrification). Both gases typically eluate close to each other, and integration routines have to be designed to account for these dynamics. Likewise, it may be difficult to integrate small O2 peaks next to large N2 peaks.
-
Calibration: Calibration should occur by internal standards, i.e. dry bottles filled with certified gas mixtures in He. By including such bottles into the sampling sequence, problems in sample admission, changes in detector sensitivity and leakage can be detected. Standard concentrations should be adopted carefully to the expected concentration ranges.
-
Temperature regulation and agitation: Automated incubation setups measure concentrations in the headspace only, and dissolved concentrations must be inferred using temperature and Henry constants, assuming equilibrium between sample and headspace. This means that the incubation units should be temperature controlled during the entire incubation which is typically achieved by thermostatic water baths or heating blocks. To facilitate the exchange of gases between headspace and sample, it is often desirable to constantly stir samples in aqueous solutions (e.g. suspensions or slurries). This can easily be achieved by placing incubation bottles equipped with Teflon-coated magnetic stirrers on submersible stirrers in a water bath. Alternatively, samples may be shaken in between sampling using programmable shaking units.
In the following sections, two automatic incubation systems will be described.
3.1.2 System 1
The system for determining anaerobic atmospheric trace gases, namely, H2, NO, N2O, CH4, and CO2 comprises a gas chromatograph (7890B GC, Agilent Technologies Inc., Santa Clara, CA, USA) equipped with a split/splitless injector as well as a micro-packed column (Shin Carbon ST 80/100, 2 m, 0,5 mm ID, Part#: 19043, Restek Corporation, Bellefonte, PA, USA) and a capillary column (RT-Molecular Sieve 5A, 30 m, 0,53 mm ID, 50 µm, Cat#: 19723, Restek Corporation, Bellefonte, PA, USA; Plate 3.1). A pulsed discharge helium ionization detector (PDHID, V1D-3-I-HP-220, Valco Instruments Company Inc. VICI AG International, Houston, TX, USA) is coupled to the outlet of the ShinCarbon column via a restriction capillary, and a thermal conductivity detector (TCD, G3440B, Agilent Technologies Inc., Santa Clara, CA, USA) is coupled to the outlet of the molecular sieve 5A column. The ShinCarbon column acts as a water-insensitive molecular sieve where CO2 and N2O elute (which is essentially impossible for “regular” molecular sieve columns that trap CO2). Baseline separation of O2 and N2 in the % range is achieved on the molecular sieve 5A after “heart cutting” of the combined peak eluting from the ShinCarbon column.

Setup of the gas chromatographic system. Switch represents a “Microfluidics Deans Switch” enabling “heart cutting” of peaks eluting from the ShinCarbon column
The TCD is used for analytes in the % range. The PDHID is a universal detector which offers high sensitivity in the higher ppb to lower % range without the need to fulfil the increasing requirements for operating a radionuclide-dependent electron capture detector (ECD) (Forsyth 2004) or to purchase a rather cost-intensive mass sensitive detector (MSD). The gas chromatograph is operated with helium 6.0 as carrier gas that is purified by two sequential helium purifiers (V1HPM-220, Valco Instruments Company Inc. VICI AG International, Houston, TX, USA). One of the outlets of either the PDHID or the TCD can be connected to an NO analyser equipped with a chemiluminescence detector (CLD; Sievers NOA 280i, Zysense LLC, Weddington, NC, USA) to increase sensitivity for NO. The digital signal of the nitric oxide analyser is sent to an analogue-to-digital converter (A/D converter ADC 39500E, Agilent Technologies). The signals of all three detectors are recorded by the software Chemstation (Chemstation Rev. C.01.07.SR2 [255], Agilent Technologies Inc., Santa Clara, CA, USA) on a Windows computer, which is likewise used to control the gas chromatograph.
A robotized incubation system (Combi PAL-xt, CTC Analytics, Zwingen, Switzerland) is connected to the gas chromatograph to complete the setup (Plate 3.2). The Combi PAL-xt is equipped with two trays for 32 vials (10 or 20 ml) each. One of the trays can be temperature controlled in the range of 4 °C–70 °C. Furthermore, it comprises a temperature-controlled agitator (temperature range: 30 °C–200 °C; agitation speed: 250–750 rpm) shaking up to six samples at one time. A Y-cable (custom-made; Autosampler Guys, Alexandria, VA, USA) is used to enable the gas chromatograph to control both the incubation system and the A/D converter of the nitric oxide analyser. This has the advantage that all subordinate devices will only start a run when the gas chromatograph is “ready”. Chronos xt 4.9 Master-Software for PAL samplers (Axel-Semrau, Sprockhövel, Germany) controls the robotized incubation system.

Photograph of the robotized incubation system (a) and configuration of the robotized incubation system, including the gas chromatograph (GC) and the nitric oxide analyser (NOA) with a chemiluminescence detector (CLD) connected via an A/D converter (ADC). “Switch” indicates an ethernet switch (b)
Altogether, this system allows for a cost-efficient online quantification of gas production and consumption during incubation experiments as well as the analysis of stored gas samples. A simultaneous quantification of H2, O2, N2, NO, CO2, CH4, and N2O is achieved. Although not in focus here, C2H2, C2H4, and C2H6 are likewise easily detectable.
3.1.2.1 Measurements of Gas Samples
Analysis parameters are adjusted according to the gases to be measured as well as their expected concentrations. The most important consideration is whether the separation of a given analyte will be sufficient on the ShinCarbon column or whether “heart cutting” and further separation on the molecular sieve column is required. Specifically, baseline separation of O2 and N2 in the % range requires passage through the molecular sieve column. However, CO2, N2O, and NO are strongly retained or trapped, preventing detection. The choice of detector is generally dependent on the concentration of the gases with the PDHID being significantly more sensitive than the TCD. However, the TCD has a broader range of linear response than the PDHID. Multiple “heart cuts” (i.e. switches) between the molecular sieve—TCD path and the restrictor—PDHID path are possible during one run, enabling the quantification of trace concentrations of certain analytes in the presence of huge concentrations of others.
The following protocol provides good separation of the gases H2, NO, CO2, CH4, N2O, O2, and N2 (Table 3.1 and Fig. 3.1). The injection volume of gas samples is 250 µl. The system operates in constant pressure mode with a pressure of 379.21 kPa. The inlet is set to 160 °C, the total flow is 40.782 ml min−1, and the split flow is 12.977 ml min−1. The septum purge flow is 3 ml min−1. Carrier gas is directed to the PDHID. A “heart cut” is performed via the Deans switch at 0.6 min to direct the flow to the TCD via the molecular sieve column, and it switches back at 0.76 min to redirect the flow to the PDHID. The outlet of the PDHID is connected to the CLD inside the nitric oxide analyser. The temperature protocol starts at 90 °C. This temperature is kept for 2 min before it is ramped at 50 °C min−1 to 120 °C and held for 2.1 min. Finally, the oven is further heated to 200 °C (50 °C min−1), and the column is baked out for 2 min at this temperature. After 10 min, the gas chromatograph is ready to start a new run. H2, CH4, CO2, and N2O are detected on the PDHID, O2 and N2 on the TCD, and NO on the CLD (Fig. 3.1). The dynamic range for most of the tested gases includes four orders of magnitude with a single detector (Table 3.1).

Chromatograms of the three detectors connected to the GC system: CLD (blue), TCD (green), and PDHID (red)
The minimum change in concentration which can be regarded significant is 6.3 ppm for H2, 5.0 ppm for CH4, 2.9 ppm for CO2, and 8.7 ppm for N2O at an initial concentration of 500 ppm. For N2, the minimum significant change in concentration is 0.2 vol.% (2000 ppm) at an initial concentration of 2%.
3.1.2.2 Possible Sample Containers
The system can be adapted to a variety of vial types. The sample seats in the provided autosampler trays accommodate 20 ml glass vials (75.5 mm × 22.5 mm with rounded bottom) or 10 ml vials with specific inserts (LEAP PAL Parts and Consumables LLC, Raleigh, NC, USA). Magnetic caps can be used to enable the autosampler to move the vials from the trays to the agitator if required. Acrylic glass blocks with milled sample seats (custom-made) can accommodate up to 100 exetainers (3 ml or 12 ml; Labco, Lampeter, UK) and other vial types offering the possibility for simple measurement of stored gas samples. Butyl septa (T.H. Geyer GmbH and Co. KG, Renningen, Germany) are sufficiently gastight after multiple punctures during incubation experiments (Fig. 3.2). However, the tightness of the septa should be carefully checked to ensure the reliability of the data obtained.

Contamination of He-filled sample vials (500 mbar overpressure) with N2 from air after multiple punctures
3.1.2.3 Semi-automated Determination of Nitrate and Nitrite
An analysis of the dissolved N-oxides NO3− and NO2− is required for the calculation of N and electron recoveries along with the determination of N-gases. We use a semi-automated, highly sensitive system based on reduction of NO3− and/or NO2− and analysis of the resulting nitric oxide. The lower detection limit for NO3− or NO2− in liquid samples is 100 nM.
The heart of the system is a nitric oxide analyser (Sievers 280i, Zysense LLC, Weddington, NC, USA) with a chemical reactor reducing N-oxides to NO (“purge vessel”). The purge vessel contains a reducing agent and is heated by a circulating water bath (Grant Instruments, Cambridge, UK). Up to 96 samples can be placed in a Combi PAL-xt autosampler (CTC Analytics, Zwingen, Switzerland) equipped with a large wash module and standard tray holders for 2 ml HPLC vials. A small volume of liquid sample (10–100 µl) is injected into the purge vessel. The evolving nitric oxide is purged out of the vessel by a stream of nitrogen and reaches the CLD inside the NOA. 50 mg of potassium iodide dissolved in 0.5 ml of double-distilled water and added to 4.5 ml of glacial acetic acid are used as reducing agent for NO2− in the purge vessel. Nitrate and NO2− are both reduced to NO when using 40 mg of vanadium chloride in 5 ml hydrochloric acid as reducing agent. The purge vessel is heated to 45 and 90 °C by a circulating water bath for NO2− only and combined NO3−/NO2− analyses, respectively. Water vapour is condensed before escaping the vessel by a second circulating water bath, set at 4 °C to cool the upper part of the vessel. In addition, a propylene filter is installed in the tubing which connects the purge vessel outlet to the nitric oxide analyser to prevent any liquids from reaching the CLD inside the NOA. A sodium hydroxide trap filled with 1 M NaOH is installed in line with the purge vessel in case of the combined NO3−/NO2− analysis to neutralize any HCl before it reaches the CLD (Fig. 3.2). The data is transferred to a Windows computer which also controls the autosampler via the software Cycle Composer Pal1 (CTC Analytics, Zwingen, Switzerland).
Before the analysis a calibration is performed using NO2− or NO3− standards in the range of 100 nM to 100 mM. The standards as well as the samples are placed in 2 ml HPLC glass vials (neoLab Migge GmbH, Heidelberg, Germany), fitting into the sample tray and are sealed with septa and caps. The autosampler can perform up to 50 injections before the reducing agent needs to be replaced manually. Each measurement takes 60 s, and the needle is flushed with double-distilled water twice in the large wash modules of the Combi PAL autosampler before taking the sample and after injecting.
The concentrations of NO2− can be calculated directly from the peak areas using the calibration curve. In the case of NO3−, the actual concentration is obtained by subtracting the NO2− concentration from the combined NO3−/NO2− concentrations obtained (Plate 3.3).

Schematic diagram of the semi-automated NO3− and NO2− analysis system
3.1.2.4 Application of the Automated Incubation System to Determine Denitrification Kinetics
The following protocol can be applied for the estimation of denitrification potentials by use of soil slurry incubations and simultaneous analysis of denitrification associated trace gases. The determination of N2O production and N2O consumption rates are used to derive NO3− dependent apparent Michaelis Menten parameters for soil denitrification.
General Experimental Setup
Soil slurries are set up and pre-incubated under anoxic conditions to reduce the background of soil endogenous N-oxides. Pre-incubated soil slurries are supplemented with substrate and spiked with different concentrations of NO3−. Acetylene (10% v/v) is added to the gas phase of one of two subsets of samples to inhibit N2O reduction (acetylene inhibition technique; Yoshinari and Knowles 1976). Subsequently, the concentration of N2O in the headspace is monitored via gas chromatographic analyses while shaking the samples on a regular basis. The amount of N2 which is produced in the non-inhibited samples can be estimated from the difference of the N2O concentrations of the two subsets for low concentrations of supplemental NO3− or measured directly when high concentrations of NO3− in the mM range are supplemented.
Materials
-
Duran glass bottles (DWK Life Sciences GmbH, Mainz, Germany)
-
Butyl stopper (Ochs Glasgerätebau, Bovenden, Germany)
-
Helium bottle (5.0, Linde AG, Munich, Germany) with tube connection
-
Vacuum pump RV 3 with tube connection (Edwards, Burgess Hill, UK)
-
1 ml gastight syringe (Trajan Scientific and Medical, Ringwood, Australia)
-
100 µl micro-volume liquid syringe (Trajan Scientific and Medical, Ringwood, Australia)
-
Disposable needles 0.45 mm (Braun GmbH, Kronberg, Germany)
-
1.5 ml tubes (Eppendorf, Hamburg, Germany)
-
20 ml glass vials with crimp top, rounded bottom, 75.5 × 22.5 mm with magnetic crimp caps and butyl septa (T.H. Geyer GmbH & Co. KG, Renningen, Germany)
-
Crimping tool
-
Filtropur S02 syringe filtration unit (Sarstedt AG and Co. KG, Nümbrecht, Germany)
-
Acetylene (≥99.5%, Linde AG, Munich, Germany)
-
Centrifuge Megafuge 8R (Heraeus, Hanau, Germany)
-
Spectrophotometer Infinite M Plex (Tecan Austria GmbH, Grödig, Austria)
-
Roller mixer (Ratek Instruments Pty Ltd, Boronia, Australia).
Experimental Procedure
For the pre-incubation, the soil is diluted 1:5 with distilled water in a Duran glass bottle. The bottle is closed with a butyl stopper, and the slurry is made anoxic by applying alternating cycles of vacuum and helium for five times.
A minor overpressure of helium (200 mbar) is applied to avoid mass flow of air into the bottle. The soil slurry is pre-incubated on a roller mixer until endogenous N-oxides are depleted. Incubation times are soil-dependent. 1 ml samples of the soil slurries are taken every 24 h using a helium-flushed syringe and needle to monitor combined NO3−/NO2− concentrations with the semi-automated NO3−/NO2− analysis system. The sample is centrifuged for 5 min at 16,000 × g and 4 °C before the supernatant is filtered through a 0.2 µm pore size membrane to remove microorganisms as well as any remaining particles. This filtrate is then used for NO3−/NO2− analysis (Plate 3.4). Alternatively, colorimetric methods can be used. The pre-incubation is stopped when NO3− and NO2− are depleted.

Flushing of incubation vials containing soil slurry
The pre-incubated soil slurry is aliquoted into 20 ml glass vials. Each vial receives 15 ml of the soil slurry via small funnels while shaking continuously by hand to avoid sedimentation. Acetate is added as C-source for denitrification as it is one of the most important fermentation products found in soils potentially fuelling denitrification. The vials are crimp-sealed with butyl septa and magnetic caps. To restore anoxic conditions in the samples, the septa are pierced by a thin needle and the headspace is evacuated and helium-flushed five times leaving an overpressure of 620 or 800 mbar in the subsets that will or will not receive acetylene, respectively. Those vials with 620 mbar overpressure receive 0.9 ml acetylene via a helium flushed gastight syringe with a thin needle, resulting in 800 mbar overpressure and 10% (v/v) acetylene in the headspace.
Finally, all vials receive the respective concentration of NO3− ranging from 20–1000 µM from anoxic stock solutions via a 100 µl micro-volume liquid syringe. After placing the vials in the temperature-controlled tray holders of the incubation system, the gas chromatographic analysis starts. Samples are transferred from the trays to the temperature-controlled agitator and vigorously shaken for 2 min before each analysis to allow for equilibration between slurry and headspace. The autosampler syringe is flushed with helium for 60 s before piercing the septum. It draws a 250 µl sample of the headspace and directly injects it into the gas chromatograph. Each vial is sampled 4 to 8 times during the incubation period depending on the initial NO3− concentration and the denitrification potential.
In addition to the gas analysis, NO3−, NO2− and ammonium can be analysed from t0 as well as from tend samples to check for N-recovery. For NO3− and NO2− analysis, the samples are centrifuged as stated above and analysed with the semi-automated NO3− and NO2− analyser (Plate 3.4). Ammonium binds to the cation exchange sites of soil particles. Thus, extraction with 2 M potassium chloride is performed to extract ammonium prior to quantification. 1 ml samples are mixed with 1 ml of 2 M potassium chloride in an Eppendorf tube before being centrifuged for 15 min at 16740 × g and 4 °C. The supernatant is then used for a colorimetric assay (Berthelot reaction). In brief, 100 µl of supernatant is mixed with 50 µl of 2% sodium phenoxide, 25 µl of 0.005% sodium nitroprusside, and 25 µl of sodium hypochlorite. The absorption of the formed indophenol is measured at 630 nm in the spectrophotometer after 30 min of incubation at 30 °C in the dark.
Data Evaluation
The gas chromatographic data is evaluated by firstly transforming peak areas into ppm values with the help of calibration curves. These ppm values are then corrected for the pressure in the vial headspace, which decreases with the number of samplings. It is important to account for the soluble fraction of CO2 and N2O in water. The temperature-dependent Bunsen coefficients of these gases can be used to estimate the physically dissolved fraction. For nitrous oxide, the Bunsen coefficient is 0.85 at 10 °C, 0.71 at 15 °C, 0.61 at 20 °C, and 0.52 at 25 °C. For carbon dioxide, it is 0.99 at 10 °C, 0.85 at 15 °C, 0.74 at 20 °C, and 0.65 at 25 °C (Blachnik 1998). Physically dissolved CO2 reacts in a pH-dependent manner with water to H2CO3 that further reacts to bicarbonate and carbonate. This pH-dependent chemically dissolved fraction also needs to be considered.
Calculation of decreasing headspace pressure after sampling:
in the likely case that p1 = ps, this equation simplifies to:
Symbol | Unit | |
|---|---|---|
p0 | Total pressure in the headspace before sampling | Pa |
n0 | Amount of substance in the headspace before sampling | mol |
Vh | Volume of the headspace | m3 |
R | Gas constant | 8.314 J K−1 mol−1 |
T | temperature | K |
Vs | Sample volume | m3 |
ps | Pressure during sampling | Pa |
ns | Amount of substance sampled | mol |
p1 | Total pressure in the headspace after sampling | Pa |
Calculation of amount of substance from ppm values:
Symbol | Unit | |
|---|---|---|
ntotal | Total amount of substance | mol |
ngas | Amount of substance in gaseous phase | mol |
ndiss | Amount of substance in liquid phase | mol |
nchem | Chemically dissolved fraction | mol |
Vgas | Headspace volume | m3 |
Vliq | Slurry volume | m3 |
yi | Mole fraction | ppm × 10−6 |
ph | Headspace pressure | Pa |
R | Gas constant | 8.314 J K−1 mol−1 |
T | Temperature | K |
α | Bunsen coefficient |
3.1.3 System 2
The described robotized gas analysis system is an improved version of the system described by Molstad et al. (2007). The current system has been published by Molstad et al. (2016). The system has replaced the microGC with a traditional GC (Agilent 7890A), equipped with various valves, columns, and detectors (TCD, FID, and ECD) to analyse all relevant gases (except NO) by a single injection. The setup has two PLOT columns: a Molesieve column for separation of N2 and O2, and a PLOTQ column to separate CH4, N2O, and CO2. The system has three loop injectors–one for each PLOT column and one for injecting samples into the chemiluminescence NO detector. Short, packed Hayesep columns are used to retain and backflush water coming with the sample.
The use of ECD ensures sensitive measurements of N2O at low concentrations, and the FID detects CH4 in the sub-ppm range, thus allowing quantification of these gases at below ambient levels in the atmosphere (which are 0.35 ppmv for N2O, 1.84 ppmv for CH4). At high concentrations, N2O can be quantified via the TCD signal, which is linear, ensuring accurate determination of N2O over a wide concentration range.
The Gilson 222 (XL) autosampler of the old system has been replaced with the larger CTC GC-PAL (rebranded by Agilent), including a custom-made holder for a hypodermic needle. Up to 30 stirred and 16 non-stirred 120 ml serum flasks, incubated in a water bath, can now be monitored in one experiment (Plate 3.5).

Water bath with stirrer plates (blue) and rack (green) that can hold up to 30 stirred and 16 non-stirred bottles. The lid fits onto the tops of the bottles. Other types of bottles may be supported by making other versions of the two plates that serve as rack and lid. The autosampler (not shown) is attached behind the bath. The bath, rack, and lid can be produced from CAD drawings with a CNC router, using seven PVC plates of 2 cm thickness. The CAD drawings are available upon request
To transfer a sample from the headspace to the three sampling loops (Plate 3.6), a peristaltic pump (Gilson Minipuls 3) is used. We now use Iso-Versinic Fluoroelastomer (IF) tubing (17 cm, ID = 2 mm; product nr 770130, Saint-Gobain or product nr F117744, John Morris) instead of Marprene tubing. This is because the diffusion of N2 and O2 is slower through IF than Marprene, and because this replacement reduced the carryover of N2O and CH4 (see leaks, carryover, and dilution tests below).

Architecture of the GC system. The GC exists in slightly different versions, equipped with one or two ovens (one for the PLOTQ column and one for the Molesieve column) which are held at constant temperature
In all other aspects, the new system is equal to the original (Molstad et al. 2007) regarding the repeated sampling by peristaltic pumping, replacement of the sampled gas by reversed pumping (injecting He), and transfer of gas to a chemiluminescence NO analyser. In short, samples are taken from 10–120 ml serum flasks using an autosampler connected to a peristaltic pump. The autosampler operates a needle which pierces the septa on the flasks (never twice at the same spot), and the pump takes approximately 1 ml headspace gas from the flasks into the sample loops of the GC and NO analyser. After injection (the turning of the valves leading to the gas in the loops into the GC columns and the NO analyser), the pump is reversed, pumping helium and the portion of the up-pumped gas in the pipelines back into the flasks, thus minimizing dilution of the gas in the flasks while maintaining atmospheric pressure.
The system monitors O2, N2, N2O, NO, CO2, and CH4 (and various other gases if needed) by repeated sampling of the headspace, while sustaining the gas pressure near 1 atmosphere in the flasks due to replacement of sampled gas with He.
3.1.3.1 The GC
The flow scheme of the GC system is shown in Plate 3.6. Loop injection to the PLOT columns (Molesieve and PLOTQ) is done by Valve 3 and 4 via Hayesep backflush columns, which prevent water from entering the PLOT columns. For the Molesieve column, also CO2 is backflushed. The third loop injection valve (Valve 2) sends samples to the NO analyser. Like in Sitaula et al. (1992), Valve 1 directs the outlet from the TCD to the FID most of the time to prevent oxidation of the detector, except for a brief period when N2O is eluted from the PLOTQ column.
The flowchart for the valve positions during a run is shown in Plate 3.6, and a chromatogram of air is illustrated in Fig. 3.3. Figure 3.4 shows the valve positions, column back pressures, and substance elution times during a run. Further information about the GC is found in Table 3.2.

Chromatogram of an air sample (5.5 min). The peaks before the 3rd minute come from the PLOTQ column, the others come from the Molesieve column. CO2, N2, and O2 (at 2.1, 3.5, and 4 min) are detected on the TCD. Methane is detected, at 1.5 min from the PLOTQ column and at 5 min from the Molesieve column, on the FID, and also, for high concentrations, on the TCD. For a 1.2 min period, the gas from the TCD is switched from FID to ECD, allowing the quantification of low N2O concentrations. At high concentrations, N2O is also detected by the TCD, thus extending the linear range much beyond that of the ECD

Valve positions, column pressures, and eluted peaks during a single run (5 min)
Slightly different versions of the system have been constructed with our help elsewhere. Currently, there are five systems operating in China and three in Norway. These systems are identical in principle, the main difference being that some of the GC’s have the PLOTQ column in a separate oven. Our experience is now that this is not necessary for the compounds we are analysing. In one version, an H2 analyser is attached in parallel to the FID. A split in front of the FID directs a minor amount of the column flow into a Peak Performer 1 RCP H2 analyser.
3.1.3.2 The NO Analyser
For the analysis of nitric oxide, we use either a modified Teledyne 200 NO/NOx analyser or a Sievers NO 280i. The modification of the Teledyne analyser is held a trade secret by the Norwegian distributor of Teledyne (Industriell Måleteknikk, Oslo, Norway) but involves an improvement of the response time. Without this modification, the Teledyne analyser has proven to be unsuitable for the incubation system, which injects a short pulse of 0.5 ml gas into the analyser. (An unmodified analyser gives a one-minute response to a 0.5 ml injection; depending on the NO concentration, this response can be either a broad flat-topped peak or a sharp peak followed by one minute of noise.) Based on this, it is recommended to either buy the Teledyne instrument from the Norwegian distributor or acquire a different NO analyser, like the (much more sensitive) Sievers NOA 280i, which is also supported by the software, when building the incubation system.
3.1.3.3 Leaks, Carryover, and Dilution of Headspace by Sampling
The new system has another type of injection (loop injection versus the solenoid-valve system of the microGC), and this leads to less dilution of the headspace per sampling and lower leaks. We have also replaced the Marprene tubing with Iso-Versinic Fluorelastomer tubing, which is more gastight, though less durable.
By sampling repeatedly from 120 ml flasks containing air (no liquid), we have found that the dilution of the headspace per sampling is 0.71%, which corresponds to replacing 0.85 ml of gas with helium. This test was conducted with active sampling to the NO analyser; without this (i.e. without turning Valve 2), the dilution per sampling was 0.36% or 0.43 ml. These volumes are lower than the sum of the volumes of the sample loops because they are heated to 100 °C. The theoretical loss for a 1 ml sample loop should be approximately 1.0 ml*293 K/373 K = 0.785 ml if the temperature in the bottle is 293 K. The dilution is slightly higher than this due to the mixing of sample gas with helium in the pump tubing and pipes.
We have also tested the leak of O2 and N2 into the flasks per sampling by repeated sampling from flasks with He (1 atm) at 20 °C. This was done with two types of tubing, Marprene (ID = 1.6 mm), and Iso-Versinic Fluorelastomer (ID = 2 mm).
The results for Marprene are
-
N2: 10 ppmv/sampling = 50 nmol N2/sampling
-
O2: 5 ppmv/sampling = 25 nmol O2/sampling
These leak estimates were obtained by running the pump at a speed resulting in a flow of 9 ml min−1, sample pumping for 20 s = 3 ml total volume taken into the loops. If using slower pumping speed and longer times (3 ml min−1, 60 s pumping), the leaks were approximately doubled.
The results for Iso-Versinic Fluorelastomer, pumping 12 ml min−1 (due to larger ID) for 15 s:
-
N2: 2.3 ppmv/sampling = 11 nmol N2/sampling
-
O2: 1.2 ppmv/sampling = 6 nmol O2/sampling
The leaks can be further reduced by enclosing the pump in a helium chamber. A simple plastic cover over the pump head, flushed with He (10 ml min−1), reduced the leaks of N2 and O2 to 1.6 and 0.8 ppmv/sampling.
The carryover of N2O and CH4 from one sample to the next was measured by sampling from a standard containing 151 ppmv N2O and 1 vol.% CH4 before sampling in sequence five bottles containing only helium. The results (Table 3.3 and Fig. 3.5) show that, compared to Marprene, the Iso-Versinic tubing has less than half the carryover for N2O, and less than one-fifth for CH4. With Iso-Versinic, the carryover to the first helium sample yielded 0.055 ppmv N2O (0.037% of the previous sample) and 0.15 ppmv CH4 (0.0015% of the previous sample). The carryover of O2 and N2 was measured in a similar way by sampling from a helium bottle after sampling from a bottle with 20% O2 and 79% N2. The carryover of O2 and N2 was not detectable with Iso-Versinic; with Marprene, it yielded approximately 7 ppmv O2 and 16 ppmv N2. For CO2, a bottle with 8.5% CO2 yielded a carryover of 13.3 ppmv (0.016%) for Marprene and 7.3 ppmv (0.0086%) for Iso-Versinic

Leak test: Results of repeated sampling from two 120 ml bottles containing helium. The O2 (blue dots) and N2 (red dots) concentrations increase on average by 1.2 ppmv and 2.3 ppmv per sampling, respectively. The insert shows the leaks, the difference between consecutive measured ppmv values. The leaks do not appear to increase over the 60 samplings (These measurements have been adjusted for the dilution caused by replacing the sampled gas with helium; unadjusted measurements are shown in grey)
3.1.3.4 Software
The system is controlled by Python software which checks the readiness state of the chromatography software, controls the autosampler and the peristaltic pump, triggers the GC, records the output of the NO analyser, and sorts and tabulates the results. The bottles may be given individual rates of sampling, and the sampling rate of each bottle may be easily changed during an experiment (The problem of collisions of sampling time is solved by putting the bottles in a priority queue and linking together bottles that must be sampled in sequence). It is also possible to let the sampling rates automatically depend on the measured concentrations during a run (for example, measuring more often when the oxygen concentration runs low), but this requires some programming from the experimenter. For the inspection of results during an experiment, a graphical user interface is provided which lets the experimenter quickly plot time series of raw data or estimated concentrations for chosen substances and bottles.
3.1.3.5 Data Handling and Analysis
The system produces large amounts of data, requiring adequate handling. We have developed software that compiles the data as a result matrix for each gas (vial nr * time), and calculation routines for estimation of gas transport between headspace and liquid, concentrations of the gases in the liquid, and the kinetics of transformations (Molstad et al. 2007). The software has been used in many studies of N2O/N2 gas kinetics, NO scavenging kinetics, analyses of oxic and anoxic respiration, etc. (see publication list of NMBU Nitrogen Group https://www.nmbu.no/en/research/groups/nitrogen/publications). The software is provided free of charge on request.
3.1.3.6 Documentation
This description of the robotic incubation system is modified from a technical report (Molstad et al. 2016) which is available on Researchgate. The report should be referred to by its DOI, together with Molstad et al. (2007), when publishing results obtained with this incubation system.
3.2 Automated Chamber Systems for Field Measurements
3.2.1 Field Techniques Using GC Systems
Accurate measurement of GHG flux from soils is challenging because of the high temporal and spatial variabilities in the processes contributing to GHG emissions (Butterbach-Bahl et al. 2016), which result in a variability of soil surface fluxes often spanning 2–3 magnitudes. Addressing these two components of variability remains a challenge.
Automated chamber systems are increasingly used to address temporal variability. These systems consist of a set of automated chambers, mostly the closed chamber type if fluxes of rather non-reactive trace gases such as CH4, CO2, or N2O are measured, or of the dynamic chamber type, if soil–atmosphere fluxes of reactive gas species such as NO, NO2, or NH3 are measured (e.g. Butterbach-Bahl et al. 1997).
One of the first times an automated closed chamber system was used was for the quantification of CH4 fluxes from rice paddy fields in Italy (Schütz et al. 1989). These authors showed that CH4 fluxes vary strongly on diurnal to seasonal scales and, by using automated chamber systems with sub-daily measurements of fluxes, provided for the first time an accurate quantification of cumulative seasonal CH4 fluxes. The development of automated measuring chambers continued over years, and Papen and Butterbach-Bahl (1999), Gasche and Papen (1999) and Butterbach-Bahl et al. (2002) provided examples of how such systems can be used for quantifying the soil–atmosphere exchange of N2O, NO, NO2, and CH4 continuously over entire years (Plate 3.7). These measurements revealed that even during freeze–thaw periods forest soils are extremely strong sources for the atmospheric GHG N2O (Papen and Butterbach-Bahl 1999) and that in years during which such freeze–thaw N2O fluxes occur, the annual flux magnitude might be a factor of 2–10 times higher than in years without freeze–thaw events (Plate 3.7).

Schematic drawing of an automated chamber system for measurements of soil GHG fluxes with five chambers and a gas chromatograph equipped with an Electron Capture detector (for N2O analysis) and Flame Ionization Detector (for analysis of CH4 plus CO2 [the latter requires use of a methanizer]) (right panel). The right panel show a picture of an automated chamber with pistons to close/open chambers by compressed air (top), a gas chromatograph fixed to a small box (middle), and a dataset of mean daily N2O and NO fluxes as obtained by the use of an automated closed chamber system (N2O) and an automated dynamic chamber system. Further details describing these measurements are provided in Papen and Butterbach-Bahl (1999) and Gasche and Papen (1999)
Only by using automated chamber systems, Wolf et al. (2010) were able to accurately quantify N2O fluxes during freeze–thaw events in steppe ecosystems of Inner Mongolia. In this study, not only high seasonal, but also diurnal flux variations were observed. Meanwhile, to the knowledge of the authors, approx. 20–30 groups worldwide are using automated chamber systems, i.e. still in a minority of GHG studies are such systems used for quantifying GHG emissions from upland and lowland agricultural, forest, or grassland ecosystems, while installations are spanning various climate zones, i.e. from the tropics to alpine systems. Most recently, those automated closed chamber systems were combined with automated lysimeter systems, so that environmental N and C losses can be quantified at the same time (Fu et al. 2017, Plate 3.8). At the same time, the use of laser spectroscopy instead of gas chromatography allowed to lower the flux detection limit significantly, so that closing times for chambers could be reduced (Savage et al. 2014).

Automated GHG flux measurements at a grassland site in Bavaria by the Institute of Meteorology and Climate Research, Atmospheric Environmental Research (IMK-IFU), Garmisch-Partenkirchen, Germany, using a closed chamber robot system in combination with lysimeters. For this installation, undisturbed soil monoliths (1 m diameter, 1.8 m depth) were taken at various locations along a climate gradient and re-installed at a common site. This setup allows to monitor short- and long-term effects of climate change (changes in rainfall and temperature) on ecosystem CO2, N2O, and CH4 fluxes as well as on groundwater recharge and leaching of nitrogen compounds (DON, NO3−, NH4+). Photo: courtesy of Dr. Ralf Kiese, who is also leading this research programme at IMK-IFU, which is part of the German Helmholtz TERENO initiative
But how important are high-frequency measurements of GHG emissions from soils? Does this indeed significantly improve the accuracy of annual flux estimates? Or do weekly or monthly measurements suffice to conclude on annual exchange rates of GHG between soils and the atmosphere? These questions were addressed in a study by Barton et al. (2015) using 28 published datasets of sub-daily N2O fluxes from a variety of different terrestrial ecosystems across the globe. The aim of the study was (a) to provide guidelines for sampling frequency to estimate annual N2O fluxes using manual chambers and (b) to quantify the uncertainty if the sampling frequency is lowered to 2–3 times per week, weekly, or monthly or if the sampling frequency is varied, with intensive measurements during episodes with expected high fluxes (e.g. following fertilization events or first rainfalls following a dry period), but lower frequency measurements during periods with expected low fluxes (e.g. dry periods). The analyses by Barton et al. (2015) show that “Nitrous oxide emissions need to be measured daily to accurately determine annual N2O flux in environments where data has not previously been collated”, and that daily sampling was required to achieve annual N2O fluxes within 10% accuracy compared to sub-daily measurements with automated chambers. Lowering the measuring frequency to weekly, two weekly or monthly measurements result in an under- or over-estimation of annual N2O fluxes by up to 93.5%. Overall, the authors suggested that the current uncertainty in global terrestrial N2O budgets associated with the upscaling of field-based datasets can be decreased significantly using adequate sampling frequencies (daily, every second day, or by using automated measuring systems). For further reading the paper of the Global Research Alliance by Grace et al. (2020) on automated techniques is recommended.
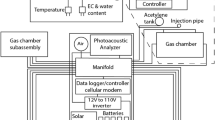
3.2.2 Combination of Automatic Chamber System and CRDS Analyser for Field GHG Flux Measurements
The LI-8100A Automated Soil CO2 Flux System (Licor, Inc.) is designed to measure CO2 efflux with an IR gas analyser (IRGA) from soils and low vegetation using up to 16 automated chambers with the closed non-steady-state transient approach controlled by a multiplexer unit LI-8150. This system allows the combination with a CRDS analyser (see Chap. 2, Sect. 2.7), capable of measuring other trace gases, including CH4, N2O, and NH3. They can be interfaced with the LI-8100A System and the SoilFluxPro data processing software (freely downloadable from the LI-COR website) to compute fluxes for the additional gases. There is an application note available that describes how to integrate a third-party analyser like the Picarro G2508, which can measure CO2, CH4, N2O, NH3, and H2O simultaneously, into the system and the procedure used to compute fluxes of additional gases in SoilFluxPro software (Licor 2016).
There are three options of combining the two systems (see Plates 3.9 and 3.10); the first two options (Plate 3.9a, b) connecting the CRDS analyser to the primary sampling loop or as a subsampling loop are described in the application note and need adjustments of the flow and a calibration of the IRGA (see application note, Licor 2016). The third mode of gas flow path does not need these adjustments, and the LI-8100A might also be used without the CRDS analyser without the need for readjustments.

Schematic modes of combining the LI-8100A automated chamber system including up to 16 clear or opaque chambers with a CRDS Picarro five-gas analyser G2508 for simultaneous flux measurement of CO2, CH4, N2O, NH3, and H2O: a entire measurement gas flow enters the IRGA first, then the CRDS analyser, b only part of the measurement gas flow from the IRGA Outlet enters the CRDS analyser, and c the IRGA unit is only used to control the pump and valve system of the multiplexer but not for CO2 flux measurement. The multiplexer has 16 ports and can be connected to clear and opaque chambers, to soil air probes or aboveground sampling ports

The combined automatic chamber system with CRDS analyser for field GHG-flux measurement in the Giessen climate change experiment combining air Temperature warming of +2 °C and free-air CO2 Enrichment of +20% (T-FACE). In the left-hand box, the Picarro analyser is installed including its monitor and pump. The LI-8150 Multiplexer (big yellow box) is connected to seven automated chambers and eight soil air probes and switches every 5 to 30 min from one port to the next controlled by the LI-8100A Analyzer Control Unit (small yellow box). The screen (lower left side) shows stable N2O concentrations (zero flux, 1st panel above), CH4 uptake (2nd panel), stable H2O concentrations (3rd panel), and ecosystem CO2 respiration (4th panel on the screen)
If the system is used in humid and cold regions, a water trap or nafion tube to dry the measurement gas flow is recommended to protect the CRDS analyser from condensed water. In the LI-COR control software, LI-8100A the total gas volume of the system (chamber, tubes, analyser internal tubing, water trap) has to be defined for correct flux calculations. With the LI-8100A software, the valve system of the Multiplexer and the measurement sequence of the 16 automated chambers can be controlled.
It is important to synchronize the system clocks of the IRGA and the Picarro frequently (daily–weekly); otherwise, the SoilFluxPro software cannot match the LI-COR and the Picarro dataset for flux calculations properly. Matching of the LI-COR and the Picarro dataset in the SoilFluxPro software is also explained in the application note (Licor 2016).
For long-term field measurement campaigns, the Picarro Analyser should be placed in a water proof box. If this system is used in a hot environment, the housing box for the Picarro Analyser has to be cooled so that temperature in the box stays below 35 °C; otherwise, the analyser will go into error. A cooling solution with a small air condition unit is possible and is described in the Picarro Application Note AN041 (Anthony and Silver 2020).
This combination of an automatic chamber system and CRDS Analyser allows to set up a high-frequency measurement sequence including up to 16 ports. A combination of clear and opaque chambers, soil air probes (e.g. Accurel© tubes installed in different soil depths), and aboveground air sampling ports is possible to create detailed data on GHG concentrations in different soil depths and aboveground and the resulting gas fluxes. The LI-8100A system allows to install and measure soil temperature and moisture probes at each automated chamber to record the top soil conditions during flux measurement. The result of such data sets allows a better understanding of hot moments of GHG emission peaks during long-term measurement campaigns and the hotspots of GHG production within the soil profile.
References
Anthony TL, Silver WL (2020) Field deployment of Picarro G2508 and Eosense eosAC/eosMx in perennially or periodically flooded soils. Picarro Application Note (AN041)
Bakken LR, Bergaust L, Liu B, Frostegård Å (2012) Regulation of denitrification at the cellular level: a clue to the understanding of N2O emissions from soils. Philos Trans R Soc B Biol Sci 367:1226–1234
Barton L, Wolf B, Rowlings D, Scheer C, Kiese R, Grace P, Stefanova K, Butterbach-Bahl K (2015) Sampling frequency affects estimates of annual nitrous oxide fluxes. Sci Rep 5:16912
Bedard C, Knowles R (1989) Physiology, biochemistry, and specific inhibitors of CH4, NH4+, and CO oxidation by methanotrophs and nitrifiers. Microbiol Rev 53:68–84
Bergaust L, Mao Y, Bakken L, Frostegard A (2010) Denitrification response patterns during the transition to anoxic respiration and posttranscriptional effects of suboptimal pH on nitrogen oxide reductase in Paracoccus denitrificans. Appl Environ Microbiol 76:6387–6396
Bergaust L, van Spanning RJM, Frostegård Å, Bakken LR (2012) Expression of nitrous oxide reductase in Paracoccus denitrificans is regulated by oxygen and nitric oxide through FnrP and NNR. Microbiol 158:826–834
Blachnik R (1998) Taschenbuch für Chemiker und Physiker Ed. 4 Vol 3. Springer, Heidelberg
Braker G, Conrad R (2011) Chapter 2- Diversity, Structure, and Size of N2O-Producing Microbial Communities in Soils—What Matters for Their Functioning? In, Laskin AI, Sariaslani S, Gadd GMBT-A. in A.M. (eds). Academic Press, pp 33–70
Brenzinger K, Dörsch P, Braker G (2015) pH-driven shifts in overall and transcriptionally active denitrifiers control gaseous product stoichiometry in growth experiments with extracted bacteria from soil. Front Microbiol 6
Butterbach-Bahl K, Gasche R, Breuer L, Papen H (1997) Fluxes of NO and N2O from temperate forest soils: impact of forest type, N deposition and of liming on the NO and N2O emissions. Nutr Cycl Agroecosyst 48:79–90
Butterbach-Bahl K, Rothe A, Papen H (2002) Effect of tree distance on N2O- and CH -4 fluxes from soils in temperate forest ecosystems. Plant Soil 240:91–103
Butterbach-Bahl K, Sander B, Pelster D, Díaz-Pinés E (2016) Quantifying Greenhouse Gas Emissions from Managed and Natural Soils. Methods for Measuring Greenhouse Gas Balances and Evaluating Mitigation Options in Smallholder Agriculture, pp 71–96
Dörsch P, Braker G, Bakken LR (2012) Community specific pH response of denitrification: experiments with cells extracted from organic soils. FEMS Microbiol Ecol 79:530–541
Forsyth DS (2004) Pulsed discharge detector: theory and applications. J Chromatogr A 1050:63–68
Fu J, Gasche R, Wang N, Lu H, Butterbach-Bahl K, Kiese R (2017) Impacts of climate and management on water balance and nitrogen leaching from montane grassland soils of S-Germany. Environ Pollut 229:119–131
Gasche R, Papen H (1999) A 3-year continuous record of nitrogen trace gas fluxes from un- treated and limed soil of a N-saturated spruce and beech forest ecosystem in Germany: 2. NO and NO2 fluxes. J Geophys Res 104:18505–18520
Grace PR, Van der Weerden TJ, Rowlings DW, Scheer C, Brunk C, Kiese R, Butterbach-Bahl K, Rees RM, Robertson GP, Skiba U, (2020) Global Research Alliance N2O chamber methodology guidelines: Considerations for automated flux measurement. J Environ Qual 49:1126–1140
Hink L, Lycus P, Gubry-Rangin C, Frostegård Å, Nicol GW, Prosser JI, Bakken LR (2017) Kinetics of NH3-oxidation, NO-turnover, N2O-production and electron flow during oxygen depletion in model bacterial and archaeal ammonia oxidisers. Env Microbiol 19:4882–4896
Horn MA, Ihssen J, Matthies C, Schramm A, Acker G, Drake HL (2005) Dechloromonas denitrificans sp. nov., Flavobacterium denitrificans sp. nov., Paenibacillus anaericanus sp. nov. and Paenibacillus terrae strain MH72, N2O-producing bacteria isolated from the gut of the earthworm Aporrectodea caliginosa. Int J Syst Evolut Microbiol 55(3): 1255–1265
Horn MA, Mertel R, Gehre M, Kästner M, Drake HL (2006) In vivo emission of dinitrogen by earthworms via denitrifying bacteria in the gut. Appl Environ Microbiol 72(2):1013–1018
Keppler F, Hamilton JTG, Braß M, Röckmann T (2006) Methane emissions from terrestrial plants under aerobic conditions. Nature 439:187–191
Liu B, Frostegard A, Bakken LR (2014) Impaired reduction of N2O to N2 in acid soils is due to a posttranscriptional interference with the expression of nosZ. mBio 5: 1383–1314
Licor (2016) Capturing and processing soil GHG fluxes using the LI-8100A—Picarro. Application Note. Licor, Linoln, Nebraska
Lycus P, Lovise Bøthun K, Bergaust L, Peele Shapleigh J, Reier Bakken L, Frostegård Å (2017) Phenotypic and genotypic richness of denitrifiers revealed by a novel isolation strategy. ISME J 11:2219–2232
McCrackin ML, Elser JJ (2010) Atmospheric nitrogen deposition influences denitrification and nitrous oxide production in lakes. Ecology 91:528–539
Molstad L, Dörsch P, Bakken LR (2007) Robotized incubation system for monitoring gases (O2, NO, N2O, N2) in denitrifying cultures. J Microbiol Methods 71:202–211
Molstad L, Dörsch P, Bakken L (2016) Improved robotized incubation system for gas kinetics in batch cultures. ResearchGate
Palmer K, Drake HL, Horn MA (2010) Association of novel and highly diverse acid-tolerant denitrifiers with N(2)O fluxes of an acidic fen. Appl Environ Microbiol 76(4):1125–1134
Palmer K, Biasi C, Horn MA (2012) Contrasting denitrifier communities relate to contrasting N2O emission patterns from acidic peat soils in arctic tundra. ISME J 6(5):1058–1077
Palmer K, Horn MA (2015) Denitrification activity of a remarkably diverse fen denitrifier community in Finnish Lapland is N-oxide limited. PLoS ONE 10(4):e0123123–e0123123
Palmer K, Kopp J, Gebauer G, Horn MA (2016) Drying-rewetting and flooding impact denitrifier activity rather than community structure in a moderately acidic fen. Front Microbiol 7:727
Papen H, Butterbach-Bahl K (1999) A 3-year continuous record of nitrogen trace gas fluxes from untreated and limed soil of a N-saturated spruce and beech forest ecosystem in Germany, 1. N2O emissions. J Geophys Res 104:18487–18503
Savage K, Phillips R, Davidson E (2014) High temporal frequency measurements of greenhouse gas emissions from soils. Biogeosci 10:2709–2720
Schlüter S, Zawallich J, Vogel HJ, Dörsch P (2019) Physical constraints for respiration in microbial hotspots in soil and their importance for denitrification. Biogeosci 16:3665–3678
Schmidt O, Hink L, Horn MA, Drake HL (2016) Peat: home to novel syntrophic species that feed acetate- and hydrogen-scavenging methanogens. ISME J 10:1954–1966
Schütz H, Holzapfel-Pschorn A, Conrad R, Rennenberg H (1989) A 3-year continues record on the influence of daytime, season, and fertilizer treatment on methane emission rates from an Italian rice paddy. J Geophys Res 941:16405–16416
Sitaula BK, Luo J, Bakken LR (1992) Rapid analysis of climate gases by wide bore capillary gas chromatography. J Environ Qual 21(3):493–496
Taylor AE, Taylor K, Tennigkeit B, Palatinszky M, Stieglmeier M, Myrold DD, Schleper C, Wagne M, Bottomley PJ (2015) Inhibitory effects of C2 to C10 1-alkynes on ammonia oxidation in two Nitrososphaera species. Appl Environ Microbiol 81:1942–1948
Vollack KU, Zumft WG (2001) Nitric Oxide Signaling and Transcriptional Control of Denitrification Genes in Pseudomonas stutzeri. J Bacteriol 183:2516–2526
Wolf B, Zheng X, Brüggemann N, Chen W, Dannenmann M, Han X, Sutton MA, Wu H, Yao Z, Butterbach-Bahl K (2010) Grazing-induced reduction of natural nitrous oxide release from continental steppe. Nature 464:881–884
Yoshinari T, Knowles R (1976) Acetylene inhibition of nitrous oxide reduction by denitrifying bacteria. Biochem Biophys Res Commun 69:705–710
Zhu J, Mulder J, Solheimslid SO, Dörsch P (2013) Functional traits of denitrification in a subtropical forest catchment in China with high atmogenic N deposition. Soil Biol Biochem 57:577–586
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
The opinions expressed in this chapter are those of the author(s) and do not necessarily reflect the views of the International Atomic Energy Agency, its Board of Directors, or the countries they represent
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 3.0 IGO license (http://creativecommons.org/licenses/by/3.0/igo/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the International Atomic Energy Agency, provide a link to the Creative Commons license and indicate if changes were made.
Any dispute related to the use of the works of the International Atomic Energy Agency that cannot be settled amicably shall be submitted to arbitration pursuant to the UNCITRAL rules. The use of the International Atomic Energy Agency's name for any purpose other than for attribution, and the use of the International Atomic Energy Agency's logo, shall be subject to a separate written license agreement between the International Atomic Energy Agency and the user and is not authorized as part of this CC-IGO license. Note that the link provided above includes additional terms and conditions of the license.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Zaman, M. et al. (2021). Automated Laboratory and Field Techniques to Determine Greenhouse Gas Emissions. In: Zaman, M., Heng, L., Müller, C. (eds) Measuring Emission of Agricultural Greenhouse Gases and Developing Mitigation Options using Nuclear and Related Techniques. Springer, Cham. https://doi.org/10.1007/978-3-030-55396-8_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-55396-8_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-55395-1
Online ISBN: 978-3-030-55396-8
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)