
Abstract
Several species of unicellular green algae, such as the model green microalga Chlamydomonas reinhardtii, can operate under either aerobic photosynthesis or anaerobic metabolism conditions. A particularly interesting metabolic condition is that of “anaerobic oxygenic photosynthesis”, whereby photosynthetically generated oxygen is consumed by the cell’s own respiration, causing anaerobiosis in the culture in the light, and induction of the cellular “hydrogen metabolism” process. The latter entails an alternative photosynthetic electron transport pathway, through the oxygen-sensitive FeFe-hydrogenase, leading to the light-dependent generation of molecular hydrogen in the chloroplast. The FeFe-hydrogenase is coupled to the reducing site of photosystem-I via ferredoxin and is employed as an electron-pressure valve, through which electrons are dissipated, thus permitting a sustained electron transport in the thylakoid membrane of photosynthesis. This hydrogen gas generating process in the cells offers testimony to the unique photosynthetic metabolism that can be found in many species of green microalgae. Moreover, it has attracted interest by the biotechnology and bioenergy sectors, as it promises utilization of green microalgae and the process of photosynthesis in renewable energy production. This article provides an overview of the principles of photobiological hydrogen production in microalgae and addresses in detail the process of induction and analysis of the hydrogen metabolism in the cells. Furthermore, methods are discussed by which the interaction of photosynthesis, respiration, cellular metabolism, and H2 production in Chlamydomonas can be monitored and regulated.
Similar content being viewed by others


Introduction: photobiological hydrogen production by unicellular green algae
In view of decreased availability of fossil fuels and the climate changes caused by anthropogenic rise of the atmospheric CO2 concentration, the recovery of renewable fuels has become more and more important. Molecular hydrogen (H2) is thought to be the ideal fuel for the future because of its high energy content and its clean combustion to water (H2O). Nature has created biological reactions that use sunlight for the oxidation of water (oxygenic photosynthesis), and enzymes that use electrons for the generation of H2 (hydrogenases).
In 1939, the German plant Physiologist Hans Gaffron discovered this hydrogen metabolism in green algae (Gaffron 1939). Cyanobacteria and green algae are so far the only known organisms with both an oxygenic photosynthesis and a hydrogen production (Schütz et al. 2004). While H2 production in cyanobacteria is mostly coupled to nitrogen fixation, unicellular green algae utilize photosynthetically generated electrons for H+ reduction.
Thus, one interesting, recent extension of photosynthesis research entails the development of methods for a sustained photobiological hydrogen H2 gas production in green microalgae such as Chlamydomonas reinhardtii (Melis et al. 2000; Ghirardi et al. 2000; Melis and Happe 2001, 2004; Melis 2007). This extension is of interest as it couples an extremely oxygen (O2)-sensitive enzyme, the FeFe-hydrogenase, to the photosynthetic electron transport pathway that generates O2 during its normal function. The hydrogenase pathway enables these microalgae to dissipate electrons from the photosynthetic electron transport chain in the form of molecular H2 (Hemschemeier et al. 2008), a volatile and harmless gas for the algae, but an attractive energy carrier for humans (Melis and Happe 2001).
In general, H2 metabolism is widespread among microorganisms. In the majority of cases, enzymes called hydrogenases catalyze either production or oxidation of molecular H2 (Vignais et al. 2001). Hydrogenases are categorized according to the metal cofactors found in their catalytically active site, and are classified as NiFe-, FeFe- or Fe-hydrogenases (Vignais and Billoud 2007). Only the NiFe- and FeFe- hydrogenases are prevalent among microorganisms (Vignais and Billoud 2007). In contrast, Fe-hydrogenases (also known as H2-forming methylenetetrahydromethanopterin dehydrogenases, Hmd; Zirngibl et al. 1990) are exclusively encountered in some methanogenic archaea (Shima and Thauer 2007) and have a completely different cofactor than NiFe- or FeFe-hydrogenases as has been recently proved by the analysis of a Fe-hydrogenase crystal structure at 1.75 Å (Shima et al. 2008).
The vast majority of the hydrogenase enzymes are sensitive to molecular oxygen. It is of interest therefore, that several species of unicellular green algae have retained the genetic information and are capable of metabolizing molecular H2 (Kessler 1974; Winkler et al. 2002b, c; Skjånes et al. 2008), in spite of the fact that these microorganisms normally carry out oxygenic photosynthesis. A substantial proportion of H2 production in such microalgae clearly depends on photosynthetic activity, on electrons derived upon photosynthetic oxidation of H2O, and on the FeFe-hydrogenase enzyme that is localized in the chloroplast (Happe et al. 1994; Florin et al. 2001).
The hydrogenase enzyme and the metabolism it is involved in are best addressed in the model green microalga C. reinhardtii. Its FeFe-hydrogenase (HydA1) is a small iron-containing protein of about 48 kDa, which is localized in the chloroplast stroma with ferredoxin being the direct electron donor (Happe and Naber 1993; Happe et al. 1994). The gene encoding HydA1 was first reported by Happe and co-workers in 2001 (Florin et al. 2001; Happe and Kaminski 2002), with a second putative hydrogenase gene, HYDA2, identified soon thereafter (Forestier et al. 2003). The function of HydA2 has not been clarified yet. Isolation of hydrogenase from C. reinhardtii did always result in pure HydA1 protein (Happe and Naber 1993; Kamp et al. 2008); however, the HYDA2-gene is transcribed (Forestier et al. 2003) and recombinant HydA2 protein has hydrogenase activity (King et al. 2006). Altogether, a collection of hydrogenase genes (Florin et al. 2001; Winkler et al. 2002a, c; Kamp et al. 2008) and proteins (Kamp et al. 2008) of different green microalgal species have been isolated, showing a high degree of similarity (Melis et al. 2004).
In C. reinhardtii (Happe and Naber 1993; Happe and Kaminski 2002) and other eukaryotic microalgae (Winkler et al. 2002b; Skjånes et al. 2008) hydrogenase gene expression and hydrogenase activity can be induced upon an artificial process called anaerobic adaptation, in which cells are concentrated, flushed with inert gas like argon (Ar) or nitrogen (N2), and kept in the dark. In this induction system, HYDA1 transcript accumulation and in vitro hydrogenase activity become detectable within a few minutes (Happe and Kaminski 2002), and this method can be applied to test novel algal species with regard to their capability to express an active hydrogenase enzyme (Winkler et al. 2002b). An alternative approach is to dark adapt cells in air-tight containers, in which the culture medium becomes anaerobic via the cells’ own respiration. This approach is suitable for testing both hydrogenase gene expression and in vivo H2 evolution, even if the latter is usually very low in the dark (Gfeller and Gibbs 1984) and short-lived in the light due to photosynthetic oxygen evolution (Ghirardi et al. 1997).
A relatively high, but very transient H2 production in green algae can be observed after a sudden dark–light shift of cells which had become anaerobic in the dark and started to express the hydrogenase gene. As light is switched on, a sudden and rampant H2 evolution can be observed, which, however, lasts only for a few minutes (Mus et al. 2005). In this system, the hydrogenase accepts electrons produced by PSII until the Calvin Benson cycle is activated and the hydrogenase is inhibited by the rising O2 concentration in the medium.
Because of the very slow rates of H2 evolution in the dark, and the transient-only H2 production in the light, a meaningful role and metabolic purpose of the plastidic FeFe-hydrogenase remained unclear for around 60 years of the related research. However, a breakthrough discovery, enabling a relatively high-rate and sustained H2 production activity in illuminated C. reinhardtii cultures, was reported by Melis and co-workers (Melis et al. 2000; Ghirardi et al. 2000). A critical condition that was applied in the development of this methodology was the lowering of the rate of photosynthesis to about the level of cellular respiration, enabling the cell’s own respiration to consume photosynthetically generated O2, thereby permitting unimpeded expression and function of the FeFe-hydrogenase pathway. A balanced photosynthesis–respiration activity is currently the platform of choice for research in this field, employed in several labs in many countries. It was originally attained upon a sulphur (S) nutrient deprivation from the growth medium of the cells, the absence of which caused a slowdown of the rate of photosynthesis (Wykoff et al. 1998) to a level just lower than that of respiration (Melis et al. 2000), thereby resulting in the establishment of those preconditions necessary for H2 evolution activity. Such internally induced anaerobiosis allowed the expression of the HYDA1 gene and permitted the HydA1 enzyme to become active.
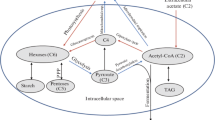
During S deprivation and H2 production, C. reinhardtii cells stop growth and down-regulate CO2 assimilation (Melis et al. 2000; Hemschemeier et al. 2008). Thus, the major photosynthetic electron sink is no longer operative. Instead, the hydrogenase pathway is activated, leading to proton reduction and H2 production, thus becoming an alternative sink for photosynthetic electron transport (Fig. 1). The latter stays active at least in the electron transport chain starting at the plastoquinone (PQ) pool (Wykoff et al. 1998; Melis et al. 2000), enabling the cells to dissipate light energy and to photoproduce adenosine triphosphate (ATP). This electron transport is driven in part by residual PSII activity, and in part by non-photochemical PQ reduction (Rumeau et al. 2007) at the expense of reducing equivalents stored as starch (Fouchard et al. 2005; Hemschemeier et al. 2008) (Fig. 1).

Schematic of photosynthetic electron transport in the unicellular green alga C. reinhardtii during normal photosynthesis (a) and H2 production during S deprivation (b). S depletion causes a drastic decrease of photosystem II (PSII) activity (indicated by the dotted line of the PSII symbol). In addition, the light harvesting complexes (LHCII) antennae are partially transferred to photosystem I (PSI) (state 2 transitions). The decreased O2 evolution at PSII results in anaerobic conditions in a respiring, sealed algal culture, so that the hydrogenase (HYD) can become active. Besides residual PSII-activity, the oxidative degradation of organic substrates such as starch is an important electron source for H2 production. The electrons derived from the latter process are probably transferred into the photosynthetic electron transport chain (PETC) by a plastidic NAD(P)H-dehydrogenase (NDH). The modified PETC of S-depleted algae allows the electron transport to continue so that the cells can generate ATP through photophosphorylation. Further abbreviations: ATP synthase (ATPase), cytochrome b 6 f complex (Cytb 6 f), ferredoxin (Fdx), ferredoxin-NADPH-reductase (FNR), plastidic terminal oxidase (PTOX), plastocyanine (PC), plastoquinone (PQ)
A precondition for a sustained H2 evolution is an adequate supply of electrons to sustain respiration and oxidative phosphorylation. The latter is provided through the regulated catabolism of starch, large amounts of which accumulate in S-deprived C. reinhardtii during the first few hours of S-nutrient limitation (Melis et al. 2000; Zhang et al. 2002; Fouchard et al. 2005).
In sum, H2 production in S-depleted C. reinhardtii cells is an elaborately complex variant of “anaerobic oxygenic photosynthesis” (Fig. 1). The study of the corresponding cellular metabolism is of interest to biotechnologists, who hope to be able to engage the microalgae as producers of H2, a clean and renewable energy carrier. In addition, this alternative “anaerobic oxygenic photosynthesis” offers an opportunity to gain insights into the flexibility and regulation of photosynthesis. This chapter aims at providing the basic knowledge on how to induce and analyze the H2 metabolism of green microalgae, with a focus on assessing the interplay between photosynthesis and H2 evolution.
Induction and analysis of the hydrogen metabolism in Chlamydomonas reinhardtii
Anaerobic adaptation and in vitro hydrogenase assay
The simplest and probably most artificial technique to induce hydrogenase activity in unicellular green algae is to flush a highly concentrated and shaded culture with an inert gas like Ar or N2. For this purpose, cells are grown in the light, either photoheterotrophically in Tris acetate phosphate (TAP) or photoautotrophically in high salt minimal (HSM) medium (Harris 1989, 2009). For all the physiological analyses of Chlamydomonas and other algae, it is important to keep the cells of precultures in the active growth phase. This means that as soon as the cultures have reached a cell density of about 1 × 107 cells ml−1, an aliquot of this culture is used to inoculate fresh medium at a cell density of about 1–2 × 104 cells ml−1. For anaerobic adaptation of C. reinhardtii, a chlorophyll content of the pre-culture of 20–25 μg ml−1 has turned out to be optimal. The chlorophyll content of C. reinhardtii is determined by mixing 200 μl of cell suspension with 800 μl acetone, letting the chlorophylls extract for several hours in the refrigerator, spinning the cells down, and measuring the absorbance of the green supernatant against 80% acetone at λ = 652 nm (Arnon 1949). At a chlorophyll content of 20 μg ml−1, which is equivalent to about 6.5 × 106 cells ml−1 in case of the C. reinhardtii wild type CC-124 (137c), the cells would have already reached the end of the exponential growth phase, but still divide. The pre-culture is then harvested by mild centrifugation (2 min at 3,500–5,000 g, room temperature) in Sarstedt (Sarstedt, Nümbrecht, Germany) or Falcon tubes and gently resuspended in about 0.2 volumes of fresh medium to reach a chlorophyll concentration of 100 μg ml−1. For reproducible and comparable results, both the respective pre-cultures and the concentrated cultures to be compared should have equivalent chlorophyll contents.
It is important for all the analyses of C. reinhardtii that the cells do not stand anywhere for more than 1 min, since they settle rapidly and establish microaerobic or even anaerobic conditions in the dense cell sediment. Accordingly, pellets of the algae should be resuspended rapidly. If the purpose of the experiments requires a real “0 h” sample, the concentrated cells may be incubated in a thin layer in Erlenmeyer flasks in the light to re-establish photosynthetic, thus O2-saturated conditions.
For anaerobic adaptation, though the highly concentrated algal culture will not carry out appreciable photosynthesis due to self-shading, the tube should be further wrapped with aluminium foil to prevent the outer layers of the cell culture to be exposed to light. The algal suspension is then flushed by Ar or N2 at 18–25°C. For this purpose, a tube (about 5 mm in diameter) connected to the gas cylinder via a pressure regulator is introduced into the flask through a suitable hole in the lid, which should have a somewhat larger diameter than the tube to allow the gas to exhaust again. The tube is then dipped into the culture so that the gas flushes the cell suspension. It is important that the gas is of high purity, i.e., with no significant contaminations of O2. Furthermore, it is essential that the bubbling of the algal suspension is not too strong to avoid shearing stress of the cells.
The success of the anaerobic induction of hydrogenase activity can be monitored by an in vitro hydrogenase activity assay. The reaction mixture of this assay contains Triton-X 100, a mild detergent which lyses the algal cells. It should be noted that some algal species have different types of cell walls which might be too resistant to Triton. The assay described here performs well in C. reinhardtii, C. moewusii, Scenedesmus obliquus, S. vacuolatus, and some other species tested to date (Winkler et al. 2002b; Kamp et al. 2008). The assay furthermore contains methyl viologen as a potent artificial electron donor to FeFe-hydrogenases and sodium dithionite (Na2S2O4) as an efficient reductant for methyl viologen.
The details: First, 1.6 ml of 60 mM potassium phosphate buffer pH 6.8, 1% Triton X-100 (0.2 ml of a 10% (v/v) stock solution in the above mentioned phosphate buffer) and 10 mM methyl viologen (of a 1 M stock solution in phosphate buffer, which can be stored in the fridge for several weeks) are mixed in a 8–10-ml edge rolls bottle (e.g., 10-ml headspace bottles ND20/ND18, cat. no. 3205550 at www.de.fishersci.com/) (Fig. 2b). The flask is then sealed by a Red Rubber Suba Seal (e.g., No. 25, cat. no. Z12,459-1 at www.sigmaaldrich.com/germany.html) and gassed with Ar (N2) for 5 min. For this purpose, a needle connected to a gas cylinder via an adequate tube is pierced through the septum, and another needle serves as gas exhaust. In parallel, a 1-M freshly prepared sodium dithionite solution is prepared in a sealed headspace bottle by injecting the required amount of phosphate buffer through the septum of the vessel, in which the required amount of sodium dithionite is already present. This solution is also flushed with Ar (N2) for 5 min. Finally, 200 μl of the anaerobic sodium dithionite stock solution is added to the pre-mix containing buffer, Triton, and methyl viologen by a syringe piercing through the rubber septum. The reaction mixture should turn deep blue to purple, an indication of methyl viologen being reduced (Fig. 2b). As an alternative to applying Ar gassing, all the reaction mixtures can be prepared in an anaerobic glove box (e.g., of Coy Laboratories, Detroit, USA).

a Development of in vitro hydrogenase activity in a concentrated C. reinhardtii culture sparged with Ar starting at 0 min. Samples of 200 μl containing the algal suspension were removed from the shaded incubation flask at the depicted time points and injected into an in vitro assay reaction mixture containing Triton X-100 used for cell lysis, and sodium dithionite reduced methyl viologen as an efficient, in vitro electron donor to FeFe-hydrogenases. After 15 min of incubation in a shaking water bath at 37°C, the headspace within the reaction vessel was analyzed by gas chromatography (GC). The detected amounts of H2 were related to the chlorophyll concentration of the cells as an indication for the cell density and the incubation time. b Edge rolls bottles sealed with red rubber Suba Seals. The two left-side bottles contain aliquots of a C. reinhardtii culture (in vivo assays), the two right-side vessels are filled with in vitro assay reaction mixture having the typical deep blue color of reduced methylviologen
Finally, 100–200 μl of the anaerobically adapted algal culture is removed from the cell suspension by a syringe and then injected into the prepared in vitro assay reaction mixture, piercing through the septum. The flask is then vortexed vigorously to lyse the cells and afterward placed into a shaking water-bath at 37°C for 15 min. The optimum temperature of C. reinhardtii HydA1 is 60°C (Happe and Naber 1993); however, to find a compromise between enzyme activity and long-term stability, 37°C was chosen as a standard temperature. If other temperatures and incubation times are chosen, it should be checked first how long the H2-evolving activity is linear by sampling the gas every 5–10 min.
After incubation, the headspace above the reaction mixture can be analyzed by GC. Gas chromatographic detection of H2 usually utilizes thermal conductivity detectors (TCDs) and argon as a carrier gas. For detailed analyses, a sensitive gas chromatograph should be at hands. Good systems are supplied by Shimadzu, Kyoto, Japan (www.shimadzu.com; e.g., GC-2010 equipped with a PLOT fused silica coating molsieve column [5 Å, 10 m by 0.32 mm] from Varian, Palo Alto, CA; www.varianinc.com). This system also allows the detection of O2, which can be valuable to detect significant O2 contaminations in the samples or to analyze the O2 consumption in S-deprived cells (see below). The hydrogenase activity of whole cells is usually defined as nmoles H2 produced per hour and μg chlorophyll (or cell number).
Anaerobic adaptation experiments commonly last for 4–6 h. In C. reinhardtii, in vitro hydrogenase activity can be detected after 5–15 min of bubbling. Hydrogenase activity rises linearly for 2–3 h and then reaches a plateau activity of around 500 nmol H2 h−1 μg Chl−1 (Fig. 2).
Photobiological hydrogen production upon sulphur deprivation
In S-deprived C. reinhardtii cultures, a very special photosynthetic metabolism develops in which the photosynthetic electron transport chain is significantly changed from what is known as photosynthesis. PSII activity is strongly down-regulated, and the oxidation of organic substances is the main source of electrons, which are proposed to be transferred to the PQ-pool via an NAD(P)H-PQ-oxidoreductase (Mus et al. 2005; Bernard et al. 2006). The electron sinks of photosynthesis change, too, since CO2 fixation becomes undetectable whereas the hydrogenase accepts electrons from the photosynthetic chain (Hemschemeier et al. 2008) (Fig. 1). This algal photohydrogen production has been studied extensively in the last few years. In this chapter, the procedures to induce the H2 metabolism in C. reinhardtii by S deprivation will be described in detail. Furthermore, some techniques to analyze the physiological status of the cells will be summarized.
Media, culture treatment, and illumination conditions
In contrast to inducing micronutrient deficiency in C. reinhardtii, which takes a lot of effort to exclude trace amounts of metal ions from the growth medium (Quinn and Merchant 1998), it is not difficult to deprive C. reinhardtii cells of the macronutrient sulphur (S). Standard TAP medium (Harris 1989, 2009) contains about 0.5 mM of sulphate. 400 μM of the latter derive from the salt solution, and about 100 μM originates from the trace element solution, which contains sulphate salts of Zn, Cu, and Fe. To prepare S-free medium, the standard TAP recipe is used, but the Beijerinck’s salt solution is prepared with MgCl2 instead of using MgSO4. Accordingly, the trace element solution contains the chloride salts of Zn, Cu, and Fe. Double distilled water should be used for the preparation of the stock solutions and the media to avoid sulphate contamination.
To induce S deprivation in C. reinhardtii, the cells are grown in standard TAP medium in the light and then transferred to S-free medium. For this purpose, the cells are centrifuged as described above, the supernatant is discarded, and the cell pellet is gently resuspended in the original volume of S-free medium. Another centrifugation step follows, the supernatant is discarded once more, and the cells are resuspended in S-free medium again. There are several philosophies on how many washing steps should be carried out. Some research groups carry out up to five washing steps (e.g., Kosourov et al. 2002), whereas others wash only once (Hemschemeier et al. 2008). It should be kept in mind that every centrifugation step affects the algal cells and may induce an anaerobic metabolism already, on the other hand, some sulphate might stick to the cells so that one washing step could be insufficient to remove any S from the cells. The procedure might be chosen according to the experimental aims.
An alternative approach to deprive algal cells of S is to inoculate them in a medium with a limited amount of sulphate (Zhang et al. 2002). We experienced that inoculating a low amount of C. reinhardtii cells grown in standard TAP medium in new medium containing 50 μM sulphate (by adding a sterile MgSO4 stock solution to S-free TAP medium) allows them to grow until they reach a chlorophyll content of about 20 μg ml−1. Then, they will pass to the S-deprived stage and induce the set of adaptations figured out below.
There is an easy method to check whether the Chlamydomonas culture already experiences S starvation. Several green algal species such as C. reinhardtii secrete a periplasmatic arylsulfatase as soon as they sense limitations of sulphate (Lien and Schreiner 1975). This enzyme is able to cleave sulphate from aromatic compounds, and its activity is easily assayed in whole cells with the chromogenic substrate 5-bromo-4-chloro-3-indolyl sulphate (XSO4), since the product of arylsulfatase action on XSO4 is a blue compound (Davies et al. 1992). Thus, to check whether C. reinhardtii cells are already in the stage of S deprivation, 1 ml of the cells is removed from the culture vessel and mixed with 10 μl of a 30 mM XSO4 stock solution in 0.1 M Tris/HCl pH 7.5. After 30–60 min, the cells are spun down at a high speed, and the supernatant, which should be visibly bluish if an arylsulfatase is active, can be analyzed photospectrometrically at λ = 650 nm.
In contrast to the simplicity of inducing S starvation in C. reinhardtii, the induction of a sustained and reproducible H2 production in these cultures is much more difficult. To understand this difficulty, the sequence of events leading to the onset of H2 production in C. reinhardtii is briefly summarized here. When the cells have been transferred to S-free medium and placed in the light, they still have a high photosynthetic activity, resulting in O2 evolution and CO2 fixation. The latter results not only in some cell growth and doubling in the beginning (Melis et al. 2000), but also in the accumulation of starch, which is a common response of nutrient starved C. reinhardtii cells (Grossman 2000). Starch levels had tripled already in the first 5 h of S depletion (Makarova et al. 2007), and after being S depleted for 24 h, the algae contain almost tenfold amounts of starch as compared with S-replete cells (Zhang et al. 2002). After several hours, PSII activity and photosynthetic O2 evolution, respectively, as well as CO2 fixation will decrease (Melis et al. 2000; Hemschemeier et al. 2008). At a certain point, the ongoing respiratory O2 uptake activity will overcome photosynthetic O2 evolution rates so that the O2 dissolved in the culture will be consumed by and by (Fig. 3). As soon as anaerobic conditions are established, the hydrogenase gene is expressed (Zhang et al. 2002) and the hydrogenase enzyme becomes active (Winkler et al. 2002b) (Fig. 3). The hydrogenase then takes over the electrons from ferredoxin, which in turn is reduced by PSI activity. The electrons arriving at PSI originate both from residual PSII activity and non-photochemical PQ-reduction (Fouchard et al. 2005; Hemschemeier et al. 2008) (Fig. 1b). The latter, again, depends on the amount of starch that was accumulated in the photosynthetic phase.

a Development of the concentrations of H2 (●), O2 ( ), and CO2 (○) as measured by MS in the headspace of an S-depleted C. reinhardtii culture incubated in squared glass bottles sealed with Suba seals upon one-site illumination as illustrated by the photograph in (b) (Hemschemeier 2005)
), and CO2 (○) as measured by MS in the headspace of an S-depleted C. reinhardtii culture incubated in squared glass bottles sealed with Suba seals upon one-site illumination as illustrated by the photograph in (b) (Hemschemeier 2005)
Having these metabolic adaptations of S-depleted algae under consideration, it is obvious that every culture parameter influencing the photosynthetic light and dark reactions as well as respiratory activity will also have an impact on establishment and dimension of the photosynthetic H2 metabolism. It has been reported that the pH of the medium (Kosourov et al. 2003), the use of synchronized versus non-synchronized cultures (Kosourov et al. 2002), certain amounts of sulphate in the medium (Zhang et al. 2002; Kosourov et al. 2002), as well as temperature and the growth phase of the pre-culture (the authors' own unpublished results) have significant effects on the time it takes for the algal culture to start producing H2 and on the amounts of H2 that are accumulated. Light intensity has a particular impact on the development of S-depleted C. reinhardtii cultures (Laurinavichene et al. 2004) similar to that the culture density has (Kosourov et al. 2002), since the latter determines the amount of light that can penetrate the cell suspension. Furthermore, the availability of carbon (C) sources strongly influences the H2 metabolism of S-deprived C. reinhardtii cultures. Standard TAP medium contains acetate, which can be used by this species as a C source both for growth and respiration. Chlamydomonas can be grown in TAP without supplemental CO2, whereas some researchers use TAP as growth medium but furthermore provide extra CO2 (up to 5%), and in some laboratories, C. reinhardtii is grown photoautotrophically in HSM medium or other minimal media (Harris 1989, 2009). For H2 production upon S deprivation, acetate is essential for the establishment of anaerobic conditions (Fouchard et al. 2005), unless PSII activity is rapidly diminished by applying light stress to the cells grown in dimmed light (Tsygankov et al. 2006; Kosourov et al. 2007). On the other hand, the attempts of several researchers to rapidly induce H2 production in illuminated algae by applying the PSII inhibitor DCMU (3-(3,4-dichlorophenyl)-1,1-dimethylurea) did not result in any H2 accumulation because of the dependence on electrons provided by organic reserves which were built up using electrons provided by PSII (Fouchard et al. 2005; Hemschemeier et al. 2008). Not in the least, the activity of the Calvin Benson cycle plays a significant role in H2 production by C. reinhardtii, since it acts as a competing electron sink. For instance, it has been shown that a Ribulosebisphosphate carboxylase/oxygenase (Rubisco)-deficient strain produces H2 in full TAP medium (Hemschemeier et al. 2008). On the other hand, C. reinhardtii transformants having a reduced ratio of photosynthetic O2 evolution and respiratory O2 uptake establish anaerobiosis and develop in vitro hydrogenase activity in full medium upon illumination, but they do not produce significant amounts of H2 unless the Calvin Benson cycle is inhibited (Rühle et al. 2008).
As a consequence of all these affecting parameters, we recommend the following to stably establish photohydrogen production in S-deprived C. reinhardtii cells: The pre-culture should have a chlorophyll content of 20–25 μg ml−1. Too thin cultures will not establish anaerobic conditions; too dense cultures will have a less efficient photosynthetic activity. Washing once with S-free medium is sufficient. However, the cells will grow a bit in the next few hours. The acetate content of the S-free medium should be at least 10 mM (standard TAP medium contains about 20 mM). For the first trials as well as for physiological or biomolecular analyses, small “photobioreactors” are suitable. We often use square narrow-neck glass bottles (e.g., Square bottles, narrow neck, DIN thread GL32, 100–500 ml; Duran cat. nos. 23 810 24 5, 23 810 36 5, and 23 810 44 5; Duran, Mainz, Germany, www.duran-group.com/) which can be sealed by Suba seals no. 37 (Z12,462-1 at Sigma-Aldrich). Depending on the diameter of the bottles, the cell suspension already transferred to S-free medium should have a chlorophyll content of at least 20 μg ml−1 (100 ml bottles) or 15 μg ml−1 (250 ml bottles), but not more than 30 μg ml−1 (100 ml bottles) or 25 μg ml−1 (250 ml bottles) when incubating the cells at a one-site light intensity of about 80 μE s−1 m−2. If the culture is too thin, the cells will produce too much O2 and hardly enter the anaerobic H2 -production phase; if the cells are too dense, they will pass into anaerobiosis very soon, only because of self-shading and not because of the effect of sulphur starvation. Furthermore, they will accumulate only small amounts of starch.
If a gaseous phase is to be left above the culture, which is necessary if the accumulating gas species are to be analyzed by GC or MS (Fig. 3), the gas–liquid ratio should not be too high. For example, we put 290 ml of cell suspension in a 250-ml bottle (which has a total volume of 320 ml) or 100 ml of cells in a 100 ml-bottle (total volume 120 ml).
However, we experienced a large variation in the metabolic responses of S-deprived C. reinhardtii cells even if the culture parameters diverged only slightly. Thus, in every lab, the optimal conditions can be somehow different, and it makes sense for everyone who wants to establish this system to try out different parameters himself or herself. If different algal species are to be examined, a standard control strain should be included to make sure that the setup is adequate. The well-studied species C. reinhardtii and Scenedesmus vacuolatus (formerly Chlorella fusca) show almost the same reactions to S depletion (Winkler et al. 2002b; Kamp et al. 2008) and are suitable to serve as control strains.
When doing biotechnologically orientated research on the H2 metabolism of green algae, one would prefer a real photobioreactor instead of using just glass bottles. A lot of different bioreactor types have been used, including tubular or flat-panel reactors applying different modes of cell mixing and light supply. However, because the development of suitable photobioreactors is a discrete research field (reviewed e.g., by Eriksen 2008), this will not be discussed in this chapter.
Online gas-exchange analyses with a mass-spectrometer
Many techniques have been applied in order to disclose the secrets of H2 production in S deprived C. reinhardtii, among which the standard techniques for the analysis of photosynthesis, such as in vivo measurements of O2 exchange rates by Clark-type electrodes, in vitro measurements of the electron flows in thylakoids isolated from S-deprived C. reinhardtii cells, PAM fluorometry, 77-K fluorescence emission, and chlorophyll fluorescence induction experiments (Wykoff et al. 1998), as well as spectrophotometric measurements of absorbance differences to determine the concentrations of functional PSII and PSI reaction centers or cytochrome f in isolated thylakoid membranes (Melis et al. 2000) (see below).
Other methods were utilized in order to analyze the H2 metabolism. The three indicators for the latter are usually in vitro hydrogenase activity assays as described above, in vivo assays (see below), and the analysis of the gas phase of the cells by GC.
However, for all these experiments, the samples are usually taken from a culture vessel to be analyzed in different devices. This always perturbs the equilibrium of the system, since a volume is extracted from the incubation vessel, and the sample itself will be exposed to air to a certain extent. Therefore, optimal experimental conditions combine the recordings of several relevant data in one sample, preferably in the culture vessel or the photobioreactor itself. Such systems, measuring pH, dissolved O2, redox potential, and chlorophyll fluorescence simultaneously in one photobioreactor have been described (e.g., Kosourov et al. 2002; Antal et al. 2003), and they allow the direct relation of photosynthetic parameters such as PSII chlorophyll fluorescence with other indicators such as dissolved O2 concentration and redox potential. Using such a photobioreactor setup, Antal et al. (2003) could demonstrate a clear relationship between a rapid drop of PSII efficiency and the onset of anaerobiosis, and could further find evidence that the hydrogenase enzyme provided a sink for electrons in the absence of the usual electron-consuming pathways such as CO2 fixation.
A very convenient system to study the exchange of gases in C. reinhardtii cell suspensions is a so-called Membrane Inlet Mass Spectrometer (MIMS). The setup is very useful to study the production or exchange of gases in liquid samples in general, e.g., in aqueous PSII solutions (Messinger et al. 1995; Shevela et al. 2008; Konermann et al. 2008), and it is described in detail in the article by Beckman et al. (On-line Mass spectrometry: Membrane Inlet sampling) in this issue.
A MIMS has been established, for instance, in a research group at CEA Cadarache, France (Laurent Cournac, Gilles Peltier, Départment d’Ecophysiologie Vegétale et de Microbiologie, CEA Cadarache, France) (Dimon et al. 1988; Lindberg et al. 2004). Here, a measuring chamber of the Hansatech DW2/2 type (Hansatech, Norfolk, England, www.hansatech-instruments.com/index.htm) is directly connected to a mass spectrometer (MS) (model MM 8-80; VG Instruments, Cheshire, United Kingdom) by a polypropylene membrane at the bottom of the chamber, which allows dissolved gases to be directly introduced into the ion source of the MS through a vacuum line (Fig. 4a). The measuring chamber is thermostat controlled by a water jacket, and the liquid is continuously mixed by a magnetic stirrer. Light can be applied by a fiber optic illuminator (e.g., from Schott, Mainz, Germany; www.schott.com) (more detailed descriptions of the setup are given in references, Jouanneau et al. 1980; Lindberg et al. 2004).

a Schematic of a measuring chamber connected to the vacuum of an MS as is set-up in the CEA Cadarache. An aliquot (ca. 1.5 ml) of the algal suspension is injected into the measuring chamber of the Hansatech type where it is stirred by a little stir bar (not shown). Light can be applied by a fiber optic cable. Inhibitors such as DCMU can be applied by a syringe through the capillary of the lid. The bottom of the chamber is sealed by a thin gas-permeable Teflon membrane supported by a stainless steel frit. Gases dissolved in the cell suspension (indicated by white circles) can diffuse through the membrane and enter the ion source of the MS by a vacuum line. The addition of heavy isotopes can be applied to differentiate between respiration (uptake of 18O2) and oxygenic photosynthesis (production of 16O2), as well as between CO2 assimilation (uptake of 13CO2) and respiratory CO2 production (12CO2). The metabolism of D2 is an indicator of the hydrogen metabolism and the hydrogenase turnover rate. b Schematic graph of the effect of DCMU on the in vivo H2 -production rate of S-depleted C. reinhardtii cells as recorded utilizing the MS system depicted in (a). A stable H2 graph indicates the instantaneous H2 evolution rate of an illuminated, S-deprived algal culture. To define the contribution of photosynthetic water splitting to the electron supply of the hydrogenase, DCMU is added. The difference of the H2-production rates before and after the addition of the PSII inhibitor is equivalent to the fraction of H2 which is generated with electrons provided by PSII. To determine the low rate of dark H2 production, light is turned off after the H2 graph has stabilized.
The merit of this set-up is that changes of the concentrations of several gases can be recorded simultaneously. The spectrometer sequentially scans the abundance of the gases of interest while measuring one mass peak takes 0.5 s in the system described by Lindberg et al. (2004). Therefore, the concentrations of gases dissolved in a cell suspension within the measuring chamber are recorded in very short-time intervals and any change in gas abundance will be observable almost immediately. Thus, this MS system allows examining different metabolic processes in real time and in parallel, allowing a direct comparison without the need to take into account different measuring conditions and set-ups (e.g., light intensity, temperature, disturbance of the system by entry of air etc.).
By utilizing a MS system, all gases of interest can be analyzed (Fig. 4a). For the analysis of photohydrogen production in C. reinhardtii, O2 (PSII activity, respiration), CO2 (CO2 assimilation, respiration, and fermentation), and H2 are the relevant gases. Moreover, mass spectrometric analyses allow differentiating between different isotopes of one element, so that O2 and CO2 production can be separated from O2 and CO2 consumption. Lindberg et al. (2004) described gas-exchange analyses in the filamentous cyanobacterium Nostoc punctiforme, in which isotopic tracing was applied. The addition of 18O2 allowed the calculation of respiratory activity, since photosynthetic activity mainly produces 16O2. In a similar manner, the uptake of 13CO2 has been used as criterion for CO2 assimilation during photosynthesis, since 12CO2 production originates mostly from the oxidation of stored carbohydrates. For the analysis of the H2 metabolism of whole cells or the activity of hydrogenase enzymes, the exchange of heavy hydrogen (D2) (HD-exchange) has been described as being a valuable tool to monitor enzyme activities within the cells (Cournac et al. 2004; Lindberg et al. 2004) or to study gas diffusion in isolated hydrogenases (Leroux et al. 2008) (in references Cournac et al. 2004 and Leroux et al. 2008; the HD-exchange technology and calculations are described in some detail).
The analysis of photohydrogen production in C. reinhardtii has also benefited from this system. For instance, the direct (real-time) effect of the PSII inhibtor DCMU on H2 evolution could be analyzed utilizing the mass-spectrometric setup (Fig. 4b), thereby allowing to show that the residual PSII activity of S-deprived algal cells only partially contributes to the ongoing in vivo H2-production rates (Hemschemeier et al. 2008). Combined with other experiments involving DCMU treatment, this observation allowed to affirm the model stated by Melis et al. (2000). This model already postulated that PSII activity in the first few hours of S deprivation is essential for H2 production since it is essential for starch accumulation, but that water-splitting becomes dispensable during the H2-production phase, since the latter occurs mainly at the expense of accumulated organic reserves (Melis et al. 2000; Fouchard et al. 2005; Hemschemeier et al. 2008). Furthermore, the application of 13CO2 permitted to verify the strong decrease of in vivo CO2 uptake activity (Hemschemeier et al. 2008), which had been concluded from the degradation of the Rubisco before (Zhang et al. 2002).
In vivo hydrogen production in microalgal cultures
If neither a MS system nor a photobioreactor equipped with several electrodes is available, key parameters of S-deprived C. reinhardtii cells have to be analyzed in independent samples. If this is the case, the measuring conditions of the utilized devices should as much as possible be adapted to the conditions of the incubation flasks. This means, for example, that the illumination applied to induce photosynthetic light reactions in a Clark-type O2 electrode setup should be the same as that used for illuminating the main culture (light intensity and quality). It should be taken into account, too, that the light path in typical measuring chambers (usually 1–2 cm) is much smaller than that in the culture vessel (5–10 cm), so that the light intensity reaching every single cell is higher due to less self-shading of the cells.
The use of O2 electrodes of the Clark type is a common technology which will not be explained here in detail. It should be noted, however, that Clark-type electrodes can easily be converted to H2 electrodes just by applying a different potential. Details of assembling and using these electrodes are given in references Wang (1980), Kuroda et al. (1991), and Takeshita et al. (1993).
An easy method to analyze in vivo H2-production rates of illuminated C. reinhardtii cells without a H2 electrode will be described here. This technique is suitable to determine the real H2-evolution rate of the cells, which can be only roughly concluded from the accumulation of H2 in the gas phase of the incubation flask or in a gas trap (see below). For this purpose, a 2-ml sample of the main culture is taken with a syringe by piercing through the septum and gently injected through the rubber seal of an 8–10 ml headspace bottle as described above, which has been gassed with Ar before. The little vessels are then placed in the light. The cell suspension has to be rocked or stirred so that the cells do not settle. A shaking water bath made from plexiglas standing on top a light source is optimal. After 10 min, a volume of the gas phase is analyzed with a gas chromatograph to determine H2 concentration. Then, the cells are incubated for 1 h, and H2 is detected again. The difference of the H2 concentration in the beginning and after 60 min is the amount of H2 that has been produced by the cells. It should be noted that the 10 min of pre-incubation is applied to let the cells adapt to the system, which will differ from the incubation conditions of the main culture in some aspects. Furthermore, during the transfer of the cells, some air (i.e., O2) might have entered the cell suspension, and the cells might have been shaded to some extent. During the pre-incubation, the algae will stabilize their H2 metabolism. The first analysis of the H2 concentration after the 10 min duration is important to take into account the H2 which has been produced during this pre-incubation phase and the gas which was introduced into the reaction vessel by the algal suspension. In the active H2-producing phase of S-deprived C. reinhardtii cultures, significant amounts of H2 are dissolved in the medium of the cells.
The above point should also be kept in mind when carrying out in vitro hydrogenase activity assays with S-depleted algae. In this case, the reaction vessel is degassed by Ar for 1 min after the cells have been injected and vortexed, so that the H2 introduced by the cell suspension is removed. On the other hand, the in vivo assays of whole cells should not be degassed for “resetting” reasons, since this will disturb the equilibrium of the cells even more.
Hydrogen yield measurements by the water displacement method in a gas trap
To determine the total amount and volume of H2 gas produced by an S-depleted algal culture, H2 gas collection can be achieved with a simple laboratory-assembled gas trap apparatus, based on the water displacement method. Flat culture bottles (usually Roux type) are fitted with an air-tight silicone or rubber stopper, perforated with a gas port (either a narrow piece of glass tubing or a Gauge 10 needle). Teflon tubing (HPLC, Aminco, Lake Forest, CA), attached to the outside-protruding portion of the gas port, is used to conduct the gas evolved by the algae in the culture bottles to an inverted burette or graduated cylinder filled with H2O (Fig. 5). The volume of the gas collected in the burette can be measured directly from the volume of water displacement. A standard GC apparatus can be used to determine the levels of N2, O2, CO2 and H2 in the headspace of the reactor.

H2-production measurements of S depleted green algae in the laboratory using gas traps. The gases produced by the algae are collected in inverted graduated cylinders via the water displacement method. Samples of the gas can be removed utilizing syringes with long and bended needles. As the cells pass into the H2-producing phase, yields of H2 can be measured directly from the volume of the water displaced in the graduated cylinders
This simple setup can be easily assembled. However, there are key methodology issues to be kept in mind. H2 is the smallest of all the molecules and a volatile gas at room temperature. It can easily escape through material that is normally impermeable to air and water, or leak through connections that are not hydrogen-tight. Accordingly, connections of tubes to bottles and stopper perforations have to be leak-proof and ultra-tight. If necessary, such connections and perforations can be additionally sealed with silicone grease or oil.
Chlorophyll fluorescence-based characterization of the photosynthetic apparatus during hydrogen production
In vivo chlorophyll a fluorescence is a powerful non-invasive technique which allows to probe and assess the functional status of the photosynthetic apparatus. As such, in vivo Chl a fluorescence has found many applications in photosynthesis research (Papgeorgiou et al. 2007). This simple measurement technique, which is described in a separate chapter in this issue (a good overview is also given by Baker 2008), offers insight into the induction of H2-production upon S-deprivation. As mentioned above, the significant H2-production capability of C. reinhardtii depends on photosynthesis. Thus, the analysis and monitoring of the photosynthetic apparatus by the non-intrusive chlorophyll fluorescence measurement can readily provide information as to the physiological status of the cells and effect of S-deprivation in terms of the functionality of the photosynthetic electron transport chain (e.g., Wykoff et al. 1998; Melis et al. 2000; Zhang et al. 2002; Antal et al. 2003; the cited articles also provide details about technical setup and culture treatment).
For example, the efficiency of PSII primary photochemistry in C. reinhardtii can readily be calculated from the variable to maximal (F v/F max) fluorescence yield ratio (Kitajima and Butler 1975). “Healthy” C. reinhardtii cells growing in TAP medium and being in the mid-exponential growth phase usually exhibit F v/F max values (F v/F max = variable fluorescence in dark-adapted cells; allows determination of maximum quantum efficiency of PSII primary photochemistry) of 0.6–0.7 (e.g., Wykoff et al. 1998; Zhang et al. 2002; Makarova et al. 2007) and ΔF/F max′ values (ΔF/F max′ = variable fluorescence in light-adapted cells; allows the determination of open reaction centers in the light) of 0.5–0.6 (e.g., Wykoff et al. 1998; Antal et al. 2003). Following a transfer of the microalgal cultures from an S-replete growth medium to a TAP-S medium, F v/F max declines exponentially in the light with a half-time of about 17 h, from about F v/F max = 0.58 at t = 0 h to about F v/F max = 0.08 at t = 60 h. At longer periods of incubation under S-deprivation (t > 60 h), F v/F max remains constant at about the 0.08 level (Zhang et al. 2002). Lack of sulphur-nutrients from the growth medium also brings about a prompt but reversible inhibition of oxygenic photosynthesis, occurring in tandem with the decline in F v/F max, with a half-time of about 17 h (Zhang et al. 2002). One reason for such inhibition under TAP-S conditions is the apparent chloroplast inability to do high rates of de novo protein biosynthesis, needed for the frequent replacement of the D1/32 kD reaction center protein in the H2O-oxidizing PSII complex (Mattoo and Edelman 1987; Melis 1999). In the absence of S, which is an essential component of cysteine and methionine amino acids, protein biosynthesis is impeded and the PSII repair cycle is severely retarded (Wykoff et al. 1998).
Application of the chlorophyll fluorescence techniques to C. reinhardtii, is subject to some peculiarities specific for this green microalga. For example, state transitions in C. reinhardtii differ from those in higher plants. In the latter, only a 15–20% fraction of light harvesting complex II (LHCII) participates in state transitions. In C. reinhardtii, a much larger fraction of PSII Chl antennae is involved in state transitions (Bassi and Wollman 1991), and a much larger decrease in PSII energy capture is observed (Delosme et al. 1994, 1996). In maximal state 2, electrons for reducing the cytochrome b 6 f complex do not originate from PSII, but from PSI (Finazzi et al. 1999), and PSII appears to be disconnected from the remainder of the electron transport chain. In fact, in C. reinhardtii cells which are in maximal state 2 condition, no photosynthetic oxygen evolution can be observed (Finazzi et al. 1999, 2002).
Furthermore, state transitions in C. reinhardtii are substantially affected by anaerobiosis. The PQ pool, whose reduction state is one of the key signals for state transitions (Wollman 2001), is maximally reduced in the absence of O2, probably because the plastidic terminal oxidase as a part of the chlororespiratory pathway cannot function (Wollman and Delepelaire 1984). In addition, oxidation of exogenously provided acetate tends to cause reduction of the PQ-pool and can result in state transitions toward state 2 in the dark (Endo and Asada 1996). Having this in mind, one has to be careful not to let the algal sample become anoxic in the dark incubation prior to the measurement, unless this is desired. On the other hand, if one takes samples from the culture container to analyze S-deprived and H2-producing C. reinhardtii cells, this might result in some aeration of the cells, causing a change in the bioenergetic status of the latter. Again, on-line measurements within a bioreactor are much better suited for the monitoring of the bioenergetic status of the photosynthetic apparatus and the cells themselves.
Screening systems for the targeted isolation of mutants with an altered H2 metabolism
Basic research on H2 metabolism and efforts to increase yields of H2 production by the microalgae make use of well-established techniques allowing forward and reverse genetics in C. reinhardtii (Galván et al. 2007). To identify genes whose products are involved in the H2 metabolism of C. reinhardtii or to create strains with optimized phenotypes regarding H2 yields, transformant libraries are created by DNA insertional mutagenesis. This is an easy and well-established method to mutagenize C. reinhardtii and tag the affected genes simultaneously (Kindle 1990). However, to identify the strains of interest, a powerful screening system must be at hand. Here, research on both algal hydrogenases and H2 metabolism has profited from the coupling of these processes with photosynthesis.
Three screening systems with different objectives have been established, all of these relying on photosynthetic activity. The first screening protocol aims at identifying algal mutant strains with any defect affecting H2 production by making use of the fact that dark-adapted and anaerobic Chlamydomonas cells show a transient but high H2-production activity after a sudden dark–light shift. This screening utilizes the characteristics of tungsten oxide, which changes its color after being reduced by hydrogen.
The second screening system has been established both for biotechnological reasons and optimizing the analysis of photosynthetic H2 production. It selectively screens for C. reinhardtii strains in which the ratio of photosynthetic O2 evolution and respiratory O2 uptake is lowered so that these strains will establish anaerobic conditions in the light, allowing them to express the hydrogenase gene and to make use of the O2 sensitive hydrogenase.
The third screening procedure was actually developed in order to identify C. reinhardtii mutant deficient in state transitions and is based on differential PSII chlorophyll fluorescence in state 1 and state 2.
Chromogenic screening system using tungsten oxide/platinum films
The chromogenic screening system makes use of the fact that tungsten oxide powder is reduced by hydrogen atoms to a blue form, a process which is reversible at room temperature. An appropriate hydrogen detector is built up from a polycrystalline tungsten oxide film with a thin catalytic overlayer. In this film, dihydrogen molecules are dissociated into hydrogen atoms on the catalyst surface, and the reducing hydrogen atoms diffuse into the interior of the tungsten oxide particles and give rise to formation of hydrogen tungsten bronzes (Ito and Ohgami 1992).
This principle can be utilized when analyzing unicellular green algae (and other H2 producing species) with regard to the H2-evolving capacity (Seibert et al. 1998; Ghirardi et al. 2000; Posewitz et al. 2004). To utilize these H2 sensors for the identification of algal mutants deficient in H2 production, an algal mutant library must first be created. This procedure is described in Kindle (1990), and several new marker genes have been identified (Lumbreras et al. 1998; Sizova et al. 2001). The algal colonies growing on selective agar plates are then transferred to grid-divided master plates. In order to be prepared for the chromogenic screening using the above mentioned films, the growing clones are transferred to square Petri dishes (120 × 120 mm, e.g., from Greiner bio-one, www.gbo.com) in a 10 × 10 raster. One needs to prepare duplicates of each master plate since the screened plate will be non-sterile after the screening. The colonies on the plates are grown for 7–10 days in the light until they form green, dome-shaped colonies of about 3–5 mm in diameter (Fig. 6a). To carry out the screening procedure, the plates are placed in an anaerobic glove box in the dark and incubated there overnight. In the next morning, chromogenic films trimmed to fit in the Petri dishes are placed directly on the colonies so that the catalytic coating touches the cells (Fig. 6a). Now, the cells are illuminated for 3 min with a light intensity of 50–100 μmoles photons m−2 s−1. The light induces the photosynthetic activity of the algae and results in a transient photoproduction of H2 by the colonies unaffected in their H2 metabolism. The H2 released by the colonies will make contact with the chromogenic layer of the film and cause a blue staining just directly above the colony (Fig. 6a and b). Thus, the H2-producing activity of a certain algal colony will result in blue circles on the chromogenic film (Fig. 6b). Accordingly, Chlamydomonas clones affected in H2 evolution can be identified visually by a less-pronounced or absent coloring of the screening plate (Ghirardi et al. 2000). Once the blue circles are clearly visible on the chromogenic sensor, the latter is removed from the cells, carefully wiped to remove sticking algae and photographed.

a Schematic process of using chromogenic sensors coated with thin layers of platinum and tungsten oxide to identify C. reinhardtii transformants having defects in the H2-evolution pathway. The transformant colonies are grown until they form a dome-shaped colony of about 5 mm in diameter and are transferred into an anaerobic glove box in the dark to induce hydrogenase gene expression and activity, respectively. After 12 h, the chromogenic films are placed directly on the colonies. A short (about 3 min) illumination of the algae results in a sudden H2 evolution depending on PSII activity. The H2 gas is split by the platinum layer so that the H-atoms can interact with the tungsten oxide causing a blue color (shown in grayshade in b; photograph courtesy of Irene Kandlen). Algal clones with reduced or no H2-production activity can be identified by a less-pronounced or absent coloration (marked by a white circle in b)
However, there are several problems that could arise with this approach. First, the coated films need to be stored carefully to avoid the loss-of-function. They are wrapped in aluminium foil and stored in a dark room to avoid destruction of any molecules by light. However, to ensure that the screening system works, one should include several control strains on each plate to be analyzed. As a positive control, the C. reinhardtii wild type (e.g., strain CC-124, wild type mt-137, which is available at www.chlamy.org/strains.html) can be used, and it should be applied on the screening plate at several places. As a negative control, one could use a PSII-deficient strain (e.g., C. reinhardtii CC-1284 FUD7 mt-, which has a deletion of the plastidic psbA gene). Since the H2 production of Chlamydomonas cells anaerobically adapted in the dark and suddenly shifted to the light is, to a large part, dependent on PSII activity (Mus et al. 2005), chromogenic films above the colonies of these PSII-deficient strains should not turn blue. To be absolutely sure, one can also use PSI-deficient strains (e.g., CC-4151 FUD26 mt+); however, these are quite light sensitive and might not grow well under the normal light conditions applied to grow the Chlamydomonas clones.
A further point to which attention needs to be paid is the illumination phase of the anaerobically adapted colonies. As mentioned in the introduction, the O2 gas evolved by activated PSII will rapidly inactivate the hydrogenase enzyme. Thus, if the illumination phase is too long or the light intensity is too high, the H2-production phase of the cultures is very short and the blue staining of the chromogenic layer might not be intensive enough.
After potential strains have been identified, these have to be characterized in more detail and under more reproducible conditions. This screening can identify mutants with entirely different genetic backgrounds. It will identify photosynthetic mutants affected in the linear electron transport chain or in the chlororespiratory pathways, mutants with knockouts in genes essential for the biosynthesis and assembly of the FeFe-hydrogenase (Posewitz et al. 2004), or strains unable to carry out the necessary gene-regulatory reactions. Thus, the putative mutant strains need to be analyzed by additional screening steps as earlier described.
Attenuation of the photosynthesis/respiration (P/R) capacity ratio in green microalgae as a tool for stabilizing H2 evolution and its metabolism
A second screening system has been established in order to specifically identify C. reinhardtii mutant strains affected in the ratio of photosynthetic O2 evolution and respiratory O2 consumption (Rühle et al. 2008). Utilization of the cell’s own respiration to consume photosynthetically generated O2 has proven to be a successful strategy for initializing hydrogenase activity in the algae. The balanced interaction between the two bioenergetic organelles in S-deprived cells is currently the only available platform for the further investigation of H2 metabolism in microalgae (Melis and Happe 2001; Melis 2007), and offers the only approach available for a sustained photobiological hydrogen production. It is therefore desirable to develop transgenic microalgae in which the photosynthesis/respiration (P/R) capacity ratio of cells growing in nutrient-replete medium is genetically defined not to exceed the 1:1 ratio without altering the high-quantum yield of photosynthesis. C. reinhardtii, and other green microalgae, naturally possess a photosynthesis/respiration (P/R) capacity ratio of about 4:1 (Melis et al. 2000; Zhang et al. 2002). Attenuating the cellular P/R capacity ratio to a value that is equal to or less than unity, without altering the high-quantum yield of photosynthesis, would permit C. reinhardtii to grow photo-heterotrophically in the presence of acetate. In sealed cultures, anaerobiosis would prevail, lifting the O2-dependent suppression of hydrogenase gene expression, which is the first step to permitting a light-dependent H2 evolution. Such constitutive expression of the FeFe-hydrogenase pathway and the resulting photosynthetic H2 metabolism would occur with physiological levels of S, or other nutrients, in the chloroplast. Accordingly, genes that lower the capacity of photosynthesis and/or enhance the capacity of respiration in C. reinhardtii, without altering the high-quantum yield of photosynthesis, are of keen interest in this field.
The creation of appropriate C. reinhardtii mutants can be achieved by applying DNA insertional mutagenesis; however, the isolation of strains with the desired phenotype requires development of a specific and stringent high throughput screening protocol.
The purpose of reaching photobiological H2 production under normal growth conditions excludes the usage of C. reinhardtii strains with severe defects in photosynthetic electron transport, as the latter provides electrons for H2 generation either directly (PSII) or indirectly (starch). The common screening system which has been successfully applied to find photosynthetic mutants, the screening for acetate requiring C. reinhardtii strains (Spreitzer and Mets 1981), is therefore inappropriate for the aim of finding algae with a continuous and nutrient-independent H2-production capability.
Thus, a screening system which specifically targets algal strains with a lowered P/R ratio was developed based on the Winkler test used to determine the level of dissolved oxygen in water samples (Rühle et al. 2008). The Winkler test, which detects the presence of oxygen in four chemical reactions, can be applied to phototrophically grown green transformant microalgae to identify strains that are photosynthetically competent but do not evolve O2 as the latter is consumed by the cell’s own respiration (P/R < 1) (Rühle et al. 2008).
To carry out this screening protocol, colonies from an algal mutant library are transferred to 48-well plates (Corning incorporated, costar®; Corning New York, total well volume of 1.6 ml) containing 200 μl of TAP-medium per well. To grow the cells, the plates are exposed to low light for several days. To induce the same physiological state in each well, 800 μl of fresh TAP-medium and a sterile solid glass bead (diameter 3 mm) are added to the individual cell suspensions in order to prepare them for the screening. These glass beads are very efficient for mixing algal suspensions in multi-well plates. The plates to be screened are then placed on a shaker in the light (40–80 μE m−2 s−1) for 6 h. To “reset” the O2 concentration of each well just prior the screening procedure, the plates are transferred to an anaerobic glove box in the dark (e.g., Glove Bag™, inflatable glove chamber model “X”, I2R®/Glas-Col, www.glascol.com), which is flushed with N2, or an anaerobic tent. This anaerobic incubation of the cells in the dark results in a complete respiratory consumption of dissolved O2. To induce photosynthetic O2 evolution of the cells, the plates are then exposed to light (70–100 μE m−2 s−1) for 20–30 min. Now, the chemical reactions of the Winkler test are induced by successively adding 10 μl MnCl2 (0.34 M) and 10 μl KI/NaOH (0.24 M/1.2 M) to each well. In the alkaline solution, dissolved O2 will oxidize the Mn(II) ions to Mn(III) ions. After mixing, 50 μl H3PO4 (v/v 50%) are added in order to acidify the solution and dissolve the brown manganese precipitate. The Mn(II) cations liberated oxidize iodide (I−) to iodine (I2). All these steps are conducted while the plates are still in the anaerobic environment to avoid atmospheric O2 to diffuse into the algal suspensions and falsify the results. The subsequent steps can then be performed under aerobic conditions. In order to visualize iodine formation, 10 μl of 4% (w/v) starch solution is supplemented to the test solution resulting in a dark-blue color because of the iodine intercalating in the starch helices (Fig. 7). After this step, algal transformant strains which have produced significantly less O2 are already notable because of a less pronounced or even absent blue color. However, to determine less-pronounced variations of the O2 concentrations in each well, the suspension is further titrated with sodium thiosulfate until the blue color has disappeared. Sodium thiosulfate stoichiometrically converts I2 back into I−, so that the amount of sodium thiosulfate necessary to eliminate the blue color is equivalent to the previous concentration of O2 in the well (Rühle et al. 2008).

Photograph of a 48-well plate after treating the wells according to the Winkler test. A deep blue color indicates that normal amounts of O2 were dissolved in the culture medium, whereas the O2 concentration was lower or very low in the light-blue or uncolored wells, respectively (photograph courtesy of Thilo Rühle)
Applying this screening, several Chlamydomonas transformants establishing anaerobic conditions in full medium in the light have been isolated (Rühle et al. 2008). First physiological and biochemical analyses have shown that this procedure allows to find transformants having diverse defects of photosynthesis, but are still able to grow photosynthetically. Thus, it is a screening protocol also suited for research on photosynthesis aiming at finding genes whose knockout does not result in the loss-of-function, but in less-pronounced impairments of the photosynthetic metabolism.
Fluorescence imaging systems for the isolation of C. reinhardtii mutants deficient in state transitions
The growing knowledge about the changes of the photosynthetic electron transport chain that lead to H2 production and the status of the former during ongoing H2 generation have led to several hypotheses as to how the H2 yields of C. reinhardtii can be optimized by manipulating photosynthesis. One approach is the creation of algal transformants with reduced P/R ratios as described above (Rühle et al. 2008). Others have stated that the cyclic electron transport around PSI and the cytochrome b 6 f complex was an additional electron sink with which the hydrogenase has to compete, therefore lowering the H2 yields (Kruse et al. 2005). Especially the latter idea did benefit from a computer-aided fluorescence imaging system developed and described in detail in 1990 by Fenton and Crofts. This setup allows the recording of images of the chlorophyll fluorescence intensity from a field of view, which might cover a whole plant leaf or a whole Petri-dish with colonies of photosynthetic bacteria or microalgae. This system has been adapted to isolate C. reinhardtii mutant strains deficient in state transitions by measuring the fluorescence yield of whole algal colonies on an agar plate at room temperature (Fleischmann et al. 1999; Kruse et al. 1999). The screening is based on the fact that PSII chlorophyll fluorescence is inversely proportional to the yield of PSII photochemistry and directly proportional to the size of the light harvesting antennae associated with PSII (Butler 1978). Especially when excluding any influence of PSII photochemistry by adding DCMU, the changes of the PSII antennae size upon state transition can be directly followed by changes of chlorophyll fluorescence yields (Finazzi et al. 2001a, b).
These changes in fluorescence can be visualized by the abovementioned video imaging system, which has been described in detail, e.g., by Fenton and Crofts (1990) and by Kruse et al. (1999). This system significantly simplifies the whole screening procedure of even large Chlamydomonas transformant libraries. The generation of the latter usually begins with transformation of the cells by a selectable marker gene. The transformed cells are then plated on selective agar plates. On these first plates, successfully transformed clones grow in unorganized patterns. Most screening procedures require the transfer of every single colony to new master plates in an organized raster, so that several thousand clones have to be transferred, though only a tiny fraction of them will turn out to have the desired phenotype.
In contrast, the fluorescence imaging system allows screening the algal colonies already on the first, unorganized agar plates, given that the colonies have approximately the same size, which usually is the case.
Furthermore, the strategies used in order to force C. reinhardtii cells into state 1 or state 2 are applicable on whole agar plates. Fleischmann et al. (1999) plated the transformed cells directly on TAP agar plates containing DCMU and incubated the plates in low light (6 μE m−2 s−1). As mentioned above, the inhibition of PSII photochemistry allows to directly concluding the state from PSII fluorescence at room temperature. In these DCMU-treated algal colonies, state 1 could then easily be achieved by illuminating the cells with white light, resulting in the oxidation of the PQ pool by PSI activity. State 2 was achieved by making use of the fact that anaerobic and dark-incubated C. reinhardtii cells have a reduced PQ pool and therefore shift to state 2 (Wollman and Delepelaire 1984). With an appropriate setup, whole Petri dishes can be flushed with N2 in the dark, forcing the algal colonies into state 2 (Fleischmann et al. 1999). Applying these treatments to the agar plates harboring Chlamydomonas transformant colonies, fluorescence pictures of the whole plates can be recorded and numerically subtracted, so that the fluorescence difference of each colony provides a measure of state transition. While C. reinhardtii wild-type colonies display strong signals, strains deficient in state transitions show weak or nearly undetectable signals (Fleischmann et al. 1999).
Kruse et al. (1999) used a similar technical setup, but applied a different strategy to induce state transitions in the microalgae. They grew the algal colonies on High Salt Minimal medium (Harris 1989, 2009), which prevents the spontaneous transition to state 2 that can be observed in the presence of acetate (Endo and Asada 1996; Finazzi et al. 2001b). For the actual screening procedure, the authors made use of the well-known fact that PSII and PSI are preferentially excitable by different light qualities. The algal colonies were adapted to state 2 by preferentially exciting PSII with light of λ = 620 nm. Vice versa, the cells were forced into state 1 by exciting PSI with light of λ = 695 nm (Kruse et al. 1999).
By utilizing such a fluorescence image-based screening system to identify C. reinhardtii cells deficient in state transitions and subsequent analyses of the H2 yields of the identified strains, Kruse et al. (2005) found C. reinhardtii strain Stm6. This strain was shown to be deficient in MOC1, which is related to a mitochondrial transcription termination factor (mTERF) (Schönfeld et al. 2004). The phenotype of strain Stm6 includes, besides being blocked in state 1, sensitivity toward high light, drastic changes in composition and function of the mitochondrial respiratory chain and the accumulation of large amounts of starch (Schönfeld et al. 2004; Kruse et al. 2005). Most interestingly with regard of the purpose of this study, C. reinhardtii strain Stm6 shows a higher H2 evolution than its parental strain (C. reinhardtii CC-1618) both after a dark–light shift and upon S deprivation (Kruse et al. 2005). It is unclear yet, which of the single altered characteristics of the strain or a combination of all of them leads to the higher H2 yields. However, this study is a nice example of how the study on the H2 metabolism of photosynthetic microorganisms can benefit from techniques established in order to analyze photosynthesis.
Conclusion
Photobiological H2 production by unicellular green algae has become an important research field because of its potential to be applied in renewable energy production. In addition, the research already done has shown that the analysis of this fascinating metabolism also contributed to a deeper understanding of photosynthesis, since the latter is drastically re-directed, especially in S-deprived H2-producing microalgae. Investigations on this re-organization in bioenergetics and metabolism benefited strongly from new techniques designed in order to analyze photosynthesis, as the screening for algal mutant strains with an altered H2 metabolism mostly depends on its coupling to the photosynthetic electron transport chain. On the other hand, methods to induce and analyze H2 production in green algae described in this article might help in characterizing the photosynthetic apparatus of the cells under special environmental conditions and/or in mutant strains with useful alterations in the characteristics of their photosynthesis.
Abbreviations
- ATP:
-
Adenosine triphosphate
- GC:
-
Gas chromatography/chromatograph
- HSM:
-
High salt minimal
- LHC:
-
Light harvesting complex
- MS:
-
Mass spectrometry/spectrometer
- MIMS:
-
Membrane inlet mass spectrometer
- NAD(P):
-
Nicotinamide dinucleotide(phosphate)
- PQ:
-
Plastoquinone
- PSII, PSI:
-
Photosystem II, photosystem I
- Rubisco:
-
Ribulosebisphosphate carboxylase/oxygenase
- TAP:
-
Tris acetate phosphate
References
Antal TK, Krendeleva TE, Laurinavichene TV, Makarova VV, Ghirardi ML, Rubin AB, Tsygankov AA, Seibert M (2003) The dependence of algal H2-production on photosystem II and O2 consumption activities in sulphur-deprived Chlamydomonas reinhardtii cells. Biochim Biophys Acta 1607:153–160. doi:10.1016/j.bbabio.2003.09.008
Arnon D (1949) Copper enzymes in isolated chloroplasts and polyphenol oxidase in Beta vulgaris. Plant Physiol 24:1–5. doi:10.1104/pp.24.1.1
Baker NR (2008) Chlorophyll fluorescence: a probe of photosynthesis in vivo. Annu Rev Plant Biol 59:89–113. doi:10.1146/annurev.arplant.59.032607.092759
Bassi R, Wollman F-A (1991) The chlorophyll-a/b proteins of photosystem II in Chlamydomonas reinhardtii. Isolation, characterization and immunological cross-reactivity to higher plant polypeptides. Planta 183:423–433. doi:10.1007/BF00197742
Bernard L, Desplats C, Mus F, Cuiné S, Cournac L, Peltier G (2006) Agrobacterium tumefaciens type II NADH dehydrogenase. Characterization and interactions with bacterial and thylakoid membranes. FEBS J 273:3625–3637. doi:10.1111/j.1742-4658.2006.05370.x
Butler WL (1978) Energy distribution in the photochemical apparatus of photosynthesis. Annu Rev Plant Physiol 29:345–378
Cournac L, Guedeney G, Peltier G, Vignais PM (2004) Sustained photoevolution of molecular hydrogen in a mutant of Synechocystis sp. Strain PCC 6803 deficient in the type I NADPH-dehydrogenase complex. J Bacteriol 186:1737–1746. doi:10.1128/JB.186.6.1737-1746.2003
Davies JP, Weeks DP, Grossman AR (1992) Expression of the arylsulfatase gene from the beta 2-tubulin promoter in Chlamydomonas reinhardtii. Nucleic Acids Res 20:2959–2965. doi:10.1093/nar/20.12.2959
Delosme R, Béal D, Joliot P (1994) Photoacoustic detection of flash-induced charge separation in photosynthetic systems. Spectral dependence of the quantum yield. Biochim Biophys Acta 1185:56–64. doi:10.1016/0005-2728(94)90193-7
Delosme R, Olive J, Wollmann F-A (1996) Changes in light energy distribution upon state transitions: an in vivo photoacoustic study of the wildtype and photosynthesis mutants from Chlamydomonas reinhardtii. Biochim Biophys Acta 1273:150–158. doi:10.1016/0005-2728(95)00143-3
Dimon B, Gans P, Peltier G (1988) Mass spectrometric measurement of photosynthetic and respiratory oxygen exchange. Methods Enzymol 167:686–691. doi:10.1016/0076-6879(88)67079-0
Endo T, Asada K (1996) Dark induction of the non-photochemical quenching of chlorophyll fluorescence by acetate in Chlamydomonas reinhardtii. Plant Cell Physiol 37:551–555
Eriksen NT (2008) The technology of microalgal culturing. Biotechnol Lett 30:1525–1536. doi:10.1007/s10529-008-9740-3
Fenton JM, Crofts AR (1990) Computer aided fluorescence imaging of photosynthetic systems. Photosynth Res 26:59–66
Finazzi G, Furia A, Barbagallo RP, Forti G (1999) State transitions, cyclic and linear electron transport and photophosphorylation in Chlamydomonas reinhardtii. Biochim Biophys Acta 1413:117–129. doi:10.1016/S0005-2728(99)00089-4
Finazzi G, Zito F, Barbagallo RP, Wollman FA (2001a) Contrasted effects of inhibitors of cytochrome b6f complex on state transitions in Chlamydomonas reinhardtii: the role of Qo site occupancy in LHCII kinase activation. J Biol Chem 276:9770–9774. doi:10.1074/jbc.M010092200
Finazzi G, Barbagallo RP, Bergo E, Barbato R, Forti G (2001b) Photoinhibition of Chlamydomonas reinhardtii in state 1 and state 2 (damages to the photosynthetic apparatus under linear and cyclic electron flow). J Biol Chem 276:22251–22257. doi:10.1074/jbc.M011376200
Finazzi G, Rappaport F, Furia A, Fleischmann M, Rochaix JD, Zito F, Forti G (2002) Involvements of state transitions in the switch between linear and cyclic electron flow in Chlamydomonas reinhardtii. EMBO Rep 3:280–285. doi:10.1093/embo-reports/kvf047
Fleischmann MM, Ravanel S, Delosme R, Olive J, Zito F, Wollman FA, Rochaix JD (1999) Isolation and characterization of photoautotrophic mutants of Chlamydomonas reinhardtii deficient in state transition. J Biol Chem 274:30987–30994. doi:10.1074/jbc.274.43.30987
Florin L, Tsokoglou A, Happe T (2001) A novel type of iron hydrogenase in the green alga Scenedesmus obliquus is linked to the photosynthetic electron transport chain. J Biol Chem 276:6125–6132. doi:10.1074/jbc.M008470200
Forestier M, King P, Posewitz M, Schwarzer S, Happe T, Zhang L, Ghirardi ML, Seibert M (2003) Expression of two [Fe]-hydrogenases in Chlamydomonas reinhardtii under anaerobic conditions. Eur J Biochem 270:2750–2758. doi:10.1046/j.1432-1033.2003.03656
Fouchard S, Hemschemeier A, Caruana A, Pruvost J, Legrand J, Happe T, Peltier G, Cournac L (2005) Autotrophic and mixotrophic hydrogen photoproduction in sulfur-deprived Chlamydomonas cells. Appl Environ Microbiol 71:6199–6205. doi:10.1128/AEM.71.10.6199-6205.2005
Gaffron H (1939) Reduction of CO2 with H2 in green plants. Nature 143:204–205. doi:10.1038/143204a0
Galván A, González-Ballester D, Fernández E (2007) Insertional mutagenesis as a tool to study genes/functions in Chlamydomonas. Adv Exp Med Biol 616:77–89. doi:10.1007/978-0-387-75532-8_7
Gfeller RP, Gibbs M (1984) Fermentative metabolism of Chlamydomonas reinhardtii. I. Analysis of fermentative products from starch in dark and light. Plant Physiol 75:212–218. doi:10.1104/pp.75.1.212
Ghirardi ML, Togasaki RK, Seibert M (1997) Oxygen sensitivity of algal H2-production. Appl Biochem Biotech 63:141–151
Ghirardi ML, Zhang L, Lee JW, Flynn T, Seibert M, Greenbaum E, Melis A (2000) Microalgae: a green source of renewable H2. Trends Biotechnol 18:506–511. doi:10.1016/S0167-7799(00)01511-0
Grossman AR (2000) Acclimation of Chlamydomonas reinhardtii to its nutrient environment. Protist 151:201–224. doi:10.1078/1434-4610-00020
Happe T, Kaminski A (2002) Differential regulation of the Fe-hydrogenase during anaerobic adaptation in the green alga Chlamydomonas reinhardtii. Eur J Biochem 269:1022–1032
Happe T, Naber JD (1993) Isolation, characterization and N-terminal amino acid sequence of hydrogenase from the green alga Chlamydomonas reinhardtii. Eur J Biochem 214:475–481. doi:10.1111/j.1432-1033.1993.tb17944.x
Happe T, Mosler B, Naber JD (1994) Induction, localization and metal content of hydrogenase in Chlamydomonas reinhardtii. Eur J Biochem 222:769–775. doi:10.1111/j.1432-1033.1994.tb18923.x
Harris EH (1989) The Chlamydomonas sourcebook. Academic Press Inc, San Diego
Harris EH (2009) The Chlamydomonas sourcebook (second edition). Introduction to Chlamydomonas and Its Laboratory Use, vol 1. Academic Press, San Diego
Hemschemeier A (2005) The anaerobic life of the photosynthetic alga Chlamydomonas reinhardtii. Photofermentation and hydrogen production upon sulphur deprivation. PhD-thesis, Ruhr-University of Bochum. http://www-brs.ub.ruhr-uni-bochum.de/netahtml/HSS/Diss/HemschemeierAnjaChristine/diss.pdf
Hemschemeier A, Fouchard S, Cournac L, Peltier G, Happe T (2008) Hydrogen production by Chlamydomonas reinhardtii: an elaborate interplay of electron sources and sinks. Planta 227:397–407. doi:10.1007/s00425-007-0626-8
Ito K, Ohgami T (1992) Hydrogen detection based on coloration of anodic tungsten oxide film. Appl Phys Lett 60:938–940. doi:10.1063/1.106467
Jouanneau Y, Kelley BC, Berlier Y, Lespinat PA, Vignais PM (1980) Continuous monitoring, by mass spectrometry, of H2-production and recycling in Rhodopseudomonas capsulate. J Bacteriol 143:628–636
Kamp C, Silakov A, Winkler M, Reijerse EJ, Lubitz W, Happe T (2008) Isolation and first EPR characterization of the [FeFe]-hydrogenases from green algae. Biochim Biophys Acta 1777:410–416. doi:10.1016/j.bbabio.2008.02.002
Kessler E (1974) Hydrogenase, photoreduction and anaerobic growth of algae. In: Steward WDP (ed) Algal physiology and biochemistry. Blackwell, Oxford
Kindle KL (1990) High frequency nuclear transformation of Chlamydomonas reinhardtii. Proc Natl Acad Sci USA 87:1228–1232
King PW, Posewitz MC, Ghirardi ML, Seibert M (2006) Functional studies of [FeFe] hydrogenase maturation in an Escherichia coli biosynthetic system. J Bacteriol 188:2163–2172. doi:10.1128/JB.188.6.2163-2172.2006
Kitajima M, Butler WL (1975) Quenching of chlorophyll fluorescence and primary photochemistry in chloroplasts by dibromothymoquinone. Biochim Biophys Acta 376:105–115. doi:10.1016/0005-2728(75)90209-1
Konermann L, Messinger J, Hillier W (2008) Mass spectrometry-based methods for studying kinetics and dynamics in biological systems. In: Aartsma TJ, Matysik J (eds) Biophysical techniques in photosynthesis research (volume II), series advances in photosynthesis and respiration, vol 26. Springer, Dordrecht, pp 167–190
Kosourov S, Tsygankov A, Seibert M, Ghirardi ML (2002) Sustained hydrogen photoproduction by Chlamydomonas reinhardtii: effects of culture parameters. Biotechnol Bioeng 78:731–740. doi:10.1002/bit.10254
Kosourov S, Seibert M, Ghirardi ML (2003) Effects of extracellular pH on the metabolic pathways in sulfur-deprived, H2-producing Chlamydomonas reinhardtii cultures. Plant Cell Physiol 44:146–155. doi:10.1093/pcp/pcg020
Kosourov S, Patrusheva E, Ghirardi ML, Seibert M, Tsygankov A (2007) A comparison of hydrogen photoproduction by sulfur-deprived Chlamydomonas reinhardtii under different growth conditions. J Biotechnol 128:776–787. doi:10.1016/j.jbiotec.2006.12.025
Kruse O, Nixon PJ, Schmid GH, Mullineaux CW (1999) Isolation of state transition mutants of Chlamydomonas reinhardtii by fluorescence video imaging. Photosynth Res 61:43–51. doi:10.1023/A:1006229308606
Kruse O, Rupprecht J, Bader KP, Thomas-Hall S, Schenk PM, Finazzi G, Hankamer B (2005) Improved photobiological H2 production in engineered green algal cells. J Biol Chem 280:34170–34177. doi:10.1074/jbc.M503840200
Kuroda K, Silveira RG, Nishio N, Sunahara H, Nagap S (1991) Measurement of dissolved hydrogen in an anaerobic digestion process by a membrane-covered electrode. J Ferment Bioeng 71:418–423. doi:10.1016/0922-338X(91)90254-E
Laurinavichene T, Tolstygina I, Tsygankov A (2004) The effect of light intensity on hydrogen production by sulfur-deprived Chlamydomonas reinhardtii. J Biotechnol 114:143–151. doi:10.1016/j.jbiotec.2004.05.012
Leroux F, Dementin S, Burlat B, Cournac L, Volbeda A, Champ S, Martin L, Guigliarelli B, Bertrand P, Fontecilla-Camps J, Rousset M, Léger C (2008) Experimental approaches to kinetics of gas diffusion in hydrogenase. Proc Natl Acad Sci USA 105:11188–11193. doi:10.1073/pnas.0803689105
Lien T, Schreiner O (1975) Purification of a derepressible arylsulfatase from Chlamydomonas reinhardtii. Biochim Biophys Acta 384:168–179
Lindberg P, Lindblad P, Cournac L (2004) Gas exchange in the filamentous cyanobacterium Nostoc punctiforme strain ATCC 29133 and its hydrogenase-deficient mutant Strain NHM5. Appl Environ Microbiol 70:2137–2145. doi:10.1128/AEM.70.4.2137-2145.2004
Lumbreras V, Stevens DR, Purton S (1998) Efficient foreign gene expression in Chlamydomonas reinhardtii mediated by an endogenous intron. Plant J 14:441–447
Makarova VV, Kosourov S, Krendeleva TE, Semin BK, Kukarskikh GP, Rubin AB, Sayre RT, Ghirardi ML, Seibert M (2007) Photoproduction of hydrogen by sulfur-deprived C. reinhardtii mutants with impaired photosystem II photochemical activity. Photosynth Res 94:79–89. doi:10.1007/s11120-007-9219-4
Mattoo AK, Edelman M (1987) Intramembrane translocation and posttranslational palmitoylation of the chloroplast 32 kDa herbicide-binding protein. Proc Natl Acad Sci USA 84:1497–1501. doi:10.1073/pnas.84.6.1497
Melis A (1999) Photosystem-II damage and repair cycle in chloroplasts: what modulates the rate of photodamage in vivo? Trends Plant Sci 4:130–135. doi:10.1016/S1360-1385(99)01387-4
Melis A (2007) Photosynthetic H2 metabolism in Chlamydomonas reinhardtii (unicellular green algae). Planta 226:1075–1086. doi:10.1007/s00425-007-0609-9
Melis A, Happe T (2001) Hydrogen production. Green algae as a source of energy. Plant Physiol 127:740–748. doi:10.1104/pp.010498
Melis A, Happe T (2004) Trails of green alga hydrogen research—from Hans Gaffron to new frontiers. Photosynth Res 80:401–409. doi:10.1023/B:PRES.0000030421.31730.cb
Melis A, Zhang L, Forestier M, Ghirardi ML, Seibert M (2000) Sustained photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the green alga Chlamydomonas reinhardtii. Plant Physiol 122:127–135. doi:10.1104/pp.122.1.127
Melis A, Seibert M, Happe T (2004) Genomics of green algal hydrogen research. Photosynth Res 82:277–288. doi:10.1007/s11120-004-2050-2
Messinger J, Badger M, Wydrzynski T (1995) Detection of one slowly exchanging substrate water molecule in the S3 state of photosystem II. Proc Natl Acad Sci USA 92:3209–3213. doi:10.1073/pnas.92.8.3209
Mus F, Cournac L, Cardettini V, Caruana A, Peltier G (2005) Inhibitor studies on non-photochemical plastoquinone reduction and H2 photoproduction in Chlamydomonas reinhardtii. Biochim Biophys Acta 1708:322–332. doi:10.1016/j.bbabio.2005.05.003
Papgeorgiou GC, Tsimilli-Michael M, Stamatalis K (2007) The fast and slow kinetics of chlorophyll a fluorescence induction in plants, algae and cyanobacteria: a viewpoint. Photosynth Res 94:275–290. doi:10.1007/s11120-007-9193-x
Posewitz MC, King PW, Smolinski SL, Zhang L, Seibert M, Ghirardi ML (2004) Discovery of two novel radical S-adenosylmethionine proteins required for the assembly of an active [Fe] hydrogenase. J Biol Chem 279:25711–25720. doi:10.1074/jbc.M403206200
Quinn JM, Merchant S (1998) Copper-responsive gene expression during adaptation to copper deficiency. Methods Enzymol 297:263–279. doi:10.1016/S0076-6879(98)97020-3
Rühle T, Hemschemeier A, Melis A, Happe T (2008) A novel screening protocol for the isolation of hydrogen producing Chlamydomonas reinhardtii strains. BMC Plant Biol 8:107. http://www.biomedcentral.com/1471-2229/8/107. doi:10.1186/1471-2229-8-107
Rumeau D, Peltier G, Cournac L (2007) Chlororespiration and cyclic electron flow around PSI during photosynthesis and plant stress response. Plant Cell Environ 30:1041–1051. doi:10.1111/j.1365-3040.2007.01675.x
Schönfeld C, Wobbe L, Borgstadt R, Kienast A, Nixon PJ, Kruse O (2004) The nucleus-encoded protein MOC1 is essential for mitochondrial light acclimation in Chlamydomonas reinhardtii. J Biol Chem 279:50366–50374. doi:10.1074/jbc.M408477200
Schütz K, Happe T, Troshina O, Lindblad P, Leitao E, Oliveira P, Tamagnini P (2004) Cyanobacterial H2 production—a comparative analysis. Planta 218:350–359. doi:10.1007/s00425-003-1113-5
Seibert M, Flynn T, Benson D, Tracy E, Ghirardi M (1998) Development of selection/screening procedures for rapid identification of hydrogen-producing algal mutants with increased oxygen tolerance. In: Zaborski OR (ed) Biohydrogen: proceedings of the international conference of biological hydrogen production. Plenum Publ Corp, New York
Shevela D, Su JH, Klimov V, Messinger J (2008) Hydrogencarbonate is not a tightly bound constituent of the water-oxidizing complex in photosystem II. Biochim Biophys Acta 1777:532–539. doi:10.1016/j.bbabio.2008.03.031
Shima S, Thauer RK (2007) A third type of hydrogenase catalyzing H2 activation. Chem Rec 7:37–46. doi:10.1002/tcr.20111
Shima S, Pilak O, Vogt S, Schick M, Stagni MS, Meyer-Klaucke W, Warkentin E, Thauer RK, Ermler U (2008) The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site. Science 321:572–575. doi:10.1126/science.1158978
Sizova I, Fuhrmann M, Hegemann P (2001) A Streptomyces rimosus aphVIII gene coding for a new type phosphotransferase provides stable antibiotic resistance to Chlamydomonas reinhardtii. Gene 277:221–229
Skjånes K, Knutsen G, Källqvist T, Lindblad P (2008) H2 production from marine and freshwater species of green algae during sulfur deprivation and considerations for bioreactor design. Int J Hydrogen Energy 33:511–521. doi:10.1016/j.ijhydene.2007.09.040
Spreitzer RJ, Mets L (1981) Photosynthesis-deficient mutants of Chlamydomonas reinhardii with associated light-sensitive phenotypes. Plant Physiol 67:565–569. doi:10.1104/pp.67.3.565
Takeshita T, Tanaka K, Ishizaki A, Stanbury PF (1993) Development of a dissolved hydrogen sensor and its application to evaluation of hydrogen mass transfer. J Ferment Bioeng 76:148–150. doi:10.1016/0922-338X(93)90073-H
Tsygankov A, Kosourov S, Tolstygina I, Ghirardi ML, Seibert M (2006) Hydrogen production by sulfur-deprived Chlamydomonas reinhardtii under photoautotrophic conditions. Int J Hydrogen Energy 31:1574–1584. doi:10.1016/j.ijhydene.2006.06.024
Vignais PM, Billoud B (2007) Occurrence, classification, and biological function of hydrogenases: an overview. Chem Rev 107:4206–4272. doi:10.1021/cr050196r
Vignais PM, Billoud B, Meyer J (2001) Classification and phylogeny of hydrogenases. FEMS Microbiol Rev 25:455–501
Wang TW (1980) Amperometric hydrogen electrode. Methods Enzymol 69:409–413. doi:10.1016/S0076-6879(80)69040-5
Winkler M, Heil B, Heil B, Happe T (2002a) Isolation and molecular characterization of the [Fe]-hydrogenase from the unicellular green alga Chlorella fusca. Biochim Biophys Acta 1576:330–334
Winkler M, Hemschemeier A, Gotor C, Melis A, Happe T (2002b) [Fe]-hydrogenases in green algae: photo-fermentation and hydrogen evolution under sulfur-deprivation. Int J Hydrogen Energy 27:1431–1439. doi:10.1016/S0360-3199(02)00095-2
Winkler M, Maeurer C, Hemschemeier A, Happe T (2002c) The isolation of green algal strains with outstanding H2-productivity. In: Miyake J, Igarashi Y, Roegner M (eds) Biohydrogen III. Elsevier Science, Oxford, pp 103–115
Wollman FA (2001) State transitions reveal the dynamics and flexibility of the photosynthetic apparatus. EMBO J 20:3623–3630. doi:10.1093/emboj/20.14.3623
Wollman F-A, Delepelaire P (1984) Correlation between changes in light energy distribution and changes in thylakoid membrane polypeptide phosphorylation in Chlamydomonas reinhardtii. J Cell Biol 98:1–7. doi:10.1083/jcb.98.1.1
Wykoff DD, Davies JP, Melis A, Grossman AR (1998) The regulation of photosynthetic electron transport during nutrient deprivation in Chlamydomonas reinhardtii. Plant Physiol 117:129–139. doi:10.1104/pp.117.1.129
Zhang L, Happe T, Melis A (2002) Biochemical and morphological characterization of sulfur-deprived and H2-producing Chlamydomonas reinhardtii (green alga). Planta 214:552–561. doi:10.1007/s004250100660
Zirngibl C, Hedderich R, Thauer RK (1990) N5, N10-Methylenetetrahydromethanopterin dehydrogenase from Methanobacterium thermoautotrophicum has hydrogenase activity. FEBS Lett 261:112–116. doi:10.1016/0014-5793(90)80649-4
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hemschemeier, A., Melis, A. & Happe, T. Analytical approaches to photobiological hydrogen production in unicellular green algae. Photosynth Res 102, 523–540 (2009). https://doi.org/10.1007/s11120-009-9415-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-009-9415-5