
Abstract
Over the last decades, animal models have become increasingly important in therapeutic drug development and assessment. The use of these models, mainly mice and rats, allow evaluating drugs in the real-organism environment and context. However, several molecular therapeutic approaches are sequence-dependent, and therefore, the humanization of such models is required to assess the efficacy. The generation of genetically modified humanized mouse models is often an expensive and laborious process that may not always recapitulate the human molecular and/or physiological phenotype. In this chapter, we summarize basic aspects to consider before designing and generating humanized models, especially when they are aimed to test antisense-based therapies.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Keywords
- Humanized models
- Model systems
- Mouse model generation
- Splicing defects
- Antisense oligonucleotides
- In vivo drug testing
1 Introduction
Even though in 2020 the Food and Drug Administration (FDA) approved 53 new drugs (their second biggest approval number ever) [1], the mean annual number of drug approvals was 41 between 2010 and 2018 (approximately 15% of all evaluated drugs until 2019) [2]. It could be argued that this limited success is mainly caused by the insufficient recapitulation of the physiological and pathological of human disease in the preclinical phase. Studying human diseases is often difficult due to the restrictions and ethical concerns regarding the manipulation of human samples or tissues and the use of animal models allows the evaluation of drugs within an entire organism. However, these models are different species, with a different DNA sequence and different behavior, that may not recapitulate the complete human phenotype. Among all animal models, mice and rats are the most frequently used models in early preclinical stages. This is due to their relatively small size, ease of handling and maintenance, short reproductive cycle, and the similarities with regard to the genomic and physiological properties compared to humans, as well as the easy genetic manipulation that can be conducted, especially in mice [3].
Antisense oligonucleotide (AON)-based therapies are sequence-specific approaches aimed to either interfere with the splicing mechanism or regulate gene expression, protein translation, or RNA/protein binding [4]. All these processes are highly regulated in cells through different mechanisms such as epigenetics and interactions between mRNA and proteins, which can differ between species [5]. Unfortunately, if the sequence of interest is not completely conserved, the molecule designed to target the human sequence cannot be assessed in the animal model. In this case, the humanization of the models offers the possibility to (partially) replace mouse genes by human counterparts, insert a human copy of the gene in the mouse genome, as well as harbor human tissues [6, 7]. This allows to have the human sequence in an in vivo model. However, before generating such a model, it is crucial to first evaluate if the effect of the mutation of interest is also recapitulated in mouse [8]. In this chapter, we discuss the most important considerations to need to be taken into account before generating a genetically modified humanized mouse model to assess the therapeutic effect of AON molecules.
2 Human and Mouse : Species Are Similar But Not Equal
During the last decades, mouse models have served as valuable organisms for investigating human biology and disease as mouse and human are very similar to each other at the cellular as well as the biochemical level. Furthermore, most of the human cellular pathways are conserved in mouse , at the genetic and molecular level [5]. Despite these similarities, mice and humans can also differ in several aspects at multiple levels. For instance, it is described that around 1% of the human genes do not have an ortholog in mouse [9], which means that not all genes are present in both genomes. Furthermore, some data indicate that there is a higher variability of gene expression between the same tissue of two different species, compared to two different tissues within the same species [5]. These findings support the hypothesis that regulatory pathways, like epigenetics, can be responsible for the interspecies differences [5]. Within epigenetics, histone modifications , such as methylation and acetylation of the promoter region, play a crucial role in expression, leading to most of the differences between both species [10] (Fig. 1a). In addition, differences at the individual gene level give rise to different protein isoforms. Finally, the conservation rate of amino acids generates some differences in protein function or substrate recognition [9] (Fig. 1b). These differences can also cause different response to drug administration [11]. All these discrepancies can make that a defective gene or pathological mutation in humans may have a completely different outcome when the orthologous gene is mutated or truncated in mice [12,13,14] (Fig. 1c).

Summary of some aspects that can differ between human and mouse . (a) Epigenetics. Organs in which the gene of interest is expressed are represented in color while those organs in which the gene is not expressed because of epigenetics regulation (such as methylation, me3) are in gray. As the pattern of gene expression between both organisms (inside the blue circle) is different, the functional role of the gene in both may also be different. (b) Amino acid conservation rate. As an example, part of the human and mouse Histone 1 amino acid sequences is represented. Discrepancies between amino acids are indicated with different symbols (: or .) while conserved regions are marked with an asterisk (*). (c) Mutation effect. Not always the same mutation has the same effect on both species. In humans, the mutation can underlay a pathological defect (left) that is not recapitulated in mice (right). (d) Splicing. The recapitulation of the normal splicing after humanization is indicated on the left. Mouse regions are indicated in purple and human regions in yellow. On the right, the presence of the splicing-related mutation of interest, located in an intron sequence, causes the insertion of a pseudoexon (PS) in the mRNA transcript . In this situation the splicing defect is recapitulated after the humanization
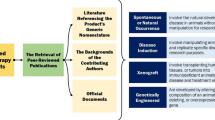
AONs are often used to modulate splicing and correct aberrant splicing. It has been reported that the consensus splice site sequences in mouse and human are highly conserved and comparable even though some small changes in these patterns have been described [15]. That could mean that a human splicing defect may not be completely reproduced in mouse . Some differences between both species at the splicing level have been already described (Fig. 1d). One example is the variant c.315-48T>C in the FECH gene, a modifier mutation for erythropoietic protoporphyria [14]. This mutation generates an aberrant splice acceptor site that results in an intron inclusion of 63 nucleotides upstream of exon 4. However, the generated mouse model not only presents this mis-splicing effect but also a strong skipping of the partially humanized exon 3 [14]. This example illustrates how a hybrid gene (or humanized mouse gene) can have undesirable effects on splicing. Another example is the low recognition of the pseudoexon insertion caused by a deep-intronic mutation in the CEP290 gene (c.2991+1655A>G) in the humanized mouse model carrying this specific variant [13]. Further studies showed that the recognition of the pseudoexon seemed to be specific of primates, and it was less efficient in other species such as pig, dog, mouse , or Drosophila. In addition, as shown in FECH, additional splicing events not detected in humans were found in the humanized Cep290 model [16]. Based on all these studies, we recommend performing several in vitro validation steps before generating a humanized mouse model (Fig. 2).

Schematic representation of the different checkpoints for generating a humanized mouse model
3 Checkpoints Before Generating a Humanized Mouse Model
3.1 Literature Research
An important aspect is to look first in literature or in mouse databases (such as the Mouse Genome Informatics (MGI, http://www.informatics.jax.org/) and the International Mouse Phenotyping Consortium (IMPC, https://www.mousephenotype.org/)) whether any genetically modified animal model for the gene of interest has already been obtained. With this information, one can avoid the generation of a model already available. In addition, if a humanized model is aimed to study disease, the availability of a knockout/mutant model will contribute to predict the possible phenotype of the potential new humanized model. For instance, mutations in ABCA4 generate a non-functional protein involved in the visual cycle that leads to retinal degeneration and subsequent loss of vision, causing Stargardt disease in humans [17,18,19]. However, the Abca4−/− mouse model shows a mild late onset phenotype, despite the fact that no ABCA4 protein is encoded [20]. That means that the defect caused by the absence of this protein is not completely comparable to the human phenotype. Thus, depending on the purpose of study, a humanized mouse model may not be suitable to evaluate antisense therapies for a particular gene.
3.2 Comparison of the Gene Sequence
If the model is not available, the first question that needs to be answered is whether the gene of interest is present in the mouse genome. The probability is high, since only 1% of human genes do not have a mouse orthologue [9]. In addition, it is important to check the structure of the gene, to ensure it is similar and no relevant exons for the study or protein function are missing. To do so, databases such as Genome Browser (https://genome.ucsc.edu) or Ensembl (https://www.ensembl.org) can be useful. If the gene name does not coincide, a blast (https://blast.ncbi.nlm.nih.gov) at DNA or protein level can be done to search for similar genes or proteins in other species.
3.3 Assessment of Gene Expression in Mouse
Although mouse and human are similar at cellular and molecular levels, some discrepancies between both species have been already reported, including variation in expression levels in different tissues. Therefore, assessing that the gene of interest is expressed in the proper murine tissue (or at least the tissue of study) is crucial. For instance, it is already known that one of the most important pathways in cancer and apoptosis, the phosphoinositide 3-kinase (PI3K) signaling cascade [21], shows high differences in the expression of its two key genes, mTOR and AKT2, between both species [22]. Thus, mouse is not the best model to conduct oncological studies that aim to investigate this pathway in vivo. In addition, it has been published that muscle , liver, and neuronal cells show a strong similarity of gene expression profiles between both species while other tissues such as testis, lung, and pancreas showed more differences due to the evolution [22]. Therefore, it is important to check expression levels in the tissue(s) of interest in mouse , to ensure that the generation of the humanized model will be useful for future experiments.
3.4 Humanization Feasibility
In general, there are two main ways to introduce the human gene into the mouse genome:
-
Introduce the human gene of interest (or part of it) in the mouse genome.
-
Replace the corresponding mouse gene (or part of it) by the human one.
In the first option, the complexity lays on where to exactly insert the gene and how to regulate its expression. The second option requires a more in-depth study on the recombination possibilities, the conservation rate between human and mouse , and the resulting humanized gene and protein. For that, first it is important to identify the mouse exons that correspond to the human ones. It is also important to check whether they encode for the same part of the protein and how conserved it is. This can be done with the previously mentioned freely available databases. Finally, the overall procedure needs to be technically feasible, by either homologous recombination in embryonic stem cells or genome editing techniques using a donor template. These steps need to be discussed with experts on these techniques either at other academic institutes or companies specialized in generating animal models.
3.5 In Vitro Validation
As indicated previously, AONs can be used for splicing modulation. Thus, it is important to study whether the splicing machinery in mouse will allow proper:
-
Splicing of the humanized transcript or human gene.
-
Recapitulation of the human splicing defect caused by a specific mutation in the mouse molecular background.
One way to perform this validation is by using artificial systems (e.g., mini-/midigenes) harboring the human region of interest either with the “wild-type” sequence or the mutated one [16]. In that way, it can be assessed whether they behave in a mouse-specific environment as expected based on observations in humans. For that, conventional mouse-derived cell lines can be a good start [23]. The data obtained from these studies are crucial to assess if the mouse might be a good animal for the generation of a humanized model.
3.5.1 In Vitro Splicing Assays Considerations
The easiest way for a first screening is to develop human mini-/midigenes with and without the mutation [24] and deliver them to conventional mouse cell lines easy to transfect, such as iMCD3, B16-F10 or N2A. However, depending on the tissue of interest, this information might not be completely accurate. For instance, in human cells, we and others have found differences in pseudoexon recognition, depending on how similar the molecular background was compared to the one of the cell/tissue of interest [24,25,26], probably due to tissue-specific splicing regulatory elements [27]. Therefore, it is important to choose an appropriate cell line. For instance, if the gene is implicated in neurological disorders affecting the brain, the mouse cell line N2A (derived from mouse brain) might be a better option than using B16-F10, derived from mouse skin or iMCD3, derived from kidney [28]. It is always good to test in several different cell lines: if the splicing pattern observed in humans is well recapitulated in multiple cell lines, most probably the humanized model will also show the same splicing patterns. In case the splicing pattern is not recapitulated at all, we recommend to stop with the generation of the humanized mouse model and try to find alternative models.
Next step, if the splicing defect is recapitulated, is to determine whether the AONs of interest previously designed and tested in vitro human models can also rescue the splicing in the murine molecular background. With that, it is possible to evaluate if those molecules will also be successful in the mouse molecular background. The generation and validation of mini-/midigenes and AON rescue can be done in 3–4 weeks as previously reported [24, 29] and can contribute significantly to reduce the number of animals by performing initial screenings in vitro [24].
If all the aforementioned points are met successfully (Fig. 2), the chances of success are higher. However, it can still happen that even though the molecular defect is recapitulated, the model does not show a similar phenotype. Unfortunately, this is difficult to predict in vitro. In addition, in order to discern whether the humanization of the model may influence the potential phenotype, especially in those cases where a mutation has been introduced, it is strongly advisable to generate two humanized models, one with only the human “wild-type” region and another one with the mutated sequence.
4 Current Examples of Humanized Mouse Models
Different approaches have been used to generate humanized mouse models, ranging from introducing the entire gene or part of it or replacing specific parts the gene by the human counterpart. Some of these models have also been used to test AON-based therapies for different types of disease.
4.1 Insertion of the Entire Human Gene
One of the approaches frequently used is the introduction of the entire human gene into the mouse genome. This method also takes into account the endogenous expression of the corresponding mouse gene and whether it can affect the development of the desired phenotype. One example of this is the humanized Tg32 FcRn mouse , which harbors the complete human FCGRT gene (around 11 kb) plus 5′ and 3′ untranslated regions (around 10 kb). This transgene is introduced into C57BL/6J oocytes with a null mutation in the FcRn mouse gene, which is involved in IgG and serum albumin turnover. As a result, only the human FcRn is expressed under the control of its natural human promoter. Besides introducing the “wild-type” sequence in the mouse genome, Anderson and colleagues also generated a defective allele [30]. In this, a neomycin-resistance cassette was inserted, replacing the human “wild-type” located between the promoter end and the beginning on exon 2. This study revealed that the FcRn+/− model recapitulated the “wild-type” condition indicating that only one allele is sufficient to regulate the IgG levels and transport [31,32,33]. However, the defective mouse model (FcRn−/−) successfully recapitulated the defective neonatal transport phenotype [30]. This model has already been used in drug preclinical studies [34]. Most of these studies focused on the evaluation and the pharmacokinetics and pharmacodynamics of human IgG [35], but it may also be useful for other therapeutic approaches, such as AONs [36].
Retinitis pigmentosa (RP) is the most frequent inherited retinal disease, for which no therapy is currently available. In this disorder, most of the studies have been focused on the mutation p.P23H of rhodopsin (RHO), a recurrent mutation causing autosomal dominant RP [37, 38]. To study this mutation , several animal models have been developed, including the generation of humanized models in either mouse [39] or rats [37, 40]. In all of them, the transgene segregated in an autosomal dominant allele manner and the lines were maintained within the murine wild-type rhodopsin background (Rho+/+). However, they differed in the expression levels of the murine and human rhodopsin mRNA . These studies revealed that a higher number of copies of the mutant allele induces a more severe RP phenotype, but also that overexpression of the wild-type allele has a detrimental effect, causing similar RP abnormalities [39, 41,42,43]. Some of these models have been used to successfully test AON-based therapies aiming to degrade the mutated transcript [44]. These and other results led to a recently started clinical trial (NCT04123626, http://www.clinicaltrials.gov).
AON-based therapies have had a major development in neuromuscular disorders [45]. Several mouse models recapitulating muscular dystrophy phenotypic traits have been generated. For instance, the mdx model, which harbors a nonsense mutation in exon 23 of the mouse Dmd gene causing loss of the functional protein expression, presents a moderate-severe phenotype with an early onset of skeletal muscle degeneration and impairment in muscle functions [46]. However, this model can only be targeted with mouse-specific AONs to the mutation in exon 23 [47,48,49]. To solve this limitation, the hDMD/mdx mouse model was generated by crossing the mdx model with a model carrying the entire human DMD gene (hDMD), allowing the expression of the “wild-type” human dystrophin in the mdx model [50]. However, the expression of hDMD hampered the development of the dystrophic phenotype caused by the mutation present in the mouse Dmd gene. In 2018, the group of Prof. Aartsma-Rus overcame these limitations by generating a new humanized mouse model (del52hDMD/mdx mice). This model carries both human and murine DMD genes with pathogenic mutations previously described. As a consequence of the nonsense mutation in exon 23 in the mouse Dmd gene and the deletion of exon 52 in the hDMD gene, none of the genes produce functional dystrophin protein [51]. The absence of dystrophin expression caused a more severe muscular dystrophy phenotype than the one observed in other models. As a result, this model was suitable to test human-specific AONs, and thereby improve AON-based therapies for Duchene muscular dystrophy patients by enabling studies at the mRNA , protein and functional level.
4.2 Replacing Part of the Mouse Gene by the Human Gene
Another way to generate a humanized mouse model is by replacing a specific region in the mouse genome by the human counterpart. One example is the humanized mouse model generated to study the mutation c.2991+1655A>G in the CEP290 gene. This mutation is the most recurrent mutation underlying Leber congenital amaurosis, accounting up to 15% of all cases in some populations [25, 52]. Two mouse models were generated in which the mouse sequence from exon 25 to exon 26 was replaced by the corresponding human exon 26, intron 26 (with or without harboring the mutation of interest), and exon 27 [13]. In the model Cep290lca/lca, human intron 26 harbors the mutation of interest while the model Cep290hum/hum does not. In both the models, Cep290 expression levels and regular splicing of the gene were maintained when compared to “wild-type” mice. However, the splicing defect was barely present in the Cep290lca/lca model, the retina being the only tissue where the exon was recognized at detectable levels, but by far not enough to lead to the human phenotype [25]. Subsequent studies, revealed that the pseudoexon recognition strongly correlates with evolutionary distance, being highly recognized in primates, and hardly recognized in lower species such as rodents and fly [16].
5 Conclusions
In summary, the suggestions given in this chapter highlight the importance of performing an accurate study to increase the chances of success when generating a humanized mouse model, e.g., to assess AON-based molecules. Despite similarities, human and mouse are different species and therefore differences are expected at all levels. When aiming to generate a humanized model, it is crucial to determine first if the gene of interest is present in the genome, has a similar structure, and if it is expressed in the same tissue(s) as in humans. Subsequently, when working with splicing defects, it is important to validate that these will be recognized in the murine molecular background, which can be easily done using “artificial systems” in vitro. If everything is conserved, the technical part is also important, is it feasible to humanize the model? If so, the chances of success are high. However, in some cases, it is not feasible, or a mouse model is not an option. In those cases, other models can be explored such as zebrafish or cellular models based on organ-on-a-chip technology. Following these suggestions may increase the success of the generation of humanized models and reduce the number of animals used in research. However, although all these in silico and in vitro tests can help to predict the recapitulation of a molecular defect in a humanized model, it is important to keep in mind that such models may not recapitulate the entire human phenotype.
References
Mullard A (2021) 2020 FDA drug approvals.Nat Rev Drug Discov. 2021 Feb;20(2):85–90. https://doi.org/10.1038/d41573-021-00002-0
Darrow JJ, Avorn J, Kesselheim AS (2020) FDA approval and regulation of pharmaceuticals, 1983-2018. JAMA 323(2):164–176. https://doi.org/10.1001/jama.2019.20288
Walsh NC, Kenney LL, Jangalwe S et al (2017) Humanized mouse models of clinical disease. Annu Rev Pathol 12:187–215. https://doi.org/10.1146/annurev-pathol-052016-100332
Sardone V, Zhou H, Muntoni F et al (2017) Antisense oligonucleotide-based therapy for neuromuscular disease. Molecules 22(4):563. https://doi.org/10.3390/molecules22040563
Lin S, Lin Y, Nery JR et al (2014) Comparison of the transcriptional landscapes between human and mouse tissues. Proc Natl Acad Sci U S A 111(48):17224–17229. https://doi.org/10.1073/pnas.1413624111
O’Connell AK, Douam F (2020) Humanized mice for live-attenuated vaccine research: from unmet potential to new promises. Vaccines (Basel) 8(1):36. https://doi.org/10.3390/vaccines8010036
Ito R, Takahashi T, Ito M (2018) Humanized mouse models: application to human diseases. J Cell Physiol 233(5):3723–3728. https://doi.org/10.1002/jcp.26045
Hammond SM, Aartsma-Rus A, Alves S et al (2021) Delivery of oligonucleotide-based therapeutics: challenges and opportunities. EMBO Mol Med 13(4):e13243. https://doi.org/10.15252/emmm.202013243
Mouse Genome Sequencing C, Waterston RH, Lindblad-Toh K et al (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420(6915):520–562. https://doi.org/10.1038/nature01262
Dong X, Greven MC, Kundaje A et al (2012) Modeling gene expression using chromatin features in various cellular contexts. Genome Biol 13(9):R53. https://doi.org/10.1186/gb-2012-13-9-r53
Blevitt JM, Hack MD, Herman K et al (2016) A single amino acid difference between mouse and human 5-lipoxygenase activating protein (FLAP) explains the speciation and differential pharmacology of novel FLAP inhibitors. J Biol Chem 291(24):12724–12731. https://doi.org/10.1074/jbc.M116.725325
Garanto A, Vicente-Tejedor J, Riera M et al (2012) Targeted knockdown of Cerkl, a retinal dystrophy gene, causes mild affectation of the retinal ganglion cell layer. Biochim Biophys Acta 8:1258–1269. https://doi.org/10.1016/j.bbadis.2012.04.004
Garanto A, van Beersum SE, Peters TA et al (2013) Unexpected CEP290 mRNA splicing in a humanized knock-in mouse model for Leber congenital amaurosis. PLoS One 8(11):e79369. https://doi.org/10.1371/journal.pone.0079369
Barman-Aksozen J, Wiek PC, Bansode VB et al (2017) Modeling the ferrochelatase c.315-48C modifier mutation for erythropoietic protoporphyria (EPP) in mice. Dis Model Mech 10(3):225–233. https://doi.org/10.1242/dmm.027755
Abril JF, Castelo R, Guigo R (2005) Comparison of splice sites in mammals and chicken. Genome Res 15(1):111–119. https://doi.org/10.1101/gr.3108805
Garanto A, Duijkers L, Collin RW (2015) Species-dependent splice recognition of a cryptic exon resulting from a recurrent intronic CEP290 mutation that causes congenital blindness. Int J Mol Sci 16(3):5285–5298. https://doi.org/10.3390/ijms16035285
Molday LL, Rabin AR, Molday RS (2000) ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet 25(3):257–258. https://doi.org/10.1038/77004
Sun H, Smallwood PM, Nathans J (2000) Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genet 26(2):242–246. https://doi.org/10.1038/79994
Charbel Issa P, Barnard AR, Herrmann P et al (2015) Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin a dimerization. Proc Natl Acad Sci U S A 112(27):8415–8420. https://doi.org/10.1073/pnas.1506960112
Weng J, Mata NL, Azarian SM et al (1999) Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 98(1):13–23. https://doi.org/10.1016/S0092-8674(00)80602-9
Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27(41):5497–5510. https://doi.org/10.1038/onc.2008.245
Monaco G, van Dam S, Casal Novo Ribeiro JL et al (2015) A comparison of human and mouse gene co-expression networks reveals conservation and divergence at the tissue, pathway and disease levels. BMC Evol Biol 15:259. https://doi.org/10.1186/s12862-015-0534-7
Tsai KW, Tarn WY, Lin WC (2007) Wobble splicing reveals the role of the branch point sequence-to-NAGNAG region in 3′ tandem splice site selection. Mol Cell Biol 27(16):5835–5848. https://doi.org/10.1128/MCB.00363-07
Garanto A, Collin RWJ (2018) Design and in vitro use of antisense oligonucleotides to correct pre-mRNA splicing defects in inherited retinal dystrophies. Methods Mol Biol 1715:61–78. https://doi.org/10.1007/978-1-4939-7522-8_5
Garanto A, Chung DC, Duijkers L et al (2016) In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum Mol Genet 25(12):2552–2563. https://doi.org/10.1093/hmg/ddw118
Parfitt DA, Cheetham ME (2016) Targeting the proteostasis network in rhodopsin retinitis pigmentosa. Adv Exp Med Biol 854:479–484. https://doi.org/10.1007/978-3-319-17121-0_64
Lopes-Ramos CM, Paulson JN, Chen CY et al (2017) Regulatory network changes between cell lines and their tissues of origin. BMC Genomics 18(1):723. https://doi.org/10.1186/s12864-017-4111-x
Jiang Z, Cote J, Kwon JM et al (2000) Aberrant splicing of tau pre-mRNA caused by intronic mutations associated with the inherited dementia frontotemporal dementia with parkinsonism linked to chromosome 17. Mol Cell Biol 20(11):4036–4048. https://doi.org/10.1128/mcb.20.11.4036-4048.2000
Sangermano R, Khan M, Cornelis SS et al (2018) ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res 28(1):100–110. https://doi.org/10.1101/gr.226621.117
Roopenian DC, Christianson GJ, Sproule TJ et al (2003) The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol 170(7):3528–3533. https://doi.org/10.4049/jimmunol.170.7.3528
Petkova SB, Akilesh S, Sproule TJ et al (2006) Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol 18(12):1759–1769. https://doi.org/10.1093/intimm/dxl110
Roopenian DC, Akilesh S (2007) FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 7(9):715–725. https://doi.org/10.1038/nri2155
Zalevsky J, Chamberlain AK, Horton HM et al (2010) Enhanced antibody half-life improves in vivo activity. Nat Biotechnol 28(2):157–159. https://doi.org/10.1038/nbt.1601
Stein C, Kling L, Proetzel G et al (2012) Clinical chemistry of human FcRn transgenic mice. Mamm Genome 23(3–4):259–269. https://doi.org/10.1007/s00335-011-9379-6
Ward ES, Ober RJ (2009) Chapter 4: multitasking by exploitation of intracellular transport functions the many faces of FcRn. Adv Immunol 103:77–115. https://doi.org/10.1016/S0065-2776(09)03004-1
Chappell AE, Gaus HJ, Berdeja A et al (2020) Mechanisms of palmitic acid-conjugated antisense oligonucleotide distribution in mice. Nucleic Acids Res 48:4382. https://doi.org/10.1093/nar/gkaa164
LaVail MM, Nishikawa S, Steinberg RH et al (2018) Phenotypic characterization of P23H and S334ter rhodopsin transgenic rat models of inherited retinal degeneration. Exp Eye Res 167:56–90. https://doi.org/10.1016/j.exer.2017.10.023
Gorbatyuk MS, Knox T, LaVail MM et al (2010) Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A 107(13):5961–5966. https://doi.org/10.1073/pnas.0911991107
Olsson JE, Gordon JW, Pawlyk BS et al (1992) Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron 9(5):815–830. https://doi.org/10.1016/0896-6273(92)90236-7
Lewin AS, Drenser KA, Hauswirth WW et al (1998) Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat Med 4(8):967–971. https://doi.org/10.1038/nm0898-967
Mao H, James T Jr, Schwein A et al (2011) AAV delivery of wild-type rhodopsin preserves retinal function in a mouse model of autosomal dominant retinitis pigmentosa. Hum Gene Ther 22(5):567–575. https://doi.org/10.1089/hum.2010.140
Frederick JM, Krasnoperova NV, Hoffmann K et al (2001) Mutant rhodopsin transgene expression on a null background. Invest Ophthalmol Vis Sci 42(3):826–833
Orhan E, Dalkara D, Neuille M et al (2015) Genotypic and phenotypic characterization of P23H line 1 rat model. PLoS One 10(5):e0127319. https://doi.org/10.1371/journal.pone.0127319
Murray SF, Jazayeri A, Matthes MT et al (2015) Allele-specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Invest Ophthalmol Vis Sci 56(11):6362–6375. https://doi.org/10.1167/iovs.15-16400
Wurster CD, Ludolph AC (2018) Antisense oligonucleotides in neurological disorders. Ther Adv Neurol Disord 11:1756286418776932. https://doi.org/10.1177/1756286418776932
Wu B, Wang M, Shah S et al (2018) In vivo evaluation of dystrophin exon skipping in mdx mice. Methods Mol Biol 1828:231–247. https://doi.org/10.1007/978-1-4939-8651-4_14
Goyenvalle A, Vulin A, Fougerousse F et al (2004) Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science 306(5702):1796–1799. https://doi.org/10.1126/science.1104297
Fletcher S, Honeyman K, Fall AM et al (2007) Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol Ther 15(9):1587–1592. https://doi.org/10.1038/sj.mt.6300245
Wu B, Lu P, Cloer C et al (2012) Long-term rescue of dystrophin expression and improvement in muscle pathology and function in dystrophic mdx mice by peptide-conjugated morpholino. Am J Pathol 181(2):392–400. https://doi.org/10.1016/j.ajpath.2012.04.006
t Hoen PA, de Meijer EJ, Boer JM et al (2008) Generation and characterization of transgenic mice with the full-length human DMD gene. J Biol Chem 283(9):5899–5907. https://doi.org/10.1074/jbc.M709410200
Veltrop M, van Vliet L, Hulsker M et al (2018) A dystrophic Duchenne mouse model for testing human antisense oligonucleotides. PLoS One 13(2):e0193289. https://doi.org/10.1371/journal.pone.0193289
Coppieters F, Lefever S, Leroy BP et al (2010) CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat 31(10):1097–1108. https://doi.org/10.1002/humu.21337
Acknowledgments
We thank Dr. Rob W.J. Collin for the critical reading of this chapter. The group is financially supported by the Foundation Fighting blindness (PPA-0517-0717-RAD to A.G.), the Curing Retinal Blindness Foundation (to A.G.) as well as the Landelijke Stichting voor Blinden en Slechtzienden, Stichting Oogfonds Nederland (who contributed through UitZicht 2019-17), together with the Rotterdamse Stichting Blindenbelangen, Stichting Blindenhulp, and Stichting Dowilvo (to A.G). The funding organizations had no role in the design or conduct of this research. They provided unrestricted grants. All the figures were made with BioRender.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Vázquez-Domínguez, I., Garanto, A. (2022). Considerations for Generating Humanized Mouse Models to Test Efficacy of Antisense Oligonucleotides. In: Arechavala-Gomeza, V., Garanto, A. (eds) Antisense RNA Design, Delivery, and Analysis. Methods in Molecular Biology, vol 2434. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2010-6_18
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2010-6_18
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2009-0
Online ISBN: 978-1-0716-2010-6
eBook Packages: Springer Protocols