
Abstract
Chaetothyriales is an ascomycetous order within Eurotiomycetes. The order is particularly known through the black yeasts and filamentous relatives that cause opportunistic infections in humans. All species in the order are consistently melanized. Ecology and habitats of species are highly diverse, and often rather extreme in terms of exposition and toxicity. Families are defined on the basis of evolutionary history, which is reconstructed by time of divergence and concepts of comparative biology using stochastical character mapping and a multi-rate Brownian motion model to reconstruct ecological ancestral character states. Ancestry is hypothesized to be with a rock-inhabiting life style. Ecological disparity increased significantly in late Jurassic, probably due to expansion of cytochromes followed by colonization of vacant ecospaces. Dramatic diversification took place subsequently, but at a low level of innovation resulting in strong niche conservatism for extant taxa. Families are ecologically different in degrees of specialization. One of the clades has adapted ant domatia, which are rich in hydrocarbons. In derived families, similar processes have enabled survival in domesticated environments rich in creosote and toxic hydrocarbons, and this ability might also explain the pronounced infectious ability of vertebrate hosts observed in these families. Conventional systems of morphological classification poorly correspond with recent phylogenetic data. Species are hypothesized to have low competitive ability against neighboring microbes, which interferes with their laboratory isolation on routine media. The dataset is unbalanced in that a large part of the extant biodiversity has not been analyzed by molecular methods, novel taxonomic entities being introduced at a regular pace. Our study comprises all available species sequenced to date for LSU and ITS, and a nomenclatural overview is provided. A limited number of species could not be assigned to any extant family.
Similar content being viewed by others

Introduction
Chaetothyriales is an ascomycetous order within Eurotiomycetes of the subphylum Pezizomycotina (Gueidan et al. 2014; Wijayawardene et al. 2020). The order is renowned for containing so-called black yeasts and their filamentous relatives, among which are numerous opportunistic agents of disease in humans and cold-blooded vertebrates. Well-known genera are Cladophialophora, Exophiala, Fonsecaea and Phialophora (Herpotrichiellaceae), but the order is much more diverse, in the literature of the last decades containing about 42 genera (with frequent additions) arranged in six families as summarized in the taxonomic browser of NCBI and 56 genera belonging to 11 families by Wijayawardene et al. (2020). A family that has been linked to Chaetothyriales, viz. Coccodiniaceae, contains four genera with few epilithic and epiphytic species having nondescript morphology and which are difficult to cultivate and sequence. Most researchers hypothesize an affinity to Capnodiales in Dothideomycetes (Hyde et al. 2013; Réblová et al. 2013; Kirk et al. 2001; Winka et al. 1998). Other families included in the order by Barr (1987a, b), i.e. Metacapnodiaceae, Microtheliopsidaceae, Strigulaceae and Trichopeltidaceae, have also been suggested to belong to Dothideomycetes or are incertae sedis (Reynolds 1985). Pyrenotrichaceae (Herrera-Campos et al. 2005) has been suggested to belong to Chaetothyriales Wijayawardene et al. (2020) but no sequence data are available to support this. Also for family Lyrommataceae, containing the single genus Lyromma producing ascomata with antler-shaped appendages and pycnidia with filiform conidia (Flakus and Farkas 2013) no sequence data is available. Some other described families will be discussed below. The five remaining families that are currently accepted in the order are Chaetothyriaceae, Cyphellophoraceae, Epibryaceae, Herpotrichiellaceae, and Trichomeriaceae (Réblová et al. 2013; Gueidan et al. 2014; Chomnunti et al. 2012a; Barr 1976; Barr and Makkai 1987).
A major problem in Chaetothyriales is that only a fraction of the extant species is likely to be known, as shown for other groups of fungi in Hyde et al. (2018). Members of Herpotrichiellaceae have limited morphological features and should be distinguished by molecular parameters observed on asexual states in culture. Conversely, species in the large family Chaetothyriaceae have classically been distinguished by the morphology of their sexual state in nature, and for most no sequence data are available. Further, new monophyletic lineages of lichen-inhabiting fungi have been reported for fungi close to Chaetothyriales (Muggia et al. 2015, 2016, 2017, 2020), for which the exact taxonomic position still needs to be determined. Therefore, taxonomic boundaries at lower and higher levels are likely to appear unstable in the future when more extant biodiversity will be added and adequately described.
In summary, the taxonomy in Chaetothyriales is unsettled, for three reasons: (1) fragmentary representation of extant species, (2) limited availability of sequence data, and (3) conflicting concepts between traditional and modern taxonomic methods. As a consequence, the order is in need of detailed taxonomic revision. In view of taxonomic stability, the present paper aims to develop a polyphasic approach to classification, at least at the family level, since strict phylogenetic taxonomy may be subject to change as soon as new aspects of biodiversity of the order are revealed.
Materials and methods
Strains and sequences
Sequences of strains used in this study were retrieved from an in-house black yeast database maintained for research purposes at Westerdijk Fungal Biodiversity Institute, supplemented with data from GenBank (Table 1). In view of optimal resolution of phylogenetic relationships, all species sequenced to date were included with a single sequence per species. Up to now (April 2020), GenBank records at NCBI list the order Chaetothyriales with 6 families, 42 genera and 273 species. Sequences in the present study comprise of 8 described families, 33 genera, 209 species, 45 undescribed species and two outgroup species. Species not included are either missing sequences for the LSU or ITS or both, or these sequences are obvious errors in NCBI. Undescribed species are mainly inhabitants of ant nests (carton-building ants or domatia) (Voglmayr et al. 2011; Nepel et al. 2014), or rock-inhabiting species of which a significant share belong to Chaetothyriales (Ruibal et al. 2018; Muggia et al. 2020); only a selection of these has been included.
Phylogeny
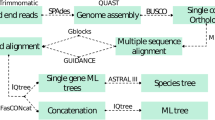
To assess the phylogenetic position of Chaetothyriales, phylogenetic analyses of the ITS and LSU loci were performed for 254 sequences representing this order. Multiple sequence alignments were made by MAFFT v7 (http://mafft.cbrc.jp/) and optimized manually using MEGA v7.2 (Kumar et al. 2012) and BIOEDIT v7.2 (Hall 1999). Missing data for part of the sequences for some taxa were coded as ‘missing’, but still could be used in the final matrix (Wiens 2006, 2008).
Models of DNA sequence evolution for each locus partition were selected with jModelTest v.2.0 (Darriba et al. 2012), using the Akaike information criterion (AIC, Akaike 1974). To detect possible topological conflicts among loci, the CADM test (Campbell et al. 2011; Legendre and Lapointe 2004) was performed using the function ‘CADM. global’ implemented in the package ‘ape’ of R (Paradis et al. 2004). With congruence, three alignments, ITS, LSU and ITS combined with LSU were used to run the tree. Three algorithms, i.e. maximum likelihood (ML), Bayesian inference (BI) and Neighbor-Joining (NJ) were employed on phylogenetic analyses. Capnodium coffeae, CBS 147.52 and Capnodium salicinum, CBS 131.34 were taken as outgroups in most of the trees.
ML trees were obtained using RaxML-VI-HPC as implemented on the CIPRES portal web server (http://www.phylo.org/). Neighbor Joining (NJ) was performed by MEGA v6 (Tamura et al. 2013) with Kimura 2-parameter model and statistical bootstrapping procedure involving 1000 replicates. Bayesian command files were prepared using MESQUITE v2.75 (Maddison and Maddison 2007), and the analysis was done in MRBAYES v3.1.2 implemented in the CIPRES web server (http://www.phylo.org/). Two parallel runs with four Markov chain Monte Carlo (MCMC) simulations for each run were set for 10,000,000 generations and the result was checked using TRACER v1.5 (Rambaut and Drummond 2009) for effective sample size (ESS). The run was then extended for another 10,000,000 generations with a sample frequency of 1,000 per generation.
Of the 209 described species, four species were not recorded in the NCBI Taxonomy browser, but were described as incertae sedis by Wijayawardene et al. (2020), who listed ten genera as incertae sedis. We tested these as possible out- or in-groups by comparing the resulting bootstrap values. Similarly, species with long branches were re-analyzed as outgroup in ML trees that were run accordingly. All branches with bootstrap values ≥ 70 % were collapsed, starting with the first group containing >1 members. The absolute and relative numbers of collapsed clades were taken as a parameter of confidence, the ratios (supported/unsupported clades) were calculated (Table 2), criteria of quality of trees being a low number of unsupported trees, as well as a low number of collapsed trees indicating high support of branches of the backbone. Phylogenetic trees were edited using TREEVIEW v1.6.6 and completed with Adobe ILLUSTRATOR CS v5. The alignment was deposited in TREEBASE under accession number 26209.
Divergence time and evolutionary rate estimation
Fossil-calibrated phylogeny was calculated by the BEAST2 tutorial (https://beast2-dev.github.io). The concatenated data set was used as a primary input to BEAST2 analyze and the choice of the GTR substitution model was based on pre-analysis using jModelTest v2.0 (Darriba et al. 2012) and the substitution rate was estimated. The divergence time estimation was executed with a strict clock and birth-death models. Fossil data of the taxonomic group closest to Eurotiomycetes, i.e. the class Sordariomycetes studied e.g. by Pérez-Ortega et al. (2016), Liu et al. (2017) and Samarakoon et al. (2019) was used as calibration point (mean: 136 Mya; sigma: 0.5; credibility interval: 95 %). Tree sequences from Sordariomycetes were included: Meliola centellae, Cordyceps agriota, and Colletotrichum agaves caricis forcing the monophyletic mode. Similarly, a calibration point of 100 Mya to the order Capnodiales was included. Default MCMC options were used. The results were analyzed using TRACER v1.7.1 and to generate a maximum confidence of clades in the tree, TREEANNOTATOR v2.6.0 (burn-in option of 10%) and BEASTv.2.6.1. The tree was visualized by FIGTREE v1.4.4 and the geological axis was added using the GEOSCALEPHYLO function from the STRAP R package (https://cran.r-project.org/web/packages/strap) (Fig. 1).

Divergence time of the order Chaetothyriales based on ITS and LSU sequences. The bottom scale presents the main geological and periods and eras
Ecology
We investigated broad ecological trends of 254 species by consulting original literature, NCBI database, Westerdijk database, MycoBank (www.MycoBank.org), and Index Fungorum (www.indexfungorum.org); additional information to extend hypothesized ecological trends per species was abstracted from specific literature where available. Average ecologies were summarized as a single symbol per species, and quantified relative to the number of species recognized per families (Table 1; Fig. 2). This aimed to extract broad evolutionary trends per family, which was used to strengthen or to falsify clades generated by ribosomal data.

Approximate types of ecology distributed over different families of Chaetothyriales, normalized to 100%. Pink represents ‘epilithic/lichenolytic’, orange ‘bryophytic’, black ‘ant domatium-associated’, blue ‘ant-made carton-associated’, green ‘epiphytic’, red ‘opportunistic’; brown unites ‘other’, remaining categories, mainly soil, water and fungus
Ancestral character state reconstruction
Trends in evolutionary ancestry and its impact on lineage and species diversification was analyzed in the following steps: (i) simulation of quantitative traits among the phylogenetic tree, (ii) stochastical character mapping and inference of a multi-rate Brownian motion model fitting and its visualization, (iii) calculation of phylogenetic signals, and (iv) ecological disparity, and comparison to (v) lineage diversification over time. Discretely valued ecological character traits are listed in Table 1. Discrete characters were converted into continuous states to estimate their evolution along the previously inferred phylogeny based on ITS and LSU gene sequences (Fig. 3). Stochastic character mapping onto the phylogeny was done according to Hulsenbeck et al. (2003) with subsequent fitting of a multi rate Brownian motion model (Likelihood test for rate variation in a continuous trait) to estimate evolutionary rates (= Sigma parameter) for each character and to infer the ancestral state at the root node (O’Meara et al. 2006). Quantitative traits (= ecologies) were simulated among the phylogeny and were plotted as phenogram to visualize trait dynamics. To assess the phylogenetic signal of our data we computed the K statistic (Blomberg et al. 2003) and λ (Pagel 1999) to assess resolution quality of our dataset. Disparity relative to lineage diversification was calculated to assess success of species cladogenesis according to Pybus & Harvey (2000). Analyses were done with R statistical software (https://www.r-project.org/), employing mainly the packages APE (Paradis and Schliep 2019), PEGAS (Paradis 2010), GEIGER (Pennell et al. 2014), MAPS (https://cran.r-project.org/web/packages/maps/index.html), TAXONOMIZR (https://cran.r-project.org/web/packages/taxonomizr/index.html), PHYTOOLS (Revel 2011), and all their reverse dependencies. We used the functions ‘MAKE.SIMMAP’ for stochastically map characters (i), ‘BROWNIE.LITE’ to model in the Brownian motion process (ii), and to reconstruct the ancestral character state. Quantitative trait simulation (iii), was conducted via the ‘TRAIGRAM’ and ‘PHENOGRAM’ functions as well to plot the Brownian motion process of character evolution and to visualize the phenotype to the phylogeny (iv). Phylogenetic signal for K and λ were computed via the function ‘PHYLOSIG’ (vi). To assess lineage diversification rates we plotted a ‘lineage through time’ (LTT) via the function ‘LTT’ plot to define the relative time ratio required for the Chaetothyriales to give rise to its present lineages. Subsequently, to assess morphological disparity we calculated and plotted a disparity through time (DTT) distribution via the function ‘DTT’ (vii). Details of the analysis and compiled scripts are available upon request.



Phylogenetic tree of Chaetothyriales based on ITS and LSU sequences, obtained by Bayesian analysis and maximum likelihood (values of ≥ 95% for Bayesian probability and ≥ 70 % for maximum likelihood shown in bold branches). Capnodium coffeae and Capnodium salicinum were used as outgroup
Results
Phylogeny
The single gene ITS and partial LSU, and combined sequences of ITS with partial LSU of 254 strains of black fungi were applied to determine phylogenetic trees of the entire order Chaetothyriales, using Capnodium salicinum and Capnodium coffeae as outgroups taxa. The alignment contained 522 characters for ITS, 497 for LSU, 1019 for combined sequences. The alignment of combined sequences had the following base frequencies: f (A) = 00.243, f (T) = 00.247, f (C) = 00.234, f (G) = 00.275, among which 642 were variable and 548 parsimony-informative sites. When separate trees of LSU and ITS were compared with the tree based on the concatenated alignment, bootstrap values in the combined tree on average were higher than those found in single-gene trees. Some families did not form supported clades in single gene trees, but obtained higher bootstrap support in combined trees. The non-collapsed NJ tree showed that this algorithm is not suitable for analysis of Chaetothyriales at ordinal level, judging from the low number of supported branches. With Bayesian analysis (BA) (Fig. 3) the combined tree contained a total of 153 supported clades (posterior probabilities PP ≥ 95%), and with maximum likelihood (ML) 123 supported clades (bootstrap support BS ≥ 70 %). A total of 120 clades were recognized consistent in the two algorithms; in Fig. 3 both types of support are indicated by thickness of the branches.
For the reconstruction of the possible evolution of the order Chaetothyriales, the order of appearance of recognized groups is significant. In most literature on Chaetothyriales, topologies of phylogenetic inferences suggest the existence of six families (Réblová et al. 2013; Gueidan et al. 2014; Teixeira et al. 2017). The family Phaeosaccardinulaceae was introduced by Batista and Ciferri (1962) and is represented by three species (Wijayawardene et al. 2020). The recently described families Strelitzianaceae and Paracladophialophoraceae have four and two species in the tree, respectively. Three more groups were added in recent studies exploring novel habitats (Muggia et al. 2020; Wang unpublished data). These groups were mostly recognized as separate clades supported with high bootstrap in the bi-locus tree with all algorithms applied.
Six species, Atrokylindriopsis (Ma et al. 2015), Lichenodiplis (Hawksworth and Dyko 1979), Melnikomyces (Crous et al. 2014), Bacillicladium (Réblová et al. 2016), Muellerella (Muggia et al. 2020) and Uncispora (Sinclair 1979), mentioned as having an uncertain phylogenetic position by Wijayawardene et al. (2020), were included in the ML analysis. Atrokylindriopsis setulosa and Uncispora in Clade 1 had bootstrap support of 72 %. When Neostrelitziana acaciigena was added to the tree, it clustered in Clade 6, almost all species of this clade were described as Trichomeriaceae, with bootstrap support remaining at 100%. Paracladophialophora formed a sister clade to a cluster of undescribed ant-domatia associated fungi. Lichenodiplis, for which only an LSU sequence was available, formed a sister clade to a group of endolichenic fungi (Muggia et al. 2020). Bacillicladium was monophyletic next to Trichomeriaceae with low bootstrap support. The tree including the genera above is shown in Fig. 3. The genera Melnikomyces and Muellerella seemed remote from Chaetothyriales and were excluded from further analysis.
In the literature, the following fungi are treated as members of Chaetothyriales, at least by some authors, but were found at relatively long branches in the ML tree: Epibryon hepaticola, Capronia villosa, Cladophialophora modesta, Cladophialophora hostae, Cladophialophora scillae, Paracladophialophora spp., Coccodinium bartschii, Arthrophiala arthreospora, Capronia nigerrima, Bacillicladium dematidis, Rhinocladiella mackenziei, and Strelitziana spp., of which Coccodinium has been surmised to be dothideaceous (Hyde et al. 2013). Species were individually rearranged as outgroups and the effect on statistical support of resulting ML trees was compared with the supposition as to whether these are members or non-members, the bootstrap values should change significantly. Supported and unsupported clades were calculated; trees with highest ratios supported vs. unsupported clades at a low number of supported clades in the backbone were considered to be optimal. The ratio of the combined ML tree including all incertae sedis above is 1.30 (Table 2). The highest ratios (1.75) were obtained when Epibryon hepaticola, Capronia villosa, Cladophialophora modesta, Cladophialophora hostae and Cladophialophora scillae, Paracladophialophora spp., Bacillicladium dematidis, or Coccodinium bartschii were used as outgroup, the ratios increased slightly compared to the reference tree (1.30, with Capnodium as outgroup); these species were consequently regarded as incertae sedis. Four of the items tested as outgroups, i.e. Capronia nigerrima (0.72), Rhinocladiella mackenziei (0.72), Arthrophiala arthreospora (1.04), and Strelitziana spp. (1.04) had a negative impact on the tree and taken as belonging in Chaetothyriales. Bacillicladium dematidis, Cladophialophora modesta, and Capronia villosa, similar to dothidealean Coccodinium bartschii, appeared as single-species branches in the tree, could not be affiliated to any of the known families and are therefore regarded as incertae sedis. Whether or not these species are members of Chaetothyriales could not be established. The complete tree including these species was compared to the same tree without these species, which led to drop of the ratio to 0.63. The complete tree with Paracladophialophora as outgroup remained the optimal tree, with a high ratio (1.75) of supported/unsupported branches and with a relatively low number of clades. This suggest that the group (Clade 3) represents a separate family, as proposed by Crous et al. (2016).
The best-fit models of evolution obtained for the different datasets were ITS = TVM+I+G, LSU = GTR+I+G, combined sequences = TIM2+I+G. No topological conflicts between the datasets were detected. The ML tree was constructed with GTRGAMMA + I in the CIPRES webserver. Robustness of trees was tested by comparing different algoritms on the individual datasets of LSU and ITS, and the combined dataset, placing accent on the backbone by collapsing all supported clades. The best tree is judged to be the one with the most resolved backbone, i.e. an optimal ratio of supported/unsupported branches, combined with high support values for all clades, starting at the outermost position (lowest value) which was variably taken by Capronia villosa or Cladophialophora modesta (Fig. 4; Table 3). With these criteria, the Bayesian tree of the combined dataset appeared to be optimal. Nine well-supported clades were recognized, which represent five existing families and several uncharacterized groups.

Outgroup test for long branches base on ML tree. a Cladophialophora modesta as outgroup; b Paracladophialophora sp. as outgroup; c Rhinocladiella mackenziei as outgroup; d all species without incertae sedis; e all species including incertae sedis
Clade 7 (Chaetothyriaceae) was relatively heterogeneous with low support, most likely caused by undersampling of sequence data as compared to the large diversity described on the natural substrate. One of the two Chaetothyrium species defining family and order, C. brischoficola, was found in this clade in several datasets (Fig. 3). The families Phaeosaccardinulaceae and Strelitzianaceae were found as part of the Chaetothyriaceae cluster; their family status is doubtful. Clade 1 (Herpotrichiellaceae) were also found to be diverse and resolved into two groups in some of the trees. The remaining families Cyphellophoraceae (Clad 2), Epibryaceae (Clade 8), and Trichomeriaceae (Clade 6) had consistent support. Three further clades had consistently high support values, i.e. a group of ant-domatia associated species (Clade 4) and two clusters of endolichenic species (Clade 5 and Clade 9).
Family Trichomeriaceae (Clade 6) comprised 50 strains, ten of which represented as yet undescribed species from an ant carton. Two species, Metulocladosporiella musicola and M. musae, were originally thought to belong to Herpotrichiellaceae (Crous et al. 2006), but in our tree clustered in Trichomeriaceae. The type strains of three species, Cladophialophora pucciniophila, Cladophialophora proteae and Cladophialophora eucalypti also clustered in this clade, although the type species of Cladophialophora, C. ajelloi (= C. carrionii) is a member of Herpotrichiellaceae. Exophiala placitae and Exophiala encephalarti should morphologically belong to Herpotrichiellaceae, but cluster in Trichomeriaceae.
Clade 4 comprised a total of 15 strains originating from ant domatia inside plant stems, known as domatia. Species typically produce sympodial conidia with flat conidial scars, and sometimes have additional catenate conidial states (Wang unpublished data). The clade has sufficient support and ecological homogeneity to be recognized as a separate family. Two species, reported as causing leaf spots on different plant hosts (Crous et al. 2007), described after their plant hosts as Cladophialophora scillae and C. hostae, had exclusively catenate micromorphology. They cluster in one clade with a long branch, and upon taking them as outgroups, the general support values of tree improved (ratio rise from 1.30 to 1.75); consequently, Cladophialophora scillae and C. hostae are listed here as incertae sedis.
Clade 2 with 100% (ML/BI) bootstrap support contains 25 species belonging to family Cyphellophoraceae. Twenty strains described Cyphellophora species are clustered in this clade together with four Phialophora species (P. livistona, P. attae, P. capiguarae, and P. intermedia), together with a strain from the ant-made carton strain (CBS 128959). Cyphellophora and Phialophora traditionally differ by conidial shape, either lunate and septate, or subsphaerical, respectively, but the type species of Phialophora, P. verrucosa, is a member of the ‘carrionii-clade’ in Herpotrichiellaceae (de Hoog et al. 2011).
Clade 7 contains 21 species belonging to Chaetothyriaceae. The clade is well-supported in ML and BI trees (73/100). Inter-specific distances are relatively large due to incomplete taxon sampling. Members of this family have been reported since the 19th century after their ascomata on the natural substrate; culture and sequence data are available of only a fraction of these. Appropriate description of the family Chaetothyriaceae is therefore as yet impossible.
Clade 8 contains members of Epibryaceae, with 100% ML and 100% BI bootstrap support. The phylogeny of this family also suffers from a severe taxon sampling effect, as of the 47 species listed in Index Fungorum, only seven are available in GenBank. Of these, Epibryon hepaticola clusters at some distance from remaining taxa, but given the poor representation of extant biodiversity this is probably insignificant. When E. hepaticola was treated as outgroup, the ratio rose from 1.30 to 1.75; consequently, the species is listed as incertae sedis. This clade also contained three species belonging to Cladophialophora, classified as such on the basis of catenate conidia. The original strain of Cladophialophora minutissima was isolated from bryophytes, while other Epibryon species had been described on the basis of their ascomata produced inside moss thalli; obviously this Cladophialophora is a cultural state of an Epibryon species. Cladophialophora humicola and C. sylvestris were derived from soil and decaying pine needles, respectively. The cladophialophora-type of conidiation is common throughout the entire order Chaetothyriales.
Herpotrichiellaceae (Clade 1) is best represented by sequence data, because a large part of the known species was described from isolates in culture, thus only representing the asexual state. Traditionally, species were described after their ascomata on the natural substrate, classified in the genus Capronia. Index Fungorum lists 89 described species, of which 85 belong to Herpotrichiellaceae and one to Trichomeriaceae. For a total of 119 strains in Herpotrichiellaceae, sequence data were available, including 11 carton fungi. It is unknown whether these are asexual isolates of known sexual species; the connection between sexual and asexual morphs has been made only occasionally (Müller et al. 1987; Untereiner 1997). The core structure of Herpotrichiellaceae was poorly resolved. The group fell apart into several, poorly supported subclusters. On the basis of LSU-data, de Hoog et al. (2011) distinguished a number of approximate clades within the family, of which the ‘bantiana-clade’ and the ‘carrionii-clade’ could be recognized. In a third, large remainder of species, numerous novel taxa had been added since 2011; no clades or clusters could be distinguished.
Nomenclature
The order Chaetothyriales was validated by Barr (1987a, b) for epiphytic sooty molds mostly producing setose, clypeolate ascomata containing dark, multi-celled ascospores, with Chaetothyriaceae (Barr 1979) as type family. The invalidly described families Phaeosaccardinulaceae and Euceramiaceae (Batista and Ciferri 1962) were regarded as synonyms (Barr 1987a, b).
Chaetothyriaceae had provisionally been introduced by Hansford (1946) with Chaetothyrium, based on C. guaraniticum Speg., as the type species. The original dried material of the type species, described in 1888, insufficiently allows interpretation. The Index Fungorum lists 76 published names in Chaetothyrium, of which 67 are accepted as members of Chaetothyriaceae. However, GenBank contains only two sequenced species, viz. Chaetothyrium agathis (Liu et al. 2015) and C. bischofiicola (Chomnunti et al. 2012b), both isolated on a single occasion from leaves of tropical plants. It remains uncertain whether this is in accordance with the intention of Spegazzini (1888), but numerous authors maintained the ecological concept of ‘sooty moulds’, i.e. epiphytic colonizers of living plants: at least 64 of the 67 species mentioned above were described from plant leaves, generally without symptoms. In order to stabilize the nomenclatural reference of Chaetothyriales, we herewith propose Chaetothyrium agathis Hongsanan & K.D. Hyde (Liu et al. 2015) as a neotype of Chaetothyrium. Chaetothyrium agathis takes a central position in the clade of Chaetothyriaceae (Fig. 3) and is the reference point of the order Chaetothyriales. Wijayawardene et al. (2020) listed the genus Aithaloderma in the Chaetothyriaceae. Hansford (1946) reexamined the type of A. clavatisporum which displayed a Triposporium asexual state, and reclassified it in Chaetothyrium.
Chaetothyriaceae further comprises the genus Ceramothyrium. This genus is listed with 41 names in Index Fungorum, of which 39 are surmised to belong to Chaetothyriaceae. The type species is Ceramothyrium paivieae (Batista 1956), originally reported from leaves of Paivea langsdortii (= Copaifera langsdorfii; Leguminosae) in Brazil. No molecular data are available for this species. Judging from older literature, this genus is also reserved for species colonizing plant leaves, with 37 of 39 species demonstrating this ecology, including the nine species of which LSU sequences are available in GenBank. Of these, Ceramothyrium thailandicum colonizes living leaves of Lagerstroemia (Lythraceae) in Thailand. Awaiting selection of neotype material which is closer to the original type location of Batista (Batista 1956), we regard Ceramothyrium thailandicum as the reference species for Ceramothyrium in the present paper.
Phaeosaccardinula, introduced by Hennings (1905) with type species P. diospyricola on leaves of Diospyros (Ebenaceae) in Amazonian Brazil, contains 47 species in Index Fungorum, of which 41 were regarded as members of Chaetothyriaceae. The genus currently has six synonymous generic names (Table 3), all containing a very small number of species that were mostly discarded for nomenclatural reasons. In accordance with the type species P. diospyricola, nearly all authors in older literature classified plant-colonizing species in the genus. Of three species, LSU sequences are available in GenBank, í.e. P. dendrocalami and P. multiseptata (Yang et al. 2014), and P. ficus (Chomnunti et al. 2012b), all from living plant leaves, in (sub)tropical China and Thailand, respectively. In absence of sequence data of the remaining 39 species of Phaeosaccardinula, we regard these species as representative for the genus, with P. ficus as reference.
Two species are known in Vonarxia of which V. anacardii is the type species (Batista 1960). The species is in poor condition (van der Aa and von Arx 1986) and is currently judged to be of uncertain affinity (Index Fungorum), while V. vagans has been sequenced and described by several authors (Réblová et al. 2013; Crous et al. 2009). That taxon, based on Ypsilonia vagans Speg. on leaves of Spiraea cantonensis (Rosaceae) in Brazil, has setose sporodochia with splayed stauroconidia. Crous et al. (2009) epitypified the species with CBS 123533 as the type culture. Given the unclear status of the type species V. anacardii, we might regard V. vagans as a reference species for the genus Vonarxia, but it should be noted that it is also the type species of Kazulia (Raj 1977). The morphologically similar genus Fumagopsis was described by Spegazzini (1910) with F. triglifioides, on living leaves of Lucuma neriifolia (Sapotaceae) in Argentina, as the type species. Using the dried herbarium specimen of the holotype, van der Aa and van Oorschot (1985) redescribed this specimen. It is characterized by setose sporodochia bearing stauroconidia, similar to those of Vonarxia vagans but differing by the conidia being pronouncedly multicellular. Fumagopsis triglifioides has as yet not been sequenced. Three species records of Fumagopsis are listed in Index Fungorum, but only one, F. stellae, CBS 145078 from leaves of Eucalyptus (Myrtaceae) in Australia, has been deposited in NCBI. This species had similar morphology, with setose sporodochia and multicellular stauroconidia on the natural substrate, and sequences placed it in Chaetothyriales (Crous et al. 2018). Numerous other sporodochial, morphologically reminiscent genera have been described, such as Zelopelta (Sutton and Gaur 1984), Phalangispora (Nawawi and Webster 1982), which are in need of modern sequence data.
Four small genera were recently described for which sequence data are available, i.e. Aphanophora, Arthrophiala, and Camptophora. All type species of these genera (Table 3) cluster in the supported clade of Chaetothyriaceae (Fig. 3), all at relatively long branches, underlining their position as separate genera. Nullicamyces clusters amidst species of Ceramothyrium in a cluster that is however not supported (Fig. 3). Stanhughesia was described as Ceramothyrium asexual states (Constantinescu et al. 1989). Species of Microcallis have been reclassified in Chaetothyrina which is a genus of Micropeltidaceae.
Cyphellophoraceae was introduced by Réblová and Untereiner (Réblová et al. 2013) with Cyphellophora (de Vries 1962) as the type genus and C. laciniata as the type species. CBS 190.61 is available as the type strain, and the taxon has several genes in GenBank. Currently, 28 species have been described in the genus, two of which were transferred as independent genera of Chaetothyriaceae (C. eugeniae as type of Anaphora, and C. hymeloconis as type of Camptophora) and one, C. suttonii, has been excluded. Another genus of this family is Anthopsis, based on A. deltoidea as type species with CBS 263.77 as type strain (Moussa et al. 2017a, b).
Trichomeriaceae was introduced by Chomnunti et al. (2012b) with Trichomerium as type genus. This genus is based on the sooty mold Limacinia coffeicola Puttemans [non Phaeosaccardinula coffeicola (Maharachchikumbura et al. 2018)] as the type species (Puttemans 1904). Reynolds (1983) judged this species as being close to or identical to T. grandisporum, which he considered as the only recognized species in Trichomerium with a large number of synonymous names. No living ex-type material was available to recent authors (Chomnunti et al. 2012a), who consequently took T. foliicola, with sequence data, as reference for genus and family. From their extensive illustrations of the sexual state of this fungus, it appears that the ascigerous fruit bodies of Trichomerium are morphologically very similar to those of Capronia, the rather monomorphic sexual state observed in numerous species of Herpotrichiellaceae. Conidia were not observed, but several members of Trichomeriaceae [e.g. Trichomerium gloeosporum (Hongsanan et al. 2016a) and T. changmaiensis (Maharachchikumbura et al. 2018)] produce elaborate stauroconidia.
The family Epibryaceae was introduced by (Gueidan et al. (2014) with Epibryon (Döbbeler 1978, 1980) as type genus which has Epibryon plagiochilae as the type species. This species was described with molecular data by Stenroos et al. (2010a, b) in a detailed overview of the genus, and is accepted here as reference for this group of phylogenetically consistent moss endophytes.
The best-known family in the order Chaetothyriales is Herpotrichiellaceae, introduced by Munk (1953) with Herpotrichiella (Petrak 1914) as the type genus. Herpotrichiella moravica was selected as the type species, which is considered to be a synonym of Capronia pilosella (Untereiner 1997). Consequently, the currently accepted name for Herpotrichiella is that of its older synonym Capronia, introduced by Saccardo (1883) with Capronia sexdecimspora (Cooke) Sacc. as type species, characterized by setosa ascomata with asci containing 16 hyaline, 3-4-septate ascospores. As no interpretable type material of this species is available, the identity of this species remains uncertain. As yet, none of the species with 16-spored asci has been sequenced, and thus replacement of C. sexdecimspora by an extant neotype is difficult and the exact position of the reference for Capronia in the Herpotrichiellaceae remains uncertain. We propose to stabilize the nomenclature of Herpotrichiellaceae by selecting Capronia pilosella AFTOL 657 as reference for the family.
A large number of Capronia species has been subsequently described (e.g. Barr 1987a, b; Friebes 2012), of which Index Fungorum considers 81 to be of chaetothyrialean affinity. The family Herpotrichiellaceae comprises 30 generic names (Table 3), which are principally available for a future taxonomic rearrangement with phylogenetic affinity as leading principle and which therefore are in need of redefinition with reference material. The oldest name of these is Berlesiella, based on Sphaeria nigerrima Bloxam 1859, which in spite of absence of usable type material is now considered to be Capronia nigerrima (Barr 1991). Sequenced material of this species is available from Untereiner and Naveau (1999) who used strain CBS 513.69 described by Müller et al. (1987). Caproniella was introduced (Berlese 1896) with Melanomma pleiosporum as a single species, now known as Capronia pleisporum (MycoBank), but no recent material is known to be available. Berlese (1899) used Caproniella with Sphaeria sexdecimspora as the type. This generic name is superfluous as S. sexdecimspora was the type of Capronia, and Caproniella Berlese 1899 is a later homonym of Caproniella Berlese 1896. For these reasons we consider Caproniella as a nomen confusum. Moussa et al. (2017a, b) noted that Foxia and Melanchlenus were invalid due to absence of descriptions in the protologues. Most of the remaining genera are represented by extant type strains with molecular data (Table 3).
Ecology and evolution
Members of Chaetothyriales have a rich ecological diversity, with a general tendency to extremotolerance (Gostincar et al. 2019) and toxin management (Teixeira et al. 2017). The difficulty to isolate the fungi from the environment (Sudhadham et al. 2008, Vicente et al. 2008) interferes with understanding of the preferred ecological niche. Available data may provide distorted information since unspecific habitats may have been sampled thus far. For example, Cyphellophora europaea is commonly encountered colonizing human nails. It has been found in bathrooms where this fungus is likely to have been acquired by the patients. A natural habitat has not been found, but colonization of moist surfaces suggests oligotrophy. For only a small number of species of Cyphellophoraceae, environmental data are available. Numerous species have been described from a single strain on a single host plant, without indication of a specific plant-pathogenic lifestyle. We have assumed oligotrophy for these species as well, listing them as colonizers of the phyllosphere with an epiphytic lifestyle.
Ecologies of 254 strains and their relatives in Chaetothyriales were investigated (Table 1). Many species of Chaetothyriales have been described from single collections and hence epidemiological investigations are problematic. Habitat data were abstracted from the sampling sites of strains described in the original publications, supplemented with a summary of ecological trends per species abstracted from the literature. Seven categories were summarized as follows, ‘epilithic/lichenolytic’ (on bare or parasitizing on lichens), ‘epiphytic’ (colonizing plant leaves without symptoms), ‘opportunistic’ (deep, single- or multi-organ infection in humans, also infection in cold-blooded vertebrates), ‘carton’ (carton of chewed wood in ant nests), ‘domatium’ (ant nest inside living plant stem), ‘bryophytic’ (endophytic in mosses), and ‘other’ (aquatic, fungicolous, in soil). Members of the family Herpotrichiellaceae showed highly diverse ecological sources. In a total of 119 strains, five ecologies were distinguished. In the main categories, 38 strains derived from opportunistic infections, 30 from other, 36 were epiphytic, 4 were epilithic/lichenolytic, while 11 as yet undescribed strains had been isolated from carton material in ant nests.
Available information on members of Cyphellophoraceae was scant, not allowing definitive conclusions. Several species were isolated from living plants, but it remained unclear whether this was an infectious process, or epiphytic growth without notable invasion. Data are abstracted from a summary given by Feng et al. (2014). Cyphellophora europaea is the only common species of the family. It is a commensal or mild infectious agent on human skin and nails, and was repeatedly isolated in bathrooms where the fungus was suggested to be picked up (Lian and de Hoog 2010); for this reason, we prefer ‘opportunistic’ as its ecology. In total, four ecology types are observed in this family. Given the frequent plant origin without clear description of disease, we listed the main ecology as ‘epiphytic’ (44%).
Members of Trichomeriaceae are surface colonizers: 42 % of the species were isolated from rock. Knufia epidermidis was originally described as repeatedly being involved in mild nail infections (Li et al. 2008), but Zakharova et al. (2013) found the same fungus occurring as a rock colonizer with an ecology similar to remaining Knufia species. 38 % of members of Trichomeriaceae reportedly were derived as ‘sooty molds’ from plants which often had somewhat leathery leaves. Since these were single sampling events and no reports about plant disease have been published, we listed all species as being epiphytic. The species of Bradymyces had single isolation events (rock and fish) for which no common denominator could be found.
Members of Chaetothyriaceae have nearly always been reported from living plants. Detection was generally by ascomata on the natural substrate, which eventually were immersed on a stroma fixed to the undamaged host tissue. We listed those members as ‘epiphytic’; only 10% of the species were described from other habitats.
Clade 4 contains a major subclade of 15 strains that were derived exclusively from domatia of tropical ants. The second subclade contained two species with cladophialophora-like morphology which caused leaf spots on their host plants; they are known from single sampling events. Two more undescribed clades (Clades 5 and 9) were noted which all were derived from rock environments (Muggia et al. 2020).
Epibryon species are fungi forming small ascomata inside moss tissue. Index Fungorum lists 48 species, most of which have been described after material on the host and could not be included in this study for lack of sequence information. Three cladophialophora-like species clustered in the Epibryaceae, of which C. minutissima was derived from mosses without observation of the ascigerous state. The ecologies of the five cultured Epibryon species are consistently bryophilous.
Of the distinguished ecological categories, epilithic and epiphytic are commonly encountered in several families (Herpotrichiellaceae, Trichomeriaceae, Cyphellophoraceae, Chaetothyriaceae, and Clades 5 and 9). Also carton-material of ant nests and tunnels is widely distributed (Herpotrichiellaceae, Trichomeriaceae and Cyphellophoraceae). Human infection is nearly exclusively found in Herpotrichiellaceae, occasionally in Trichomeriaceae, and restricted to mild, superficial infections in Cyphellophoraceae. Infections in cold-blooded vertebrates are restricted to Herpotrichiellaceae. Dominant ecology in Clades 9 and 5 is ‘epilithic’; in Epibryaceae this is ‘bryophytic’, in Clade 4 ‘ant-domatium associated’, in Chaetothyriaceae ‘epiphytic’, and in Trichomeriaceae it is ‘epiphytic’. The overview contains 39 ant-associated strains, isolated either from carton material of nests and tunnels, or from domatia inside living plants. The latter type (15 entries) is restricted to Clade 4, while carton-associated species (24 entries) have a wide distribution in Herpotrichiellaceae, Trichomeriaceae and Cyphellophoraceae and are not found in Clade 4, confirming data of Voglmayr et al. (2011) and Nepel et al. (2014).
The evolutionary time estimation (Fig. 1) reveals that the Chaetothyriales crown order emerged in the late Devonian Period. Between the end of the Cretaceous, i.e. 151.69 Mya, the family was split, separating Clade 8, Epibryion hepaticola M10, Clade 9 and Capronia villosa from the remaining species. Fundamental speciation events occurred through the Cretaceous and Paleocene periods. The formation of the family Chaetothyriaceae (Clade 7) appears ancestral, starting about 122 Mya. The diversification of the family Herpotrichiellaceae was later, around 111 Mya (Fig. 1).
Ancestral character state reconstruction
In a first step, ecological traits were plotted model-free on to the phylogeny via the function ‘phenogram’ over time, in order to determine approximate number of ancestral trait changes (Fig. 5). Seven major directions of trait evolution are obvious (1–7 in Fig. 5). Although the phenogram does not indicate the exact ancestral state to the Chaetothyriales, particularly the traits ‘epiphytic’ and ‘epilithic-lichenicolous’ (branching point 1) suggests ancestry as a ‘epilithic-lichenicolous-epiphytic’ type. This assumption is strongly supported by absence of early overlapping trait changes. Branching point 1 gave rise to at least 3 major traits (branching points 2–4), which subsequently led to a strong lineage diversification and occupation of vacant ecological space leading to extant traits. During this process, most traits underwent multiple trait shifts visualized by overlapping branches, which is apparent for the epiphytic (branching points 2, 3 and 6) and the opportunistic characters (branching points 6, 7). Particularly the opportunistic trait appears to have sourced its extant trait from a strong random walk of ancestral intermediate traits (strong branch/line overlap). Traits ‘carton’, ‘domatia’ and ‘bryophytic’ have a non-random distribution. ‘Carton’ has diversified from early ancestral branching point (5), with almost no overlap to other traits.

Quantitative trait simulation among the phylogeny inferred via the R package ‘PHYTOOLS’. The plot depicts phenotypic distribution over the phylogeny and its associated changes over time. While it is similar the Brownian motion phenogram, the quantitative trait simulation does not depict the stochastically mapped character on to the phylogeny, and with that the phenotypic changes estimated for each branch, neither the relative evolutionary rate ratio (σ parameter) for each phenotype. Instead, it visualizes trait changes, uniformity and discreteness of such changes over time in a more comprehensive way. Branching points indicate approximated major directions of phenotypic changes at the root node to the entire order Chaetothyriales. X-axis depicts relative time for the phenotype (ecology) to evolve given the underlying phylogeny. Y-axis depicts relative phenotypic categories
To assess trait transitions, we calculated conditional likelihood for each character state at each node of the phylogenetic tree, including the root and simulated ancestral states at each internal node by sampling from the posterior distribution of states (stochastical character mapping). The waiting times between substitutions are drawn from an exponential distribution with the rate being the diagonal elements of the model’s instantaneous rate matrix (Q), conditioned on the current state to infer a character transition matrix (Table 6). This matrix served as input for the Brownian motion model fitting to estimate evolutionary rate changes (Table 4), and are visualized by mapped character changes along each individual branch (Fig. 6). The plotted Brownian motion process (Fig. 6) indicated that three traits can be considered as ancestral: ‘epilithic’, ‘carton’, and, at a more derived position, ‘epiphytic’. Short sections of ‘opportunistic’ among initial branches indicate that these traits, although later subject of a diversification burst, had precursor traits at a very early stage of evolution (as a result of the stochstically mapped characters). Conversely, the Brownian null model inferred the ancestral state as being quantitatively in between ‘epilithic’ and ‘epiphytic’, with tendency towards ‘epilithic’ (recoded discrete character ‘epiphytic’ = 1, ‘epilithic’ = 2, Brownian null model ancestral state = 1.71). The more ancestral characters ‘epilithic’ and ‘epiphythic’, as well the highly derived character ‘domatia’ have low evolutionary rates (Brownian rates σ2); remaining traits, which are randomly distributed over the taxa, experience a strong rate burst. Paucity of niche shifts on internal branches decreases covariances among tips relative to the neutral expectation and repress phylogenetic signals; conversely, an initially high rate of niche differentiation which decreases towards the present, tends to increase phylogenetic signal relative to the neutral expectation. Thus the most drastic niche shifts are concentrated near the root of the tree during early evolutionary history. The likelihood matrix for individual character transitions is given in Table 6, where higher values indicate a higher likelihood that the character was derived from another entity. The monophyletic character ‘domatia’ has a single character origin likelihood shared by ‘opportunistic’ (Table 6). The derived characters with high evolutionary rates (i.e. ‘domatia’ and ‘bryophytic’) expose a likelihood for a single character origin. Testing for the resolution of our dataset, assessment over the phylogenetic signal for the K and λ (lambda) statistic was performed. While the K statistic indicates a low phylogenetic signal for the global dataset, which is equivalent to a ‘tip-swap’ model, it is obvious from the Brownian motion process (Fig. 6), that character evolution towards the tips cannot be fully resolved given the current phylogeny comprising solely ribosomal gene data, which indicate (e.g. as for ‘opportunistic’ or ‘carton’) a strong random distribution (Brownian random walks) towards the tips. In contrast, Pagels’ lambda indicates a higher phylogenetic signal, which taken together as a result of the K statistic and the Brownian distribution, equates to a medium resolution quality of the dataset (phylogenetic signal, discrete character evolution), similar to Munkemueller et al. (2012) and Ackerly (2009), supporting validity over the assumptions on trait transitions above (Table 5).

Brownian motion phenogram inferred via the R package ‘PHYTOOLS’. Brownian rate parameter σ was set to 0.1 to simulate trait evolution under Brownian motion. X-axis depicts relative time for the phenotype (ecology) to evolve given the underlaying phylogeny. Y-axis depicts relative phenotypic variation under the Brownian rate parameter. Color coding for the various ecologies derived from stochastical character mapping: epiphytic (blue), epilithic/lichenolytic (red), opportunistic (brown), domatium (yellow), other (orange), carton (green) and bryophytic (pink). If lines do not cross, vertically and or horizontally the phenotype (= character = ecology) does not tend to be randomly distributed (the situation towards the base, ancestral state), while phenotypic changes towards the tips of the phenogram underlay in many cases (not all eg. ‘domatium’ or ‘bryophytic’) a strong random distribution. Topological distribution is equivalent to a late-burst model of phenotypic evolution

Disparity through time (DTT) plot inferred via the R package ‘GEIGER’. The plot depicts morphological disparity between phenotypes over time. X-axis depicts relative time for the lineages to disparate given the underlaying phylogeny and phenotypic data (ecologies). Y-axis depicts relative disparity between phenotypes (ecologies). Remarkably, and in concordance with the Brownian motion phenogram and the ‘LTT’ plot, morphological disparity peaked very early in Chaetothyrialian evolution prior to most lineages being born. This indicates that major phenotypic innovations occurred very early in Chaetothyrialian evolution, with a significant slow down in novelty and evolutionary rates (as of the Brownian model) to mostly the ancestral phenotypes and increase towards more derived phenotypes (e.g. ‘domatium’). Multiple small phenotypic disparate fluctuations (birth/extinctions) equates to the rise of the extant species
Lineage diversification and disparity distribution over time was demonstrated with a lineage through time plot (LTT; Fig. 8). Given the late burst of lineage diversification in Chaetothyriales, major phenotypic innovations occurred very early in the evolution. Highest disparity coincides with earliest diversification events, and with novelty for vacant niches (Fig. 7). After the late burst of diversification, beyond the greatest disparity peak, niche conservatism limits further character innovation; major trait innovation is subsequently declining. However, significantly positive Pybus’s γ statistics (+2.384) indicates that cladogenesis increases over time leading to pronounced and continuous species diversification in existing niches (Table 5).

Lineage through time (LTT) plot inferred via the R package ‘PHYTOOLS’. The plot depicts lineage diversification (accumulation) over time. X-axis depicts relative time for the lineages to diversify given the underlying phylogeny. Y-axis depicts relative number of lineages identified from the phylogeny. In concordance with the Brownian motion phenogram, the ‘LTT’ plot equates for a late burst model of diversification since a step increase (accumulation) in lineages accelerates only late in time. While establishing niche conservatism and occupation of niches took almost as much time for the early Chaetothyrialian taxa to evolve relative to the total number of lineages, lineage diversification and speciation events for all extant taxa in the present niches equates for less than one quarter of the total time. The underlaying phylogeny, depicted as ultrametric tree where evolutionary rates are set equal is shown as background graph in blue
Discussion
The study of biodiversity
The scientific history of the order Chaetothyriales consists of two parts, i.e. a phenotypic era using the sexual state in its natural habitat, and a prevalently molecular phase mostly applying asexual states in culture. The order attained recognition particularly through the contributions of M.E. Barr (1923‒2008) on Loculoascomycetes. Studies in that time were performed by morphology of the sexual state on the natural substrate, such as lichenized rock or decorticated wood, of which dried voucher specimens were preserved in herbaria. Only very few species were studied in culture. Numerous older, previously described species became assigned to the order (www.indexfungorum.org). Among these were members of the group that had already been recognized in the 19th century. As the early descriptions have nomenclatural priority, comparison of type material would be necessary to stabilize generic and specific names in the order. However, much of this material is now lost or is otherwise inaccessible, so that we are unsure about the identity of reference material defining families, genera and species. Even the identity of the species defining the order, Chaetothyrium guaraniticum Spegazzini, described in 1888, is uncertain, and hence an epitype will be designated below.
A similar line of research, after studies of Barr and others, has been on so-called ‘sooty moulds’, i.e. black fungi colonizing plants, rock or other inert material without invasion, forming a moss-like black felt. Most of these studies (Reynolds 1982; Reynolds 1983; Reynolds 1985; Chomnunti et al. 2014) comprised members of Capnodiales as well as Chaetothyriales. Only a fraction of these species, mainly belonging to Chaetothyriaceae and Trichomeriaceae, have cultures.
In the second half of the 20th century, an independent line of research emerged that almost exclusively used living cultures. Schol-Schwarz (1968) systematically revised phialophora-like organisms, and after studies of de Hoog (1977) and Hermanides-Nijhof (1977) and the term ‘black yeasts’ became adopted in the literature. Reference material of these studies was deposited in culture collections, and today sequence data are commonly available for almost all species cultured after 1970. The great majority of novel taxa initially belonged to a single family, Herpotrichiellaceae. Later, diversity studies discovered a gamut of fungi that phylogenetically clustered in other families of the Chaetothyriales. These studies take GenBank data as reference for novelty of their isolates, neglecting older, nomenclaturally valid but unsequenced taxa.
Müller et al. (1987) and Untereiner (1997) were the first to make systematic connections between sexual and asexual phases by either bringing ascospores to germinate, or by stimulating asexual strains to produce ascomata, respectively. Today, the connection between the ascigerous state in nature and the conidial state in culture can be verified by sequence data. Remarkably, Haase et al. (1999), as confirmed in subsequent studies, noted that only very few of the sexual strains brought into culture appeared to match with any of the numerous available names of asexual herpotrichiellaceous species. In our data, only possible connections between Capronia coronata/Exophiala angulospora and Capronia semiimmersa/Phialophora americana have been confirmed by sequencing. This suggests a preponderance of clonal reproduction as a survival strategy in Herpotrichiellaceae.
The disruptive scientific history of Chaetothyriales provides an unbalanced view on the order, due to current accent on molecular data. As a result, most molecular studies focus on Herpotrichiellaceae at the expense of other families within the order. Generic circumscriptions before the year 2000 have been phenotypic, while simple forms of asexual sporulation such as catenate cladophialophora-like conidia are now known to occur widely throughout the order. For example, members of Cladophialophora (Feng et al. 2013) can be found scattered in four families, Herpotrichiellaceae, Trichomeriaceae, Epibryaceae and Clade 4. Many morphological genera thus are obviously polyphyletic. Novel species are introduced at a regular pace, phylogenetic trees suffering from a significant taxon sampling problem, and therefore redefinition of genera is recommended to be postponed until a more complete overview of extant and still-to-be-described species is obtained.
Origin and evolution of Chaetothyriales
Judging from results of divergence time estimations (Fig. 1), the order Chaetothyriales emerged about 387 Mya, during the end of Devonian (416–359 Mya), but the speciation events occurred in the Jurassic (201–145 Mya), which was initiated by the major Triassic-Jurassic extinction event, possibly as a result of rapid climate change due to volcanism or methane production during the active split into continental plates of the ancient Pangea continent (Ivanov 2007). During the early period, animal and plant life on earth became very scarce. Our hypothesis is that for a long time, the ancestral Chaetothyriales colonized rock surfaces, and under these extreme and oligotrophic conditions grew slowly without much diversification.
A significant change in diversity in Chaetothyriales was observed around 151 Mya. One possible hypothesis is that the interaction of Chaetothyriales with toxin-containing lichens and Cyanophytes on rock became a driver towards toxin-management, which opened other windows of opportunity. As a result of the subsequent Cretaceous-Paleogene extinction event (66 Mya), global species diversity greatly declined again, resulting in many vacant ecological niches. It took a long time for the ecosystem to restore general diversity (MacLeod et al. 1997; Wilf and Johnson 2004), but the Chaetothyriales, which were not significantly impacted by the extinction event, began an explosion of diversification.
Ancestral groups of Chaetothyriales had an epilithic, lichen-associated strategy (Muggia et al. 2020). This is in line with earlier assumptions, where black lichenized fungi of Verrucariales were listed in ancestral position to Chaetothyriales (Gueidan et al. 2008, 2011). Several clades with identical non-lichenized, endolichenic lifestyles (Clades 8, 5 and 9) emerged, including the basal clade, oldest clade (Clade 9). We speculate that the oldest Chaetothyriales co-evolved with lichens that live on the surface of rocks. Metabolic products are accumulated in the lichen thallus during the growth, known as lichenic acid or lichen substances (Barnes 2000). Usnic acid, a dibenzofuran derivative, is one of the most common and abundant lichen metabolites (Cochietto et al. 2002). The Cyanobacteria that occur in the lichens as photosynthetic accessory contain other toxins, such as microcystins (Oksanen et al. 2004) which are larger molecules containing benzene rings. Early fungi living under these conditions must have the ability to tolerate or to degrade these chemicals.
Cytochrome p450 genes (CYPs) play a fundamental role in primary, secondary, and xenobiotic metabolism (van den Brink et al. 1998). Some black yeasts are among the Ascomycota species with the highest number of CYPs (Teixeira et al. 2017). Also genes related to alcohol dehydrogenase (ADH), aldehyde dehydrogenase (ALDHs) and drug efflux pumps were copied in large quantity, which helps the black fungus adapt to the toxic environment better. The gene replications may have become the basis of metabolic versatility observed in modern black yeast. These genetic adaptations acquired in the common ancestor of the studied species are maintained throughout the evolution of Chaetothyriales. The apparent rapid explosion of diversification in the order, underlined by the low rate of extinction and giving rise to all within a very short time frame (Fig. 8) certainly has contributed to opening of numerous windows of opportunity for members of the order.
Chaetothyrialean main families
Herpotrichiellaceae is the largest family within the order, containing 19 recognized genera and 179 species (as per 01-10-2019); 117 species were analyzed in this study. Excluded species were those without known ITS or LSU sequences, or with obviously incorrect sequences as concluded from large distances to any of the taxa in the chaetothyrialean tree. In all trees, species published as being members of the family Herpotrichiellaceae showed instability and low bootstrap values with different algorithms. In the single-gene LSU or ITS trees, the family was not supported but deteriorated into many subclades. With LSU + ITS, they clustered together but the bootstrap values remained relatively low.
In search of common ecological features for Herpotrichiellaceae, Gostinčar et al. (2018) referred to these fungi as being polyextremotolerant, i.e. combining tolerance of e.g. temperature, dryness, toxin, and nutrient limitation. Numerous, as yet undescribed members of the family are epilithic, colonizing hard, sun-exposed rock (Ruibal et al. 2008). The medical counterpart of the meristematic ecotype on exposed habitats is the muriform cell formed in tissue of patients with chromoblastomycosis, but members of the family are known from a plethora of opportunistic diseases (de Hoog et al. 2019). Species are notoriously difficult to isolate from natural environments (Sudhadham et al. 2008; Vicente et al. 2014), but are enriched in human-created habitats, such as oil-contaminated soil (Prenafeta-Boldú et al. 2001), creosoted railway sleepers (Gümral et al. 2014), gasoline (Isola et al. 2013), dishwashers (Raghupathi et al. 2018), bathing facilities (Matos et al. 2002), or household sinks (Nishimura et al. 1987). These environments suggest, in line with suggestions of Gostincar et al. (2018), oligotrophy in addition to extremotolerance and toxin management. Infective ability seems to be consistently present in the family, as waterborne species without thermotolerance infect numerous cold-blooded vertebrates instead of humans (de Hoog et al. 2011). Quan et al. (2019) developed an isolation method based on enrichment with hydrocarbons, underlining the significance of toxicity in the biology of these fungi, while earlier authors successfully implemented experimental inoculation of environmental samples into laboratory animals (Gezuele et al. 1972; Dixon et al. 1980), high incubation temperature and low pH (Sudhadham et al. 2008), extraction with mineral oil (Satow et al. 2008; Vicente et al. 2008), and cycloheximide as suppressor of contaminants (Wang et al. 2018). Generally, low competitive ability with co-occurring saprobes has been hypothesized, as a result of which they prevalently occupy (micro-)habitats that are hostile to microbial life and are inaccessible for their competitors (Gueidan et al. 2008).
The Cyphellophoraceae were previously known as the ‘europaea-clade’ within Herpotrichiellaceae (de Hoog et al. 2011) and were raised to family level by Réblová et al. (2013). As yet, insufficient data are available to recognize an unambiguous ecological trend in the family. Several species are known from individual reports from plants, but mostly without clear information on the type of growth, whether as an endophyte, a pathogen, or a colonizer. Cyphellophora europaea is a common species causing mild infections on human skin and nails (de Hoog et al. 2000). Lian and de Hoog (2010) hypothesized a life style as an oligotrophic colonizer of moist, warm environments, where it could accidentally be picked up by human hosts. This might also hold true for other clinical Cyphellophora species.
The clade representing Trichomeriaceae contained 48 strains, in addition to some undescribed species isolated from ant nest carton and four species of the genus Strelitziana described from living plants. Strelitziana australiensis, S. eucalypti, S. albiziae and S. cliviae were all named after their host plants and have been classified in a separate family, Strelitzianaceae (Crous et al. 2015). In our study, these species cluster in the Trichomeriaceae clade albeit at rather long branches; this result is different from previous studies (Cheewangkoon et al. 2009; Crous et al. 2010) and is possibly explained by our larger dataset. Morphologically, the asexual states of the species resemble those of Cyphellophora, and their growth on decaying leaves are not suggestive for primary pathogenicity. Thus we have reason to believe that no separate family status is necessary and the genus Strelitziana might well be maintained in the family Trichomeriaceae. Members of this family are ‘sooty moulds’ colonizing inert substrates such as leathery plants (Chomnunti et al. 2014), while Knufia and relatives contains rock-colonizing species (Isola et al. 2016).
Chaetothyriaceae is the type family of the order Chaetothyriales. Although the family is monophyletic, bootstrap values were relatively low, and distances between taxa rather large. This clearly demonstrates a taxon sampling error, as explained above. The majority of species has been isolated from plants, with small populations as colonizers without significant invasion of living tissue. Their epiphytic life style is not associated with significant disease (Crous et al. 2006; Crous et al. 2007; Gueidan et al. 2014; Hongsanan et al. 2016b).
Clade 4 mainly consists of ant-domatia colonizing species. The pronounced ability to metabolize monoaromatic hydrocarbons explained the overabundance of members of Herpotrichiellaceae in human-dominated environments; in nature, this may be an association with ants (Voglmayr et al. 2011, Nepel et al. 2014), as these insects communicate with similar compounds. Their nest materials of cartons, tunnels or domatia are antimicrobial. Schlick-Steiner et al. (2008) was the first to find a relationship between ants and Chaetothyriales. Defossez et al. (2009) revealed a symbiotic tripartite of domatia-forming plant, ant and fungus. In our study, the domatia fungi all belong to a single, novel clade. Mayer and Voglmayr (2009), Voglmayr et al. (2011), Nepel et al. (2014) and Vasse et al. (2017) revealed a stunning biodiversity, of which the carton-associated species were divided over families Herpotrichiellaceae, Trichomeriaceae and Cyphellophoraceae. Attili-Angelis et al. (2014) described some species in Cyphellophoraceae from leaf-cutting ants in Brazil. Probably only a fraction of these fungi is known, because there are thousands of tropical ants which build specific tunnel structures which may carry ant-specific black yeasts.
Ancestral character state reconstruction
Phenotypic plasticity (i.e. the sum of morphotypes and growth abilities) enables a fungus to respond differentially to novel environmental conditions. Most organisms are able to survive outside their original and preferred habitat. In the theory of ecological fitting (Janzen 1985), this operational environment is known as the ‘sloppy fitness space’ (Agosta and Klemens 2008). Under conditions of survival stress, adaptation of the organism is promoted, whereas in absence thereof, or with populations occupying different environments and connected by gene flow, and thus being subjected to heterogeneous pressure, more likely leads to evolutionary stasis.
Brownian motion is an effective model as the sum of a large number of very small, random forces relative to the given trait changes. The wide species richness, the diverse and fairly consistent ecotypes and the size of our dataset provides an opportunity for modelling evolutionary transitions in comparison with the evolutionary timing of Chaetothyriales since its origin (Fig. 1). To this aim, ecological traits are plotted along phenogram branches via stochastic character mapping, revealing which traits in a population follow a uniform pattern, and which may have evolved as products of other traits and or transitions. While ecological disparity peaked very early in Chaetothyriales evolutionary history, interestingly a general trend in biology (e.g. as for animals and plants; Harmon et al. 2012; Hughes et al. 2013), it logically leads to a strong decline in occupying new niches given the combinations of ancestral phenotypes and associated genetic abilities. While a Brownian process by means of evolutionary history is driven by two indicators of a clade’s success, i.e. diversity as measured by the number of species, and disparity which is an estimate of the lineage’s occupancy of a defined ecological space (Minelli 2016; Foote 1997; McGhee 1999; Wills 2001; Erwin 2007), these attributes are not mutually exclusive. Success in disparity does not necessarily go together with success in diversity.
Ancestral, epilithic Chaetothyriales, possibly preceded by rock-colonizing groups such as Pleostigmaceae (Muggia et al. 2020) colonized harsh and extreme environments, with low diversity, while significant disparity is observed with the abrupt evolution of e.g. ‘domatia’ and ‘bryophytic’ ecotypes in highly derived lineages. After these innovations, disparity decreased, but with explosive diversification in similar habitats. As a general rule many large organism genera show high diversity with low disparity, while others are highly diverse but also exhibit high disparity, the latter being an important attribute to the Chaetothyriales (= early high disparity burst lead to large diversity). The largest taxonomic genera are often characterized by key innovations that often, but not necessarily, coincide with their diagnostic apomorphies. A key principle in the black fungi strongly supported by our data (=ecologies).
How is the evolution of fungal clades with high diversity and/or disparity be explained? It can be addressed from three main perspectives: (1) evolvability, in terms of release from previous constraints and of the presence of genetic or developmental conditions favoring multiple parallel occurrences of a given evolutionary transition and its reversal (clearly supported by the Brownian analysis); (2) phenotypic plasticity as a facilitator of speciation; and (3) modularity, heterochrony and a coupling between the complexity of the life cycle and the evolution of diversity and disparity in a clade. The possible role of saltational evolution in the origination of high diversity and/or disparity (eg. when considering our transition matrix results, indicating highly abrupt evolution of eg. ‘domatia’ or ‘bryophytic’ ecologies) in Chaetothyriales needs to be further explored. While under the simplest conditions (neutral genetic drift) there is no relationship between evolutionary rate and phylogenetic signal however, such a relationship can exist when evolution is not entirely neutral. For other circumstances, such as functional constraint, fluctuating selection, niche conservatism, and evolutionary heterogeneity, the relationship between process, rate, and phylogenetic signal is complex. While our data precisely reflects this complex case, it is due to its completeness an example for macroevolutionary modelling and stochastical effect discovery in kingdom fungi.
Conclusions
Numerous fungi with rock-inhabiting life styles have been described, but the Chaetothyriales are special by representatives with intimate relationships with lichens, which led to expansion of cytochromes providing windows of opportunity for diversification. Colonization of toxic environments is an alternative way to escape microbial competition. Toxic hydrocarbons are found in nature e.g. in ant-dominated habitats. In human-made environments, toxic hydrocarbons are present as compounds of oil- and gasoline-pollution and industrial exhaust of xenobiotic volatiles. Members of Herpotrichiellaceae have been proposed as agents of bioremediation, for example in industrial biofilters, where they survive the conditions of acidification and dryness much better than the bacteria that are currently used in air clean-up (Cox et al. 1993; Groenestijn and Kraakman 2005). The vertebrate nervous system also contains aromatic hydrocarbon neurotransmitters, which might be used by black yeasts once they are introduced into the human body. This would explain opportunism of these fungi, in analogy of hypotheses put forward in Cryptococcus (Esher et al. 2018). A summarizing diagram of the possible lines of adaptation in Chaetothyriales leading to species-rich families is given in Fig. 9.

Diagrammatic representation of evolution of Chaetothyriales over geological times. Epilithic lifestyle is ancestral, possibly emerging from groups with uncertain phylogenetic position such as Pleostigmaceae. Ecological disparity increased at an early stage, followed by diversification with several ecotypes shortly after each other and at low levels of innovation as expressed by Brownian rates (Table 4). The red lines indicate multiple origins
References
Ackerly D (2009) Conservatism and diversification of plant functional traits: evolutionary rates versus phylogenetic signal. Proc Natl Aacad Sci USA 106(2):19699–19706
Akaike H (1974) A new look at the statistical model identification. Selected papers of hirotugu akaike. Springer, New York, pp 215–222
Arzanlou M, Crous P (2006) Strelitziana africana. Fungal Planet 8:2
Arzanlou M, Groenewald J, Gams W, Braun U, Shin H-D, Crous PW (2007) Phylogenetic and morphotaxonomic revision of Ramichloridium and allied genera. Stud Mycol 58:57–93
Attili-Angelis D, Duarte A, Pagnocca F, Nagamoto N, de Vries M, Stielow JB, de Hoog GS (2014) Novel Phialophora species from leaf-cutting ants (tribe Attini). Fung Div 65(1):65–75
Badali H, Gueidan C, Najafzadeh M, Bonifaz A, Gerrits van den Ende AHG, de Hoog GS (2008) Biodiversity of the genus Cladophialophora. Stud Mycol 61:175–191
Badali H, Bonifaz A, Barrón-Tapia T, Vázquez-González D, Estrada-Aguilar L, Cavalcante Oliveira N, Sobral Filho J, Guarro J, Meis J, De Hoog G (2010) Rhinocladiella aquaspersa, proven agent of verrucous skin infection and a novel type of chromoblastomycosis. Med Mycol 48(5):696–703
Barnes J (2000) Pharmacognosy in the 21st century. Pharm J 264:701–703
Barr ME (1976) Perspectives in the Ascomycotina. Mem New York Bot Gard 28:1–8
Barr ME (1979) A classification of Loculoascomycetes. Mycologia 71(5):935–957
Barr ME (1987a) New taxa and combinations in the Loculoascomycetes. Mycotaxon 29:501–505
Barr ME (1987b) Prodromus to class Loculoascomycetes. Barr, Amherst
Barr ME (1991) Notes on and additions to North American members of the Herpotrichiellaceae. Mycotaxon 41(2):419–436
Barr M, Makkai M (1987) On representations of Grothendieck toposes. Can J Math 39(1):168–221
Batista AC (1956) Systematic revision of the genera Ellisiella Sacc. and Ellisiellina Da Camara and the new genus Ellisiopsis. Anais Soc Biol Pernambuco 14:16–25
Batista AC (1960) Lembopodia, Yamamotoa e Peresiopsis, novos gêneros de Asterinaceae. Publ Inst Micol Univ Recife 291:1–35
Batista AC, Ciferri R (1962) The chaetothyriales. Beih. Sydowia 3:1–129
Berlese AN (1896) Icones Fungorum (Abellini) 2(2–3):62
Berlese AN (1899) Icones Fungorum (Abellini) 2(4–2):177
Blomberg SPT, Garland JR, Ives AR (2003) Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57:717–745
Campbell V, Legendre P, Lapointe F-J (2011) The performance of the congruence among distance matrices (CADM) test in phylogenetic analysis. BMC Evol Biol 11(1):64
Cheewangkoon R, Groenewald J, Summerell B, Hyde K, To-Anun C, Crous PW (2009) Myrtaceae, a cache of fungal biodiversity. Persoonia 23:55–85
Chomnunti P, Schoch CL, Aguirre-Hudson B, Ko-Ko TW, Hongsanan S, Jones EG, Kodsueb R, Phookamsak R, Chukeatirote E, Bahkali AH (2011) Capnodiaceae. Fungal Divers 51(1):103–134
Chomnunti P, Bhat D, Jones EG, Chukeatirote E, Bahkali AH, Hyde KD (2012a) Trichomeriaceae, a new sooty mould family of Chaetothyriales. Fung Div 56(1):63–76
Chomnunti P, Ko TWK, Chukeatirote E, Hyde KD, Cai L, Jones EG, Kodsueb R, Hassan BA, Chen H (2012b) Phylogeny of Chaetothyriaceae in northern Thailand including three new species. Mycologia 104(2):382–395
Chomnunti P, Bhat D, Jones EG, Chukeatirote E, Bahkali AH, Hyde KD (2012c) Trichomeriaceae, a new sooty mould family of Chaetothyriales. Fungal Divers 56(1):63–76
Chomnunti P, Hongsanan S, Aguirre-Hudson B, Tian Q et al (2014) The sooty moulds. Fung Div 66(1):1–36
Cocchietto M, Skert N, Nimis P, Sava G (2002) A review on usnic acid, an interesting natural compound. Naturwissenschaften 89(4):137–146
Constantinescu O, Holm K, Holm M (1989) Teleomorph-anamorph connections in Ascomycetes. 1-3. Stanhughesia (Hyphomycetes) new genus, the anamorph of Ceramothyrium. Stud Mycol 31:69–84
Cox HHJ, Houtman JHM, Doddema HJ, Harder W (1993) Enrichment of fungi and degradation of styrene in biofilters. Biotechnol Lett 15:737–742
Crous P, Groenewald J (2011) Why everlastings don’t last. Persoonia 26:70
Crous PW, Schroers H-J, Groenewald JZ, Braun U, Schubert K (2006) Metulocladosporiella gen. nov. for the causal organism of Cladosporium speckle disease of banana. Mycol Res 110(3):264-275.
Crous PW, Schubert K, Braun U, de Hoog GS et al (2007) Opportunistic, human-pathogenic species in the Herpotrichiellaceae are phenotypically similar to saprobic or phytopathogenic species in the Venturiaceae. Stud Mycol 58:185–217
Crous PW, Braun U, Wingfield MJ, Wood AS et al (2009) Phylogeny and taxonomy of obscure genera of microfungi. Persoonia 788(22):139–161
Crous PW, Barreto RW, Alfenas AC, Alfenas RF, Groenewald JZ (2010) What is Johansonia? IMA Fungus 1(2):117–122
Crous PW, Shivas R, Wingfield MJ, Summerell BA, Rossman AY, Alves J, Adams G, Barreto R, Bell A, Coutinho M (2012) Fungal Planet description sheets: 128–153. Persoonia 29:146
Crous PW, Wingfield MJ, Guarro J, Cheewangkoon R, Van der Bank M, Swart W, Stchigel A, Cano-Lira J, Roux J, Madrid H (2013) Fungal Planet description sheets: 154–213. Persoonia 31:188
Crous PW, Shivas R, Quaedvlieg W, Van der Bank M et al (2014) Fungal planet description sheets: 214–280. Persoonia 32:184
Crous PW, Wingfield MJ, Guarro J, Hernández-Restrepo M et al (2015) Fungal planet description sheets: 320–370. Persoonia 34:167
Crous PW, Wingfield MJ, Burgess TI, Hardy GSJ et al (2016) Fungal planet description sheets: 469–557. Persoonia 37:218
Crous PW, Wingfield MJ, Burgess TI, Carnegie AJ, Hardy GSJ, Smith D, Summerell BA, Cano-Lira JF, Guarro J, Houbraken J (2017) Fungal Planet description sheets: 625–715. Persoonia 39:270
Crous PW, Luangsa-ard J, Wingfield M, Carnegie A et al (2018) Fungal planet description sheets: 785–867. Persoonia 41:238
Crous PW, Schumacher RK, Akulov A, Thangavel R, Hernández-Restrepo M, Carnegie A, Cheewangkoon R, Wingfield MJ, Summerell BA, Quaedvlieg W (2019) New and interesting fungi 2. Fungal Syst Evol 3:57
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nature Meth 9(8):772
Das K, Lee S-Y, Jung H-Y (2019) Cladophialophora lanosa sp. nov., a New Species Isolated from Soil. Mycobiology 47(2):173–179
Davey ML, Currah RS (2007) A new species of Cladophialophora (hyphomycetes) from boreal and montane bryophytes. Mycol Res 111(1):106–116
de Azevedo CM, Gomes RR, Vicente VA, Santos DW, Marques SG, do Nascimento MM, Andrade CE, Silva RR, Queiroz-Telles F, de Hoog GS (2015) Fonsecaea pugnacius, a novel agent of disseminated chromoblastomycosis. J Clin Microbiol 53(8):2674–2685
Decock C, Delgado-Rodríguez G, Buchet S, Seng J (2003) A new species and three new combinations in Cyphellophora, with a note on the taxonomic affinities of the genus, and its relation to Kumbhamaya and Pseudomicrodochium. Antonie van Leeuwenhoek 84(3):209
Defossez E, Selosse MA, Dubois MP, Mondolot L et al (2009) Ant-plants and fungi: a new threeway symbiosis. New Phytol 182(4):942–949
de Hoog GS (1977) Rhinocladiella and allied genera. Stud Mycol 15:1–140
de Hoog GS, Mayser P, Haase G, Horré R, Horrevorts A (2000) A new species, Phialophora europaea, causing superficial infections in humans. Mycoses 43(11–12):409–416
De Hoog G, Vicente V, Caligiorne R, Kantarcioglu S, Tintelnot K, van den Ende AG, Haase G (2003) Species diversity and polymorphism in the Exophiala spinifera clade containing opportunistic black yeast-like fungi. J Clin Microbiol 41(10):4767–4778
De Hoog G, Nishikaku A, Fernandez-Zeppenfeldt G, Padín-González C, Burger E, Badali H, Richard-Yegres N, van den Ende AG (2007) Molecular analysis and pathogenicity of the Cladophialophora carrionii complex, with the description of a novel species. Stud Mycol 58:219–234
de Hoog GS, Vicente VA, Najafzadeh M, Harrak M, Badali H, Seyedmousavi S (2011) Waterborne Exophiala species causing disease in cold-blooded animals. Persoonia 27:46–72
de Hoog GS, Guarro J, Gené J, Ahmed SA, Al-Hatmi AMS et al (2019) Atlas of clinical fungi. Web-edition, Utrecht
de Vries GA (1962) Cyphellophora laciniata nov. gen., nov. sp. and Dactylium fusarioides Fragoso et Ciferri. Mycopath Mycol Appl 16(1):47–54
Diederich P, Ertz D, Lawrey JD, Sikaroodi M, Untereiner WA (2013) Molecular data place the hyphomycetous lichenicolous genus Sclerococcum close to Dactylospora (Eurotiomycetes) and S. parmeliae in Cladophialophora (Chaetothyriales). Fungal Divers 58(1):61–72
Dixon DM, Shadomy H, Shadomy S (1980) Dematiaceous fungal pathogens isolated from nature. Mycopathologia 70(3):153–161
Dobbeler P (1978) Moosbewohnende ascomyceten. I. Die pyrenocarpen, Den Gametophyten Besiedelnden Arten
Döbbeler P (1980) Epibryon endocarpum sp. nov. (Dothideales), ein hepaticoler Ascomycet mit intrazellulären Fruchtkörpern. Z Mykol 46:209–216
Döğen A, Ilkit M, De Hoog GS (2013) Black yeast habitat choices and species spectrum on high altitude creosote-treated railway ties. Fungal Biol 117(10):692–696
Dong W, Hyde KD, Bhat DJ, Zhang H (2018) Introducing Aculeata aquatica gen. et sp. nov., Minimelanolocus thailandensis sp. nov. and Thysanorea aquatica sp. nov. (Herpotrichiellaceae, Chaetothyriales) from freshwater in northern Thailand. Mycol Progr 17(5):617–629
Erwin DH (2007) Disparity: morphologic pattern and developmental context. Palaeontology 50:57–73
Esher SK, Zaragoza O, Alspaugh JA (2018) Cryptococcal pathogenic mechanisms: a dangerous trip from the environment to the brain. Mem Inst Oswaldo Cruz. 113(7):e180057
Feng P, Vicente VA, Najafzadeh MJ, Gerrits van den Ende AHG et al (2013) Cladophialophora abundans, a novel environmental species in the Chaetothyriales. Mycol Prog 13(2):381–391
Feng P, Lu Q, Najafzadeh MJ, Gerrits van den Ende AHG et al (2014) Cyphellophora and its relatives in Phialophora: Biodiversity and possible role in human infection. Fung Div 65:17–45
Flakus A, Farkas E (2013) A contribution to the taxonomy of Lyromma (Lyrommataceae, lichenized Ascomycota) with a species key. Mycotaxon 124(1):127–141
Foote M (1997) The evolution of morphological diversity. Ann Rev Ecol Syst 28:129–152
Friebes G (2012) A key to the non-lichenicolous species of the genus Capronia (Herpotrichiellaceae). Ascomycete.org 4(3):55-64
Gao L, Ma Y, Zhao W, Wei Z, Gleason ML, Chen H, Hao L, Sun G, Zhang R (2015) Three new species of Cyphellophora (Chaetothyriales) associated with sooty blotch and flyspeck. PLoS ONE 10(9):e0136857
Gezuele E, Mackinnon J, Conti-Diaz I (1972) The frequent isolation of Phialophora verrucosa and Phialophora pedrosoi from natural sources. Sabouraudia 10(3):266–273
Gjessing MC, Davey M, Kvellestad A, Vrålstad T (2011) Exophiala angulospora causes systemic inflammation in Atlantic cod Gadus morhua. Dis Aquat Org 96(3):209–219
Gomes RR, Vicente VA, Azevedo CMd, Salgado CG, da Silva MB, Queiroz-Telles F, Marques SG, Santos DW, de Andrade TS, Takagi EH (2016) Molecular epidemiology of agents of human chromoblastomycosis in Brazil with the description of two novel species. PLoS Negl Trop Dis 10(11):e0005102
Gostinčar C, Zajc J, Lenassi M, Plemenitaš A et al (2018) Fungi between extremotolerance and opportunistic pathogenicity on humans. Fung Div 93(1):195–213
Gostinčar C, Turk M, Zajc J, Gunde-Cimerman (2019) Fifty Aureobasidium pullulans genomes reveal a recombining polyextremotolerant generalist. Environ Microbiol 21(10):3638–3652
Groenestijn JW, Kraakman NJR (2005) Recent developments in biological waste gas purification in Europe. Chem Eng J 113:85–91
Gueidan C, Ruibal C, de Hoog GS, Gorbushina A, Untereiner W, Lutzoni F (2008) A rock-inhabiting ancestor for mutualistic and pathogen-rich fungal lineages. Stud Mycol 61:111–119
Gueidan C, Ruibal C, de Hoog GS, Schneider H (2011) Rock-inhabiting fungi originated during periods of dry climate in the late Devonian and middle Triassic. Mycol Res 115:987–996
Gueidan C, Aptroot A, da Silva Cáceres ME, Badali H, Stenroos S (2014) A reappraisal of orders and families within the subclass Chaetothyriomycetidae (Eurotiomycetes, Ascomycota). Mycol Prog 13(4):1027–1039
Gümral R, Tümgör A, Saraçlı MA, Yıldıran ŞT, Ilkit M, de Hoog GS (2014) Black yeast diversity on creosoted railway sleepers changes with ambient climatic conditions. Microbial Ecol 68(4):699–707
Haase G, Sonntag L, Melzer-Krick B, de Hoog GS (1999) Phylogenetic inference by SSU gene analysis of members of the Herpotrichiellaceae, with special reference to human pathogenic species. Stud Mycol 43:80–97
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hansford CG (1946) The foliicolous ascomycetes, their parasites and associated fungi. Mycol Pap 15:1–240
Harmon LJ, Schulte JA, Larson A, Losos JB (2012) Tempo and mode of evolutionary radiation in inguanian lizards. Science 301:961–964
Hawksworth D, Dyko B (1979) Lichenodiplis and Vouauxiomyces: two new genera of lichenicolous coelomycetes. The Lichenologist 11(1):51–61
Hennings PC (1905) Fungi Africae orientalis IV. Bot Jahrb Syst Pflges Pflgeographie 38:102–118
Hermanides-Nijhof (1977) Aureobasidium and allied genera. Stud Mycol 15:141–177
Hernández-Restrepo M, Schumacher RK, Wingfield MJ, Ahmad I, Cai L, Duong TA, Edwards J, Gene J, Groenewald JZ, Jabeen S (2016) Fungal systematics and evolution: FUSE 2.
Herrera-Campos M, Huhndorf S, Lücking R (2005) The foliicolous lichen flora of Mexico IV: a new, foliicolous species of Pyrenothrix (Chaetothyriales: Pyrenothrichaceae). Mycologia 97(2):356–361
Hongsanan S, Hyde KD, Bahkali AH, Camporesi E, Chomnunti P, Ekanayaka H, Gomes AA, Hofstetter V, Jones EG, Pinho DB (2015a) Fungal biodiversity profiles 11–20. Cryptogam Mycol 36(3):355–380
Hongsanan S, Tian Q, Hyde K, Chomnunti P (2015b) Two new species of sooty moulds, Capnodium coffeicola and Conidiocarpus plumeriae in Capnodiaceae
Hongsanan S, Tian Q, Hyde KD, Hu D (2016a) The asexual morph of Trichomerium gloeosporum. Mycosphere 7(9):1473–1479
Hongsanan S, Sánchez-Ramírez S, Crous PW, Ariyawansa HA et al (2016b) The evolution of fungal epiphytes. Mycosphere 7:1690–1712
Hubka V, Réblová M, Řehulka J, Selbmann L, Isola D, de Hoog SG, Kolařík M (2014) Bradymyces gen. nov. (Chaetothyriales, Trichomeriaceae), a new ascomycete genus accommodating poorly differentiated melanized fungi. Antonie van Leeuwenhoek 106(5):979–992
Hughes M, Gerber S, Wills MA (2013) Clades reach highest morphological disparity early in their evolution. Proc Natl Acad Sci USA 110(34):13875–13879
Hulsenbeck JP, Nielsen R, Bollback JP (2003) Stochastic mapping of morphological characters. Syst Biol 52(2):131–158. https://doi.org/10.1080/10635150390192780
Hyde KD, Jones EG, Liu J-K, Ariyawansa H et al (2013) Families of dothideomycetes. Fung Div 63(1):1–313
Hyde KD, Norphanphoun C, Chen J, Dissanayake AJ et al (2018) Thailand’s amazing diversity – up to 96% of fungi in northern Thailand are novel. Fung Div 93:215–239
Isola D, Selbmann L, de Hoog GS, Fenice M et al (2013) Isolation and screening of black fungi as degraders of volatile aromatic hydrocarbons. Mycopathologia 175(5–6):369–379
Isola D, Zucconi L, Onofri S, Caneva G, de Hoog GS, Selbmann L (2016) Extremotolerant rock inhabiting black fungi from Italian monumental sites. Fung Div 76(1):75–96
Ivanov AV (2007) Evaluation of different models for the origin of the Siberian traps. In: Foulger GR, Jurdy DM (eds) Plates, plumes and planetary processes. Geol. Soc, Boulder
Iwatsu T, Udagawa S, Toyazaki N (1988) A new species of Phialophora. Mycotaxon 32:439–445
Kirk PM, Cannon PF, David J, Stalpers JA (2001) Ainsworth and Bisby’s dictionary of the fungi, 9th edn. CABI Publishing, Wallingford
Kiyuna T, An KD, Kigawa R, Sano C, Sugiyama J (2018) Two new Cladophialophora species, C. tumbae sp. nov. and C. tumulicola sp. nov., and chaetothyrialean fungi from biodeteriorated samples in the Takamatsuzuka and Kitora Tumuli. Mycoscience 59(1):75–84
Koukol O (2010) Revision of “Septonema ochraceum” revealed three new species of Venturiaceae and Herpotrichiellaceae. Mycol Prog 9(3):369–378
Kumar S, Stecher G, Peterson D, Tamura K (2012) MEGA-CC: computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics 28(20):2685–2686
Legendre P, Lapointe FJ (2004) Assessing congruenceamong distance matrices: single—malt scotch whiskies revisited. Austral N Z J Statistics 46(4):615–629
Lian X, de Hoog GS (2010) Indoor wet cells harbour melanized agents of cutaneous infection. Med Mycol 48(4):622–628
Liu JK, Hyde KD, Jones EG, Ariyawansa HAB et al (2015) Fungal diversity notes 1–110: taxonomic and phylogenetic contributions to fungal species. Fung Div 72(1):1–197
Liu J-K, Hyde KD, Jeewon R, Phillips AJL et al (2017) Ranking higher taxa using divergence times: a case study in Dothideomycetes. Fung Div 84:75–99
Li DM, de Hoog G, Saunte DL, Gerrits van den Ende AHG, Chen X (2008) Coniosporium epidermidis sp. nov., a new species from human skin. Stud Mycol 61:131–136
Li Y, Xiao J, De Hoog G, Wang X, Wan Z, Yu J, Liu W, Li R (2017) Biodiversity and human-pathogenicity of Phialophora verrucosa and relatives in Chaetothyriales. Persoonia 38:1
Li J, Jeewon R, Phookamsak R, Bhat DJ, Mapook A, Chukeatirote E, Hyde KD, Lumyong S, Mckenzie EH (2018) Marinophialophora garethjonesii gen. et sp. nov.: a new hyphomycete associated with Halocyphina from marine habitats in Thailand. Phytotaxa 345(1):1–12
Ma Y-R, Xia J-W, Gao J-M, Li X-Y et al (2015) Atrokylindriopsis, a new genus of hyphomycetes from Hainan, China, with relationship to Chaetothyriales. Mycol Prog 14(9):77
Maciá-Vicente JG, Glynou K, Piepenbring M (2016) A new species of Exophiala associated with roots. Mycol Prog 15(2):18
MacLeod KG, Huber BT, Ward PD (1997). The biostratigraphy and paleobiogeography of Maastrichtian inoce-ramids. In: Ryder G, Fastovsky D, Gartner S (eds.) The cretaceous-tertiary event and other catastrophes in earth history. Geol Soc America Spec Pap 307:361-373
Maddison W, Maddison D (2007) Mesquite: a modular system for evolutionary analysis. http://mesquiteproject.org.
Madrid H, Hernandez-Restrepo M, Gené J, Cano J, Guarro J, Silva V (2016) New and interesting chaetothyrialean fungi from Spain. Mycol Prog 15(10–11):1179–1201
Maharachchikumbura S, Haituk S, Pakdeeniti P, Al-Sadi A et al (2018) Phaeosaccardinula coffeicola and Trichomerium chiangmaiensis, two new species of Chaetothyriales (Eurotiomycetes) from Thailand. Mycosphere 9(4):769–778
Marincowitz S, Crous PW, Groenewald JZ, Wingfield MJ (2008) Microfungi occurring on Proteaceae in the fynbos. CBS Biodivers Ser
Matos T, de Hoog GS, de Boer A, de Crom I, Haase G (2002) High prevalence of the neurotrope Exophiala dermatitidis and related oligotrophic black yeasts in sauna facilities. Mycoses 45(9–10):373–377
Mayer VE, Voglmayr H (2009) Mycelial carton galleries of Azteca brevis (Formicidae) as a multi-species network. Proc R Soc B 276(1671):3265–3273
McGhee GR (1999) Theoretical morphology: the concept and its applications. Columbia University Press, New York
Minelli A (2016) Species diversity vs. morphological disparity in the light of evolutionary developmental biology. Ann Bot 117:781–794
Moreno LF, Vicente VA, de Hoog GS (2018) Black yeasts in the omics era: achievements and challenges. Med Mycol 56(Suppl 1):S32–S41
Moussa TA, Al-Zahrani HS, Kadasa NM, Moreno LF et al (2017a) Nomenclatural notes on Nadsoniella and the human opportunist black yeast genus Exophiala. Mycoses 60(6):358–365
Moussa TA, Gerrits van den Ende BH, Al Zahrani HS, Kadasa NM, de Hoog SG, Dolatabadi S (2017b) The genus Anthopsis and its phylogenetic position in Chaetothyriales. Mycoses 60(4):254–259
Moustafa A, Abdul-Wahid O (1990) Veronaea constricta, a new hyphomycete from Egyptian soils. Mycotaxon 38:167–171
Muggia L, Kopun T, Ertz D (2015) Phylogenetic placement of the lichenicolous, anamorphic genus Lichenodiplis and its connection to muellerella-like teleomorphs. Fung Biol 119(11):1115–1128
Muggia L, Fleischhacker A, Kopun T, Grube M (2016) Extremotolerant fungi from alpine rock lichens and their phylogenetic relationships. Fung Div 76(1):119–142
Muggia L, Kopun T, Grube M (2017) Effects of growth media on the diversity of culturable fungi from lichens. Molecules 22:824
Muggia L, Quan Y, Gueidan C, Al-Hatmi AMS et al (2020) Ancestral Chaetothyriales with two novel lineages of lichen-associated fungi. Fung. Div, (in press)
Müller E, Petrini O, Fisher P, Samuels GJ, Rossman AY (1987) Taxonomy and anamorphs of the Herpotrichiellaceae with notes on generic synonymy. Trans Br Mycol Soc 88(1):63–74
Munk A (1953) The System of the Pyrenomycetes: a contribution to a natural classification of the group Sphaeriales sensu Lindau. Munksgaard, Copenhagen
Munkemueller T, Lavergne S, Bzeznik B, Dray S et al (2012) How to measure and test phylogenetic signal. Meth Ecol Evol 3:743–756
Nai C, Wong HY, Pannenbecker A, Broughton WJ, Benoit I, de Vries RP, Gueidan C, Gorbushina AA (2013) Nutritional physiology of a rock-inhabiting, model microcolonial fungus from an ancestral lineage of the Chaetothyriales (Ascomycetes). Fungal Genet Biol 56:54–66
Nascimento MM, Selbmann L, Sharifynia S, Al-Hatmi AM, Voglmayr H, Vicente VA, Deng S, Kargl A, Moussa TA, Al-Zahrani HS (2016) Arthrocladium, an unexpected human opportunist in Trichomeriaceae (Chaetothyriales). Fungal Biol 120(2):207–218
Nascimento MM, Vicente VA, Bittencourt JV, Gelinski JML, Prenafeta-Boldú FX, Romero-Güiza M, Fornari G, Gomes RR, Santos GD, Van Den Ende AG (2017) Diversity of opportunistic black fungi on babassu coconut shells, a rich source of esters and hydrocarbons. Fungal Biol 121(5):488–500
Nawawi A, Webster J (1982) Phalangispora constricta gen. et. sp. nov., a sporodochial hyphomycete with branched conidia. Trans Br Mycol Soc 79(1):65–68
Nepel M, Voglmayr H, Schönenberger J, Mayer VE (2014) High diversity and low specificity of Chaetothyrialean fungi in carton galleries in a Neotropical ant-plant association. PLoS ONE 9(11):e112756
Nishimura K, Miyaji M, Taguchi H, Tanaka R (1987) Fungi in bathwater and sludge of bathroom drainpipes. Mycopathologia 97(1):17–23
O’Meara BC, Ane C, Sanderson MJ, Wainwright PC (2006) Testing for different rates of continuous trait evolution using likelihood. Evolution 60:922–933
Oksanen I, Jokela J, Fewer DP, Wahlsten M et al (2004) Discovery of rare and highly toxic microcystins from lichen-associated cyanobacterium Nostoc sp. strain IO-102-I. Appl Environ Microbiol 70(10):5756–5763
Overy DP, Martin C, Muckle A, Lund L, Wood J, Hanna P (2015) Cutaneous phaeohyphomycosis caused by Exophiala attenuata in a domestic cat. Mycopathologia 180(3–4):281–287
Pagel M (1999) Inferring the historical patterns of biological evolution. Nature 401:877–884
Papendorf M (1969) New south african soil fungi. Transactions of the British Mycological Society 52(3):483-IN420
Papendorf M (1976) Notes on Veronaea including V compactci sp. nov. Bothalia 12(1):119–121
Paradis E, Schliep K (2019) APE 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35(3):526–528. https://doi.org/10.1093/bioinformatics/bty633
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20(2):289–290
Park M-J, Shin H-D (2011) Cladophialophora pucciniophila, a new hyphomycete parasitizing a rust fungus. Mycotaxon 116(1):449–456
Pennell MW, Eastman JM, Slater GJ, Brown JW et al (2014) Geiger v2.0: an expanded suite of methods for fitting macroevolutionary models to phylogenetic trees. Bioinformatics 30(15):2216–2218. https://doi.org/10.1093/bioinformatics/btu181
Petrak F (1914) Beiträge zur Pilzflora von Mähren und Österr.-Schlesien. Annls Mycol 19(56):273–295
Phookamsak R, Hyde KD, Jeewon R, Bhat DJ, Jones EG, Maharachchikumbura SS, Raspe O, Karunarathna SC, Wanasinghe DN, Hongsanan S (2019) Fungal diversity notes 929–1035: taxonomic and phylogenetic contributions on genera and species of fungi. Fungal Divers 95(1):1–273
Pratibha J, Prabhugaonkar A (2015) New record of Thysanorea papuana from India. Mycosphere 6:480–485
Prenafeta-Boldú FX, Luykx D, Vervoort J, de Bont JA (2001) Fungi growing on volatile aromatic hydrocarbons as sole carbon and energy source. In: Proc 5th Int Symp Environm Biotechnol, Kyoto, pp. 253-256
Puttemans A (1904) Contribution à l’étude de la fumagine des caféiers. Bull Soc Mycol Fr 20:152–154
Pybus OG, Harvey PH (2000) Testing macro-evolutionary models using incomplete molecular phylogenies. Proc R Soc Lond B 267:2267–2272
Quan Y, Gerrits van den Ende B, Shi D, Prenafeta-Boldú FX et al (2019) A comparison of isolation methods for black fungi degrading aromatic toxins. Mycopathologia 184(5):653–660
Raghupathi PK, Zupančič J, Brejnrod AD, Jacquiod S et al (2018) Microbial diversity and putative opportunistic pathogens in dishwasher biofilm communities. Appl Environ Microbiol 84(5):e02755-02717
Raj TN (1977) Ypsilonia, Acanthotheciella, and Kazulia gen nov. Can J Bot 55(12):1599–1622
Rambaut A, Drummond A (2009) Tracer v1. 5.0. http://beast.bio.ed.ac.uk/Tracer.
Réblová M, Untereiner WA, Réblová K (2013) Novel evolutionary lineages revealed in the Chaetothyriales (Fungi) based on multigene phylogenetic analyses and comparison of ITS secondary structure. PLoS ONE 8(5):e63547
Réblová M, Hubka V, Thureborn O, Lundberg J, Sallstedt T, Wedin M, Ivarsson M (2016) From the tunnels into the treetops: new lineages of black yeasts from biofilm in the Stockholm metro system and their relatives among ant-associated fungi in the Chaetothyriales. PLoS ONE 11(10):e0163396
Revel LJ (2011) Phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol 3(2):217–223. https://doi.org/10.1111/j.2041-210X.2011.00169.x
Reynolds D (1982) Foliicolous Ascomycetes. 4. The capnodiaceous genus Trichomerium Spegazzini emend. [Fungi]. Mycotaxon 14:189–220
Reynolds D (1983) Foliicolous ascomycetes. 5. The capnodiaceous clypeate genus Treubiomyces. Mycotaxon 17:349–360
Reynolds D (1985) Foliicolous ascomycetes. 6. The capnodiaceous genus Limacinia. Mycotaxon 23:153–168
Ruibal C, Platas G, Bills G (2008) High diversity and morphological convergence among melanised fungi from rock formations in the Central Mountain System of Spain. Persoonia 21:93–100
Ruibal C, Selbmann L, Avci S, Martin-Sanchez P, Gorbushina A (2018) Roof-inhabiting cousins of rock-hnhabiting fungi: novel melanized microcolonial fungal species from photocatalytically reactive subaerial surfaces. Life 8(3):30
Ruiz RFC, Heredia G, Reyes M, Arias R, Decock C (2001) A revision of the genus Pseudospiropes and some new taxa. Cryptogam Mycol 22(1):3–18
Saccardo PA (1883) Sylloge Fungorum (Abellini) 2:288
Samarakoon MC, Hyde KD, Hongsanan S, McKenzie EHC et al (2019) Divergence time calibrations for ancient lineages of Ascomycota classification based on a modern review of estimations. Fung Div 96:285–346
Satow M, Attili-Angelis D, de Hoog G, Angelis D, Vicente V (2008) Selective factors involved in oil flotation isolation of black yeasts from the environment. Stud Mycol 61:157–163
Schlick-Steiner BC, Steiner FM, Konrad H, Seifert B et al (2008) Specificity and transmission mosaic of ant nest-wall fungi. Proc Nat Acad Sci USA 105(3):940–943
Schol-Schwarz MB (1968) Rhinocladiella, its synonym Fonsecaea and its relation to Phialophora. Antonie van Leeuwenhoek 34(1):119–152
Seyedmousavi S, Badali H, Chlebicki A, Zhao J, Prenafeta-Boldu FX, De Hoog GS (2011) Exophiala sideris, a novel black yeast isolated from environments polluted with toxic alkyl benzenes and arsenic. Fungal Biol 115(10):1030–1037
Sinclair R (1979) Notes on Hyphomycetes. XXVI. Uncispora harroldii gen. et sp. nov. Mycotaxon 8(1):140–143
Spegazzini G (1888) Fungi Guaranitici. Pug. II. An Soc Cient Argent pp. 1-72
Spegazzini C (1910) Mycetes argentinenses, series V. An Mus Nac Hist Nat B Aires 20:329–467
Stenroos S, Laukka T, Huhtinen S, Döbbeler P et al (2010a) Multiple origins of symbioses between ascomycetes and bryophytes suggested by a five-gene phylogeny. Cladistics 26(3):281–300
Stenroos S, Laukka T, Huhtinen S, Döbbeler P, Myllys L, Syrjänen K, Hyvönen J (2010b) Multiple origins of symbioses between ascomycetes and bryophytes suggested by a five-gene phylogeny. Cladistics 26(3):281–300
Stielow J, de Hoog G (2014) Novel Phialophora species from leaf-cutting ants (tribe Attini). Fungal Divers 65:6575Badali
Su L (2015) Taxonomy and ecology research of Chinese Stone Fungi. China Agricultural University (In Chinese)
Sudhadham M, Prakitsin S, Sivichai S, Chaiyarat R et al (2008) The neurotropic black yeast Exophiala dermatitidis has a possible origin in the tropical rain forest. Stud Mycol 61:145–155
Sutton B, Gaur R (1984) Zelopelta thrinacospora gen. et. sp. nov. (Pycnothyriales). Trans Br Mycol Soc 82(3):556–559
Sutton DA, Rinaldi MG, Sanche SE (2009) Dematiaceous fungi. Clinical mycology, 2nd edn. Churchill Livingstone, London, pp 329–354
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Molec Biol Evol 30(12):2725–2729
Teixeira M, Moreno LF, Stielow B, Muszewska A et al (2017) Exploring the genomic diversity of black yeasts and relatives (Chaetothyriales, Ascomycota). Stud Mycol 86:1–28
Tian Q, Doilom M, Luo ZL, Chomnunti P, Bhat JD, Xu J-C, Hyde KD (2016) Introducing Melanoctona tectonae gen. et sp. nov. and Minimelanolocus yunnanensis sp. nov. (Herpotrichiellaceae, Chaetothyriales). Cryptogam Mycol 37(4):477–492
Tibpromma S, Hyde KD, Jeewon R, Maharachchikumbura SS, Liu JK, Bhat DJ, Jones EG, McKenzie EH, Camporesi E, Bulgakov TS (2017) Fungal diversity notes 491–602: taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers 83(1):1–261
Tsuneda A, Hambleton S, Currah R (2011) The anamorph genus Knufia and its phylogenetically allied species in Coniosporium, Sarcinomyces, and Phaeococcomyces. Botany 89(8):523–536
Tsurumi Y, Van Hop D, Ando K (2018) Three new anamorph of Ceramothyrium from fallen leaves in Vietnam. Adv Microbiol. 8(04):314
Untereiner WA (1997) Taxonomy of selected members of the ascomycete genus Capronia with notes on anamorph-teleomorph connections. Mycologia 89(1):120–131
Untereiner WA, Naveau FA (1999) Molecular systematics of the Herpotrichiellaceae with an assessment of the phylogenetic positions of Exophiala dermatitidis and Phialophora americana. Mycologia 91(1):67–83
van den Brink HM, van Gorcom RF, van den Hondel CA, Punt PJ (1998) Cytochrome P450 enzyme systems in fungi. Fung Genet Biol 23:1–17
Van den Ende A, De Hoog G (1999) Variability and molecular diagnostics of the neurotropic species Cladophialophora bantiana. Stud Mycol 43:151–162
van der Aa HA, van Oorschot CAN (1985) A redescription of some genera with staurospores. Persoonia 12(4):415–425
van der Aa HA, von Arx JA (1986) On Vonarxia, Kazulia and other fungi with stauroconidia. Persoonia 13(1):127–128
Vasse M, Voglmayr H, Mayer V, Gueidan C et al (2017) A phylogenetic perspective on the association between ants (Hymenoptera: Formicidae) and black yeasts (Ascomycota: Chaetothyriales). Proc R Soc B 284(1850):20162519
Veerkamp J, Gams W (1983) Los hongos de colombia-VIII some new species of soil fungi from Colombia. Caldasia:709-717
Vicente V, Attili-Angelis D, Pie M, Queiroz-Telles F et al (2008) Environmental isolation of black yeast-like fungi involved in human infection. Stud Mycol 61:137–144
Vicente VA, Orélis-Ribeiro R, Najafzadeh M, Sun J, Guerra RS, Miesch S, Ostrensky A, Meis JF, Klaassen CH, de Hoog G (2012) Black yeast-like fungi associated with Lethargic Crab Disease (LCD) in the mangrove-land crab, Ucides cordatus (Ocypodidae). Vet Microbiol 158(1–2):109–122
Vicente V, Najafzadeh M, Sun J, Gomes R, Robl D, Marques S, Azevedo C, De Hoog G (2014) Environmental siblings of black agents of human chromoblastomycosis. Fungal Divers 65(1):47–63
Voglmayr H, Mayer V, Maschwitz U, Moog J et al (2011) The diversity of ant-associated black yeasts: insights into a newly discovered world of symbiotic interactions. Fung Biol 115(10):1077–1091
Wang XF, Cai WY, Gerrits van den Ende AHG, Zhang J et al (2018) Indoor wet cells as a habitat for melanized fungi, opportunistic pathogens on humans and other vertebrates. Sci Rep 8:7685
Wiens JJ (2006) Missing data and the design of phylogenetic analyses. J Biomed Inform 39(1):34–42
Wiens JJ (2008) Missing data and the accuracy of Bayesian phylogenetics. J Syst Evol 46(3):307–314
Wijayawardene NN, Hyde KD, Khalil Tawfeeq Al-Ani L, Tedersoo L, et al. (2020) Outline of fungus and fungi-like taxa. Mycosphere 11 (2020)
Wilf P, Johnson KR (2004) Land plant extinction at the end of the Cretaceous: a quantitative analysis of 1039 the North Dakota megafloral record. Paleobiology 30(3):347–368
Wills MA (2001) Morphological disparity: a primer. In: Adrain JM, Edgecombe GD, Lieberman BS (eds) Fossils, phylogeny, and form. Kluwer/Plenum, New York, pp 55–144
Winka K, Eriksson OE, Bång Å (1998) Molecular evidence for recognizing the Chaetothyriales. Mycologia 90(5):822–830
Woo PC, Ngan AH, Tsang CC, Ling IW, Chan JF, Leung S-Y, Yuen K-Y, Lau SK (2013) Clinical spectrum of Exophiala infections and a novel Exophiala species, Exophiala hongkongensis. J Clin Microbiol 51(1):260–267
Yang GZ, Lu J, Yu Z, Zhang K, Qiao M (2011) Uncispora sinensis, a new species from China.
Yang H, Chomnunti P, Ariyawansa H, Wu H, Hyde KD (2014) The genus Phaeosaccardinula (Chaetothyriales) from Yunnan, China, introducing two new species. Chiang Mai J Sci 41:873–884
Yang H, Hyde KD, Karunarathna SC, Deng C, Gu CH, Yang SA, Zhang ZC (2018) New species of Camptophora and Cyphellophora from China, and first report of sexual morphs for these genera. Phytotaxa 343(2):149–159
Yong LK, Wiederhold NP, Sutton DA, Sandoval-Denis M, Lindner JR, Fan H, Sanders C, Guarro J (2015) Morphological and molecular characterization of Exophiala polymorpha sp nov isolated from sporotrichoid lymphocutaneous lesions in a patient with myasthenia gravis. J Clin Microbiol 53(9):2816–2822
Zakharova K, Tesei D, Marzban G, Dijksterhuis J et al (2013) Microcolonial Fungi on rocks: a life in constant drought? Mycopathologia 175:537–547
Zeng J, Sutton D, Fothergill A, Rinaldi M, Harrak M, De Hoog G (2007) Spectrum of clinically relevant Exophiala species in the United States. J Clin Microbiol 45(11):3713–3720
Zeng XY, Wen TC, Chomnunti P, Liu JK, Boonmee S, Hyde KD (2016) Ceramothyrium longivolcaniforme sp. nov., a new species of Chaetothyriaceae from northern Thailand. Phytotaxa 267(1):51–60
Acknowledgements
We would like to thank China Scholarship Council for financial support for Y.Q. (Number 201708520100). Hein van der Lee and Marlou Tehupeiory-Kooreman are acknowledged for technical assistance. Qing Tian is thanked for double checking the ordinal level tree of the Chaetothyriales.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Quan, Y., Muggia, L., Moreno, L.F. et al. A re-evaluation of the Chaetothyriales using criteria of comparative biology. Fungal Diversity 103, 47–85 (2020). https://doi.org/10.1007/s13225-020-00452-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-020-00452-8