
Abstract
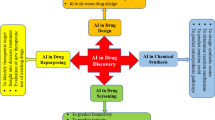
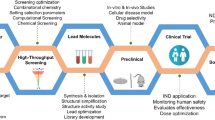
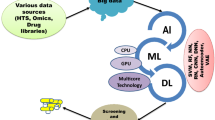
Machine Learning (ML) and Deep Learning (DL) are two subclasses of Artificial Intelligence (AI), that, in this day and age of big data provides significant opportunities to pharmaceutical discovery research and development by translating data to information and ultimately to knowledge. Machine Learning or AI is not really new but over last few years, application of better methods have emerged and they have been successfully applied for drug discovery and development. This chapter would provide an overview of these methods and how they have been applied across various work streams, e.g., generative chemistry, ADMET prediction, retrosynthetic analysis, etc. within drug discovery process. This chapter would also attempt to provide caution and pit falls in utilizing these methods blindly while summarizing challenges and limitations.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others
References
Ramesh A (2004) Artificial intelligence in medicine. Ann R Coll Surg Engl 86:334–338
Mamoshina P et al (2018) Machine learning on human muscle transcriptomic data for biomarker discovery and tissue-specific drug target identification. Front Genet 9:242
Yang Y, Siau KL (2018) A Qualitative Research on Marketing and Sales in the Artificial Intelligence Age. MWAIS 2018 Proceedings. 41
Wirtz BW (2019) Artificial intelligence and the public sector—applications and challenges. Int J Public Adm 42:596–615
Gartner’s 2018 Hype cycle for emerging technologies identifies three key trends that organizations must track to gain competitive advantage. http://www.gartner.com/newsroom/id/3412017. Available 10 April 2017
Smith RG, Farquhar A (2000) The road ahead for knowledge management: an AI perspective. AI Mag 21:17
Lamberti MJ (2019) A study on the application and use of artificial intelligence to support drug development. Clin Ther 41:1414–1426
SAS Institute. Machine learning: what it is and why it matters. Accessed 13 Feb 2020
Wilkinson M, Dumontier M, Aalbersberg I et al (2016) The FAIR guiding principles for scientific data management and stewardship. Sci Data 3:160018
Schneider P, Walters WP, Plowright AT et al (2020) Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov 19:353–364
Raymond JL, Medina JF (2018) Computational principles of supervised learning in the cerebellum. Annu Rev Neurosci 41:233–253
Duda RO, Hart PE, Stork DG (2001) Unsupervised learning and clustering. Pattern classification, 2nd edn. Wiley, New York
Breiman L (2001) Random forests. Mach Learn 45:5–32
Joachims T (1998) Text categorization with support vector machines: learning with many relevant features. In: European conference on machine learning. Springer Verlag, Heidelberg, pp 137–142
Lewis DD (1998) Naive (Bayes) at forty: the independence assumption in information retrieval. In: European conference on machine learning. Springer Verlag, Heidelberg, pp 4–15
Kohonen T (1990) Self-organizing map. Proc IEEE 78:1464–1480
Hartigan JA, Wong MA (1979) Algorithm AS 136: a k-means clustering algorithm. J R Stat Soc C-Appl 28:100–108
Johnson SC (1967) Hierarchical clustering schemes. Psychometrika 32:241–254
Brownlee J. Deep learning with time series forecasting, machine learning mastery. https://machinelearningmastery.com/machine-learning-with-python/. Accessed 1 Mar 2018
https://practicalcheminformatics.blogspot.com/2021/01/ai-in-drug-discovery-2020-highly.html
Yang X, Wang Y, Byrne R, Schneider G, Yang S (2019) Concepts of artificial intelligence for computer-assisted drug discovery. Chem Rev 119:10520–10594
Vamathevan J, Clark D, Czodrowski P et al (2019) Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 18:463–477
Patel L, Shukla T, Huang X, Ussery DW, Wang S (2020) Machine learning methods in drug discovery. Molecules 25:5277
Bajorath J et al (2020) Artificial intelligence in drug discovery: into the great wide open. J Med Chem 63:8651–8652. Special issue of JCIM
Vo A, Van Vleet T, Gupta R, Liguori M, Rao M (2020) An overview of machine learning and big data for drug toxicity evaluation. Chem Res Toxicol 33:20–37
Gómez-Bombarelli R, Wei JN, Duvenaud D, Hernández-Lobato JM, Sánchez-Lengeling B, Sheberla D, Aguilera-Iparraguirre J, Hirzel TD, Adams RP, Aspuru-Guzik A (2018) Automatic chemical design using a data-driven continuous representation of molecules. ACS Central Sci 4:268–276
Schneider G, Fechner U (2005) Computer-based de novo design of drug-like molecules. Nat Rev Drug Discov 4:649–663
Plowright AT, Johnstone C, Kihlberg J, Pettersson J, Robb G, Thompson RA (2012) Hypothesis driven drug design: improving quality and effectiveness of the design-make-test-analyse cycle. Drug Discov Today 17(1–2):56–62
Mansbach RA, Leus IV, Mehla J, Lopez CA, Walker JK, Rybenkov VV, Hengartner NW, Zgurskaya HI, Gnanakaran S (2019) Development of a fragment-based machine learning algorithm for designing hybrid drugs optimized for permeating Gram-negative bacteria. arXiv:1907.13459 [q-bio.QM]
Reymond JL, Van Deursen R, Blum LC, Ruddigkeit L (2010) Chemical space as a source for new drugs. MedChemComm 1:30–38
Prykhodko O, Johansson SV, Kotsias PC et al (2019) A de novo molecular generation method using latent vector based generative adversarial network. J Cheminform 11:74
Méndez-Lucio O, Baillif B, Clevert DA et al (2020) De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat Commun 11:10
Wan F, Zeng J (2016) Deep learning with feature embedding for compound–protein interaction prediction. bioRxiv
Le Guilloux V, Schmidtke P, Tuffery P (2009) Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics 10:168
Martin EJ, Polyakov VR, Zhu X-W, Tian L, Mukherjee P, Liu X (2019) All-assay-Max2 pQSAR: activity predictions as accurate as four-concentration IC50s for 8558 novartis assays. J Chem Informat Model 59(10):4450–4459
Wallach I, Dzamba M, Heifets A (2015) AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. ArXiv e-prints
Molinski SV, Shahani VM, MacKinnon SS, Morayniss LD, Laforet M, Woollard G, Kurji N, Sanchez CG, Wodak SJ, Windemuth A (2017) Computational proteome-wide screening predicts neurotoxic drug-protein interactome for the investigational analgesic BIA 10-2474. Biochem Biophys Res Commun. Jan 29;483(1):502–508
Callaway E (2020) It will change everything: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 588:203–204
Shah P, Kendall F, Khozin S et al (2019) Artificial intelligence and machine learning in clinical development: a translational perspective. NPJ Digit Med 2:69
Gupta RR, Gifford EM, Liston T, Waller CL, Hohman M, Bunin BA, Ekins S (2010) Using open source computational tools for predicting human metabolic stability and additional absorption, distribution, metabolism, excretion, and toxicity properties. Drug Metab Dispos 38(11):2083–2090
Morgan P, Van Der Graaf PH, Arrowsmith J, Feltner DE, Drummond KS, Wegner CD, Street SD (2012) Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving phase II survival. Drug Discov Today 17:419–424
Wang Y, Xing J, Xu Y, Zhou N, Peng J, Xiong Z, Liu X, Luo X, Luo C, Chen K (2015) In silico ADME/T modelling for rational drug design. Q Rev Biophys 48:488–515
Lombardo F, Desai PV, Arimoto R, Desino KE, Fischer H, Keefer CE, Petersson C, Winiwarter S, Broccatelli F (2017) In silico absorption, distribution, metabolism, excretion, and pharmacokinetics (ADME-PK): utility and best practices. An industry perspective from the international consortium for innovation through quality in pharmaceutical development. J Med Chem 60:9097–9113
Keefer CE, Kauffman GW, Gupta RR (2013) Interpretable, probability-based confidence metric for continuous quantitative structure-activity relationship models. J Chem Inf Model 53:368–383
Segall M (2014) Advances in multiparameter optimization methods for de novo drug design. Expert Opin Drug Discovery 9:803–817
Debe DA, Mamidipaka RB, Gregg RJ, Metz JT, Gupta RR, Muchmore SW (2013) ALOHA: a novel probability fusion approach for scoring multi-parameter drug-likeness during the lead optimization stage of drug discovery. J Comput Aided Mol Des 27(9):771–782
Gupta RR et al. (2015) AIDEAS: An Integrated Cheminformatics Solution. BioIt World Abstract and Presentation, p 25. https://www.bioitworldexpo.com/uploadedFiles/Bio-IT_World_Expo/Agenda/15/BIT-2015-Agenda.pdf
Molga K, Szymkuć S, Grzybowski BA (2021) Chemist ex machina: advanced synthesis planning by computers. Acc Chem Res. 54(5):1094–1106
Segler M, Preuss M, Waller M (2018) Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555:604–610
Ahneman DT, Estrada JG, Lin S, Dreher SD, Doyle AG (2018) Predicting reaction performance in C–N cross-coupling using machine learning. Science 360:186–190
Coley CW, Barzilay R, Jaakkola TS, Green WH, Jensen KF (2017) Prediction of organic reaction outcomes using machine learning. ACS Cent Sci 3:434–443
Lowe D. Chemical reactions from US patents (1976-Sep2016). https://figshare.com/articles/Chemical_reactions_from_US_%20patents_1976-Sep2016_/5104873. Accessed 1 Jan 2021
Szymkuć S, Gajewska EP, Klucznik T, Molga K, Dittwald P, Startek M, Bajczyk M, Grzybowski BA (2016) Computer-assisted synthetic planning: the end of the beginning. Angew Chem Int Ed 55:5904–5937
Mo Y, Guan Y, Verma P, Guo J, Fortunato ME, Lu Z, Coley CW, Jensen K (2021) Evaluating and clustering retrosynthesis pathways with learned strategy. Chem Sci., 12, 1469–1478
https://insilico.com/blog/pcc. Blog post related to insilico medicine’s use of AI to discover novel molecule for IPF
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

Cite this protocol
Gupta, R.R. (2022). Application of Artificial Intelligence and Machine Learning in Drug Discovery. In: Heifetz, A. (eds) Artificial Intelligence in Drug Design. Methods in Molecular Biology, vol 2390. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-1787-8_4
Download citation
DOI: https://doi.org/10.1007/978-1-0716-1787-8_4
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-1786-1
Online ISBN: 978-1-0716-1787-8
eBook Packages: Springer Protocols