
Abstract
Biomarker testing, which is now often the standard of care for patients diagnosed with cancer, can be used by physicians to assess cancer risk, diagnose a particular cancer, select treatment, and/or assess treatment response. In this summary of a keynote address, Edith Perez, Professor of Medicine at the Mayo Clinic, discusses basic concepts and issues of biomarker-based precision medicine in clinical trials and oncology practice. She describes the general features of well-designed biomarker-driven clinical trials and offers specific suggestions for designing clinical trials to support FDA approval. Perez predicts that in the near future, tumor sequencing will become standard clinical practice; liquid biopsies will become available to sample circulating tumor DNA (ctDNA); tumor classification will become molecular-based, and tumor-agnostic biomarker strategies may be used to manage patients; and clinical trials in oncology will use sequencing at both enrollment and follow-up. Additionally, she describes some basic concepts and challenges in the use of cancer immunotherapy (CIT) biomarkers, which are revolutionizing oncology. A new Program for Accelerated Cancer Therapies (PACT) was cited as an example of a multidisciplinary collaboration with NIH, NCI, and biopharma that aims to support the development of standardized biomarkers for immunoprofiling and exploratory biomarkers of high relevance to patient care. Finally, Perez explains why bringing biomarker-based trials to patients is challenging and predicts that large collaboratives such as PACT will move precision medicine and oncology forward by linking clinical retrospective and prospective cancer genomic and proteomic data with longitudinal clinical outcomes.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
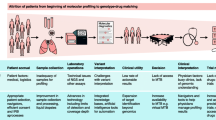
Introduction: Biomarkers in Clinical Care and Research
Biomarker-Based Precision Medicine
Biomarker-based precision medicine is now often the standard of care for patients diagnosed with cancer. Industry and government have invested heavily in the development of precision medicine, and as improved diagnostics, testing, and biomarkers become more common, existing barriers to the use of precision medicine will be eliminated. To make this happen, there must be clear scientific communication that enhances understanding and influences clinical practice. One concern is the high cost of new precision medicines available for patients, which should be offset by efficiency and overall value provided to patients with cancer. Additionally, rather than testing tumor specimens just once, there will be increased reliance on dynamic biomarkers in the continuum of cancer care. This will influence existing guidelines and procedures in many hospitals, clinical practices, and insurance companies, so that patients can access the best medicine for them .
Biomarkers for Decision Support
It is increasingly evident that the introduction of targeted therapies has revolutionized the management of patients with cancer. Integration of biomarkers, in the tumor and stroma, in addition to clinical characteristics, helps healthcare professionals optimize diagnosis and treatment recommendations. However, when should a physician consider the use of biomarkers for decision support in the continuum of cancer care? Biomarker testing can be used to help assess cancer risk, best diagnose a particular malignancy, select treatment, and/or assess the treatment response. There are many decisions that physicians must make when they use biomarker testing, not the least of which is what tests to choose from the many now available. And once the results are in, how does the physician interpret the sometimes massive amount of information and report them in an understandable way? How should the results be applied to patient care? Will patients be reimbursed for this selected therapy? Ultimately, physicians are increasingly expected to consider these issues in the context of biomarker testing in the continuum of cancer care.
Biomarker Properties
Biomarkers, which are measureable indicators of biological processes, may be prognostic or predictive. A prognostic biomarker demonstrates the relationship between the biomarker and response in a control group (patients receiving standard of care); it predicts disease aggressiveness regardless of the experimental study treatment. A prognostic biomarker can be evaluated by comparing a control group response in patients who have the positive biomarker (Dx+) with patients who do not have the positive biomarker (Dx−). A predictive biomarker demonstrates the relationship between the biomarker and treatment effect; it differentiates between patients who are likely to benefit from a particular treatment (relative to those in a control) and patients who are not. Sometimes a predictive biomarker helps identify patients who will benefit the most from a treatment; however, it does not necessarily preclude patients without the biomarker from also receiving treatment benefits. This scenario often occurs in the context of new cancer immunotherapies and the use of PD-L1 testing or tumor mutational burden biomarker analyses. These concepts are further clarified in Figs. 11.1, 11.2, 11.3, and 11.4; note that depending upon the circumstance, biomarkers may be both prognostic and predictive.

Average patient response versus the value of a pretreatment personalized healthcare biomarker (PHC) in a single arm trial. While there appears to be a correlation between response and biomarker value, it is not possible to determine whether the biomarker is prognostic or predictive without a control group for comparison

Average patient response versus biomarker value in a randomized trial. The biomarker is prognostic, because the benefit is similar for all biomarker values irrespective of the treatment

Average patient response versus biomarker value. This example shows a biomarker that is both prognostic and predictive. Patients in the treatment group (red line) with higher biomarker values showed a better response. For the control group (blue line), the biomarker gives some prognostic information and shows that patients with lower biomarker values receive the greatest benefit of treatment

Further examples clarifying the difference between prognostic and predictive biomarkers
Considerations in Biomarker-Driven Trial Design
Because the analysis of biomarker studies can be complex and challenging, it is recommended that a knowledgeable statistician be consulted for high-level biostatistics guidance. When designing a trial, it is best if all hypotheses are prespecified and ranked to guide interpretation and that appropriate analytical strategies are used to minimize bias. Also, it is best if the experimental design does not have a large number of analytical covariates, because it can decrease confidence in the results. An additional general consideration in trial design is having respect for the patients participating in the study and understanding that their time is valuable. Thus, it is critical to develop the best clinical or translational trial possible. Obviously, one size design strategy does not fit all, but strategies based on the three pillars of time, cost, and risk still serve as a good guiding principle for biomarker-driven drug development.
There are some unique challenges in biomarker research that require special consideration in trial design. Sometimes there is a need to shorten the timeline from specimen collection to having the biomarker report available for clinical decisions, because patients are waiting for the information needed to improve (as well as lengthen) their lives. In this case, it is possible to use a clinical trial design that is adaptive—having the ability to change the design or hypotheses in an ongoing study based on early results from the same study or on biomarker data from other studies. For example, when phase II and III studies run in parallel, the data from phase II may be used to adjust the phase III study design while the study is still ongoing, thus shortening the time it takes to get results. The opportunities or triggers for adaptation are prespecified in the initial design to protect the integrity of the phase III study.
Tumor Sequencing for the Near Future
Looking to the future, one projection is that tumor sequencing will become standard clinical practice in the next few years, and the question is when should this tumor sequencing occur? Should it occur right after initial diagnosis? Should it occur after patients have received initial therapy and then develop refractory disease for us to identify potential biomarkers or to think about novel approaches?
It is also likely that liquid biopsy technology will be developed. Blood tests used to follow patients in the past are based on single proteins, such as CEA, CA 125, CA 19-9, but in the last few years there has been much interest in new technology to sample circulating cell-free tumor DNA (ctDNA). The ability to detect mutations in tumor DNA from a blood sample rather than from multiple tumor biopsies would be a huge improvement for patients. The isolation and subsequent analysis of ctDNA is viewed as a powerful tool with considerable potential to facilitate and improve clinical outcomes across multiple cancer types. This technology is promising, but still has limitations such as its inability to examine a large number of genes. Accurate blood sample collection, handling, and storage procedures are essential for reliable ctDNA extraction and molecular analyses. The conditions in which the blood samples are stored and shipped, as well as the amount of time that elapses between blood drawing and plasma extraction, are just some of the factors that may influence the accuracy of ctDNA analysis. Both clinical practitioners and researchers should be particularly attuned to these steps to maximize progress. In addition to screening for cancer, circulating tumor DNA technology can become part of the assessment of patients receiving cancer therapy. A positive finding of an actionable mutation in ctDNA (if using valid procedures and assays) could represent sufficient evidence to initiate targeted therapies. If the patient, for example, has had mutations detected in their tumor with next-generation gene sequencing, the physician can sample circulating tumor DNA during and after treatment to follow these mutations and other molecular changes over time—and do so without performing invasive tumor biopsies. The role of sensitivity, specificity, and concordance rates among various techniques will be important to elucidate in the next few years.
Cancer taxonomy is expected to become molecular-based; however, it is likely that tumor classification is going to be based on a combination of tumor location and molecular diagnosis. This field is evolving quickly, for example, there are therapies that have been approved for patients with melanoma whose tumors have the BRAF mutation. Many of these medicines have already been approved by the FDA including some combinations such as a BRAF inhibitor and a MEK inhibitor that work better than monotherapy. The future may be for an increasing tumor-agnostic biomarker strategy to manage patients. Two of these approvals have been recently granted by the FDA and other regulatory agencies, based on the analysis of somatic microsatellite instability (MSI) and the other NTRK fusion alterations. Basket trials and biomarker testing in various tumor types will help us gain more insights that will be relevant to the inclusion of new therapeutic strategies by the FDA, guideline, and pathway development groups. As an example of a recent development, the Southwest Oncology Group conducted a study in which they used a BRAF inhibitor (which had only been approved by the FDA for melanoma) in patients with refractory colon cancer whose tumors have the BRAF mutation. The impressive results of that trial led to an NCCN (National Comprehensive Cancer Network) designation, so that patients with colorectal cancer whose tumors have the BRAF mutation, may gain access to BRAF treatment, even though not yet approved by the FDA for the treatment of colorectal cancer. Another type of biomarker testing is the one of germline alterations, or alterations of homologous recombination, with therapies such as PARP inhibitors being either approved or under study. For clinicians and healthcare professionals, it is significant that this type of research is being done, because of its impact to improve options for patients.
Finally, clinical trials in oncology will increasingly use sequencing at enrollment and follow-up. This is already the standard of care for many current research studies. Earlier intervention and prevention strategies will facilitate adoption of gene and protein testing and will require better trial designs and statistical plans. As a consequence, people will have more access to these genomic technologies, regulatory approval will become adaptive, and early phase clinical trials will reflect a genomic approach .
FDA Approvals of Next-Generation Gene Sequencing (NGS) Panels and In Vitro Diagnostics (IVDs)
Many companies offer next-generation gene sequencing , but in addition, many healthcare facilities have developed their own panels. There are probably about 20,000 genes in our genome, but only a portion of those actually encode protein. In the context of cancer, the question is how many genes should be tested for clinical practice versus clinical research, because they have different purposes. For example, some hospitals have a 46-gene panel, 50-gene panel, and 200-gene panel, and it is not known which is best.
The FDA now considers not only safety but also efficacy as a way to regulate approvals, and the good news is that the FDA has given regulatory approval and clearance for some next-generation sequence, multigene marker tests. On June 23, 2017, the FDA approved the Oncomine Diagnostic Target Test, a 23-gene next-generation assay for patients with non-small cell lung cancer. On November 15, 2017, the FDA gave clearance (not approval) to the Memorial Sloan Kettering Cancer Center (MSKCC) for IMPACT—a tumor profiling test based on next-generation gene sequencing. To do this, the FDA developed a mechanism to approve diagnostic tests in addition to medicines. In this case, they accredited the New York State Department of Health (NYSDOH) as an FDA third party reviewer of in vitro diagnostic (IVD) tests. This resulted in the establishment of a Class II regulatory pathway for the review of other NGS-based tumor profiling tests, making these tests eligible for the 510(k) clearance process by applying either to the FDA directly or through an accredited third party reviewer like the NYSDOH. The cleared IMPACT test analyzes 468 genes and captures somatic mutations (e.g., point mutations, insertions, deletions) and microsatellite instability. This test, however, does not detect gene copy number alterations or rearrangements.
On November 30, 2017, the FDA and the Center for Medicare approved another multigene marker test, FoundationOne from Foundation Medicine (FMI). The approval was somewhat different from the other diagnostic tests, because it is a companion diagnostic (CDx) used to predict and inform therapy decisions. This is the first time there has been an FDA approval of a test that also considers what therapeutic drug can be used based on the patient’s test results. This test analyzes 324 genes and can be used for any tumor; it detects four genomic alterations (base pair substitutions, insertions and deletions, copy number alterations, and rearrangements), tumor mutational burden, and microsatellite instability. Other tumor sequencing platforms and the so-called liquid biopsies, such as the Guardant test, have more recently received regulatory approval.
A new type of test that is available for clinical testing is tumor mutational burden (TMB)—a genomic biomarker for cancer immunotherapy that measures the number of mutations in a tumor’s genome. There is research evidence that there is a correlation between the amount of tumor mutational burden and responsiveness to checkpoint inhibitors, a fairly new class of immunotherapies for patients with cancer. Thus, TMB may be an important predictive marker for patients who are being considered for this new type of immunotherapy. In addition to strengthening the correlation between TMB and benefit to checkpoint inhibitors, further research must answer two questions. One, who can do the most accurate and reliable test? And second, what should be the cutoff for tumor mutation burden and decision for patient treatment? Most recently, tests that allow for comprehensive genomic results from a blood draw have started gaining FDA approval, which could be instrumental to help move beyond the limitations of tissue biopsies to match patients to best personalized treatments .
Designing Clinical Trials to Support FDA Approval
Suggestions for designing biomarker trials that support FDA approval include design trials that make sense in the context of the United States; ask clinically meaningful questions; select and refine drug dose; think about biomarkers early and often; and include more patient-related outcomes and do them well. What is an ideal biomarker trial? It should inform about the in vitro diagnostic (IVD), the drug, and their interaction. To determine whether an IVD can be used to select a therapy, there must be knowledge about sensitivity determined by the fraction of responders that are marker-positive; specificity determined by the fraction of non-responders that are marker-negative; positive predictive value (PPV) based on the fraction of marker-positive patients who respond; negative predictive value (NPV) determined by the fraction of marker-negative patients who do not respond; and cutoff point for positivity.
A further consideration when designing biomarker trials is to include diverse ethnic patient participation, because it provides the most comprehensive ability to apply data to the general population. The issue is what is a sufficient number of diverse patients to adequately do the analysis? Finally, one additional caveat is that limiting a study to marker-positive patients may characterize the drug/diagnostic poorly. Sometimes researchers incorrectly assume that a biomarker is required for a response and may thus deny effective therapy to marker-negative patients.
Cancer Immunotherapy (CIT) Biomarkers
General Concepts
The use of cancer immunotherapy (CIT) biomarkers for a variety of malignancies is revolutionizing oncology. The number of drugs approved by the FDA is rapidly growing, added to unprecedented opportunities to better understand biology and offer better treatments for patients with various malignancies.
Many different types of cells and molecules are necessary for the immune system to work properly. These include T-cell lymphocytes such as CD4+ and CD8+ T cells, natural killer (NK) cells, dendritic cells, macrophages, and a myriad of regulatory cells and proteins that influence immune mechanisms. Because the immune response is so complex and each individual’s response unique, there are many challenges to develop effective CIT biomarkers. For example, researchers must first be able to answer some fundamental questions such as which cells are important for the immune system, how to test whether cells are functioning properly, and how to test whether they are activated or not. It is also critical to know specifically which of the many proteins (and their spatial localization) are important for elucidating the role of biomarkers as predictors for immunotherapy, as well as how to identify patients who are most likely to benefit from these cancer immunotherapies.
There are a variety of immunotherapy biomarkers currently being researched and used in cancer management. One is a measure of tumor infiltrating lymphocytes (TILs); in many tumors, there is a correlation between infiltration of T cells into the tumor (e.g., CD8+ T cells) and patient outcome. However, validation studies are still required before incorporation for therapeutic decisions. The biomarker most advanced to help predict the likelihood of benefit to checkpoint inhibition is the expression of an immune checkpoint protein, programmed death ligand (PD-L1); although it is not a binary marker (in other words, high expression predicts for greater likelihood of response), it does not mean that responses will not be seen in patients whose tumors do not express the protein. Combined expression patterns of multiple genes such as T-effector gene expression signatures as well as mutational load across targeted genes may be used to evaluate the predictability of clinical benefit to immune therapies. One promising note is that, in the past, mutational burden was measured from tumor biopsies, but now there is the potential to measure it from blood (liquid biopsy). A future goal is to identify neoantigens—tumor-specific antigens that may be detected and targeted by T cells (e.g., CD4+ and CD8+). Whole transcriptome sequencing can now be used to identify neoantigens and T-specific subsets in a given patient. One current avenue for research is to identify neoantigens that may help predict benefit to therapy and those that may be helpful to develop new vaccines. There are multiple companies worldwide that are developing vaccines for patients with cancer, and this neoantigen approach is one of the most promising (added to focused trials using protein-based vaccine approaches).
Program for Accelerated Cancer Therapies (PACT): An Example of Multidisciplinary Collaboration to Elucidate Relevant Biomarkers in Oncology
A new Program for Accelerated Cancer Therapies (PACT) has been initiated in collaboration with NIH, NCI, and biopharma. It emerged from the importance of working together to solve the problem of understanding biomarkers in the area of cancer immunotherapy. The overall goal is to provide a systematic approach to immune and related oncology biomarker investigation in clinical trials by supporting the development of standardized biomarkers and assays. This project will leverage NCI investments in its Cancer Immune Monitoring and Analysis Centers (CIMACs) and Cancer Immunologic Data Commons (CIDC) Network to select biomarkers for uniform clinical applications, to validate and standardize biomarker assays, to incorporate biomarkers as standards in clinical trials, and to create a comprehensive database, integrating biomarker, and clinical data to enable pre-competitive correlative biomarker analyses. Also, PACT will engage the FDA in its biomarker standardization and harmonization efforts in order to enhance regulatory decision making. This important project will help develop standardized biomarkers for immunoprofiling and exploratory biomarkers of high relevance to patient care.
Challenges and Opportunities to Improve Biomarker-Based Trials
There is growing pressure on many fronts to accelerate the pace at which new medicines are launched and made available for patients, but bringing biomarker-based trials to patients is challenging. A major hurdle is that investigators are reluctant to run extensive molecular profiling panels if only a small fraction of patients will be eligible to participate in their clinical trial. Thus, there is a need for a more efficient way to connect patients with genomic information to a clinical study, because high screen failure rates make trials more costly and time consuming. For example, if a marker is present perhaps in one out of 500 patients, it is really difficult for a clinician or institution to order 500 tests to find only one patient who might be enrolled in that particular clinical trial. This has slowed down the progress. Thinking about this scenario in more detail brings to light another issue—accountability to the patient. Increasing screen fail rate in clinical trials not only makes a large number of screenings necessary but also leaves many patients with no accountability. For example, imagine a patient with refractory ovarian cancer who wants to join a clinical trial testing a promising new drug for a particular alteration, and the patient is screened only to find that her tumor is negative for the marker. This is very tough on the patient who at this point wants to try everything that might help.
One way to address both of these issues is to link patient screening to a set of trials with an algorithm that assigns patients to the most relevant study. For example, consider a scenario with five trials A through E. If it is found on screening that a patient has a particular abnormality based on the biomarker A, the patient can enroll in trial A. If a patient does not have the biomarker for trial A, the patient can be considered for one of the other trials (B, C, D, or E) with one screening and patient consent. If screened patients do not have any of the abnormalities for the five trials, they can be enrolled in an all-comer trial. Thus, this theoretical initiative is a win-win scenario because patients will always have access to a clinical trial with new options, and cancer centers and institutions will have more efficient recruitment into their clinical trials. This type of solution requires both collaboration and the analysis of thousands of annotated biospecimens. There are many of these projects in the United States, such as PACT and the GENIE program sponsored by AACR that now have data on more than 100,000 patients. A collaborative strategy will catalyze biomarker precision medicine and precision oncology by linking clinical retrospective and prospective cancer genomic and proteomic data with longitudinal clinical outcomes. Data aggregation will benefit patients through the validation of biomarkers; drug repositioning or repurposing; addition of new mutations to existing drug labels; and identification of new targets and new biomarkers enabling the development of better therapies for patients .
In summary, cancer is composed of a broad spectrum of biologically distinct subtypes with overlapping or unique molecular alterations. We will need to increasingly expand biomarker development in various populations, incorporate biomarker testing, as well as consider clinical, ethnic, and socioeconomic factors to optimize patients’ lives. We are on this path .
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this chapter

Cite this chapter
Perez, E.A. (2020). Biomarkers and Precision Medicine in Oncology Practice and Clinical Trials. In: Ramirez, A., Trapido, E. (eds) Advancing the Science of Cancer in Latinos. Springer, Cham. https://doi.org/10.1007/978-3-030-29286-7_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-29286-7_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-29285-0
Online ISBN: 978-3-030-29286-7
eBook Packages: MedicineMedicine (R0)