
Abstract
The Gram-negative bacterium Pseudomonas aeruginosa simultaneously expresses two O-antigenic glycoforms. While the O-specific antigen (OSA) is variable in composition, the common polysaccharide antigen (CPA) is highly conserved and is composed of a homopolymer of D-rhamnose (D-Rha) in trisaccharide repeating units [D-Rhaα1-2-D-Rhaα1-3-D-Rhaɑ1-3]n. We have previously reported that α3-D-Rha-transferase WbpZ transfers a D-Rha residue from GDP-D-Rha to D-GlcNAcα-O-PO3-PO3-(CH2)11-O-phenyl. Genes encoding two more D-Rha-transferases are found in the O antigen gene cluster (wbpX and wbpY). In this study we showed that WbpX and WbpY recombinantly expressed in E. coli differ in their donor and acceptor specificities and have properties of GT-B folded enzymes of the GT4 glycosyltransferase family. NMR spectroscopic analysis of the WbpY reaction product showed that WbpY transferred one D-Rha residue in α1-3 linkage to synthetic D-Rhaα1-3-D-GlcNAcα-O-PO3-PO3-(CH2)11-O-phenyl acceptor. WbpX synthesized several products that contained D-Rha in both α1-2 and α1-3 linkages. Mass spectrometry indicated that the mixture of WbpX and WbpY efficiently catalyzed the synthesis of D-Rha oligomers in a non-processive mechanism. Since O antigens are virulence factors, these findings open the door to advancing technology for antibacterial drug discovery and vaccine development.
Similar content being viewed by others

Introduction
Pseudomonas aeruginosa (PA) are opportunistic, pathogenic bacteria, commonly causing nosocomial infections in immunocompromised individuals [1, 2]. This pathogen is responsible for the majority of chronic pulmonary infections in cystic fibrosis (CF) patients and has a high incidence of morbidity [3]. It is also associated with sepsis, wound infections and antibiotic resistance. Antibiotic treatment of PA infections is particularly difficult due to many intrinsic bacterial defences such as the formation of biofilms, expression of drug efflux pumps, and low outer membrane (OM) permeability [4, 5].
The lipopolysaccharides (LPS) expressed by PA protect the organism from the external environment [6] and interact with the host immune system through their most distal polysaccharide, the O antigen. O antigens determine the physiochemical characteristics of bacteria, their interactions and binding to plastic, glass or metal surfaces [7]. In particular, the common polysaccharide antigen (CPA) of PA plays a significant role in biofilm formation [8].
Unlike other Gram-negative bacteria, PA expresses two types of LPS, differing in O antigen structure and mechanism of assembly. The O-specific antigen (OSA), formerly called the B band, is variable in composition among serotypes and serves for serotyping, while CPA, formerly known as the A band, is a D-rhamnose (D-Rha) homopolymer which is highly conserved among PA serotypes. D-Rha is a rare sugar that is not found in human glycoconjugates. However, Rha is most commonly found in its L-configuration in nature, including in plant and bacterial species and the OSA of PA. CPA contains repeated D-Rha trisaccharides [D-Rhaα1-2-D-Rhaα1-3-D-Rhaα1-3]n [9, 10]. It is possible that the chain is terminated with a 3-O-methyl-D-Rha residue. CPA is also a receptor for bacteriophage A7 that contains a rhamnosidase capable of hydrolyzing CPA [11]. PA have the ability to synthesize LPS with both L-Rha in the core oligosaccharide and in several of the OSA polysaccharides, and D-Rha in CPA. Although CPA is less immunogenic than OSA, as PA infections progress, OSA is downregulated while expression of LPS containing CPA is maintained, suggesting that CPA is a stronger virulence factor over the course of an infection [12].
The putative sugar-1-phosphate transferase WbpL is required for the initiation of OSA and CPA using the phosphate-undecaprenol (P-Und) intermediate [13]. OSA is assembled by the polymerase pathway [14], suggested by the genes encoding flippase Wzx, polymerase Wzy and chain length regulator Wzz [15, 16]. Biosynthesis of the long homopolymer chain of CPA is directed by the ATP binding cassette (ABC) transporter-dependent pathway [10]. The polymerization of the D-Rha polymer CPA occurs in the cytoplasmic compartment. The D-rhamnan chain of up to 70 sugar residues is then extruded to the periplasm by an ABC transport system encoded by genes wzt and wzm found in the CPA biosynthesis gene locus [7, 17]. Simultaneously, OSA repeating units synthesized in the cytoplasm are flipped by Wzx to the periplasm for polymerization by Wzy. The O antigens CPA or OSA are then transferred to the outer core oligosaccharide linked to lipid A by ligase WaaL [14, 18]. The attachment of the O antigen to an L-Rha residue of the outer core oligosaccharide is regulated by L-Rha-transferases (RhaT) MigA and WapR [6, 19] that use TDP-L-Rha as the donor substrate, synthesized by RmlA, RmlB, RmlC and RmlD [20] enzymes that convert D-Glc-1-phosphate to TDP-L-Rha. In PA, WapR appears to be an α1-3 L-RhaT necessary for the attachment of the O antigen to the outer core. The completed LPS is then transported to the outer membrane by an elaborate ABC transporter Lpt complex [21] that exposes the O antigens to the environment. GDP-D-Rha, the donor substrate for D-RhaTs is synthesized from GDP-D-Man by GDP-D-Man 4,6-dehydratase GMD and oxidoreductase RMD, encoded by gmd and rmd genes, respectively, in the CPA gene locus [22]. The wbpW gene encodes the GDP-D-Man guanylyltransferase required for the synthesis of GDP-D-Man.
D-Rha polymers have been found in several bacteria including the plant pathogen Pseudomonas syringae [23, 24], Campylobacter fetus [25], Xanthomonas campestris [26], as well as other human pathogenic bacteria such as Helicobacter pylori [27], the opportunistic, multidrug-resistant pathogen Stenotrophomonas maltophilia [28] and E. coli O99 [29]. Some of the O antigens seem to have a linear D-Rha sequence identical to that of CPA. Burkholderia cenocepacia is another CF pathogen that has LPS containing D-Rha polymers [30, 31]. Clearly, these bacteria express the enzymes that synthesize GDP-D-Rha as well as D-RhaTs that remain to be identified. A chlorovirus that infects green algae also has the gene encoding GMD, and α1-3-linked D-Rha is found in the N-glycan chains of viral glycoproteins [32, 33]. Bacteriocins are antibiotic proteins produced by several bacteria including PA that can kill other bacteria. Certain bacteriocins have a lectin domain at the C-terminus that recognizes D-Rha [34, 35] which plays a role in bacterial killing. Plant saponins that inhibit cancer cell growth have been shown to be linked to D-Rha [36]. However, none of the D-RhaTs involved have been biochemically identified.
Genetic studies have identified several genes in PA hypothesized to direct O antigen biosynthesis [10] including wbpX, wbpY, wbpZ suggested to encode D-RhaTs [7]. In our previous work, WbpZ has been characterized as an α1,3-D-RhaT that could transfer D-Rha in α1-3 linkage from GDP-D-Rha to either D-GalNAcα-O-PO3-PO3-(CH2)11-O-phenyl (D-GalNAcα-PP-PhU) or to D-GlcNAcα-O-PO3-PO3-(CH2)11-O-phenyl (D-GlcNAcα-PP-PhU) which are analogs of the natural PP-Und lipid intermediate. Thus WbpZ was proposed to synthesize an adaptor for D-Rha polymer synthesis. The donor substrate GDP-D-Rha was synthesized chemically and enzymatically [37, 38]. WbpZ also transferred D-Man from GDP-D-Man to both D-GalNAcα- or D-GlcNAcα-PP-PhU, and therefore has a relaxed donor and acceptor specificity. We chemically synthesized the reaction product of WbpZ, D-Rhaα1-3-D-GlcNAcα-PP-PhU [39], and used it as a substrate for further elongation of the D-Rha chain. Study of the PA14 strain harbouring WbpX showed that WbpX is required for CPA biosynthesis [40]. Furthermore, complementation of chromosomal mutants and overexpression of wbpX, wbpY, and wbpZ resulted in longer CPA chain lengths, providing further evidence that their gene products are responsible for CPA polymerization [10]. However, WbpX and WbpY have not been biochemically characterized and the mechanisms by which D-Rha polymers are synthesized have yet to be understood.
In our current study we expressed and characterized WbpX and WbpY. We demonstrated that these GTs are indeed D-RhaTs and catalyze the transfer of D-Rha from GDP-D-Rha to the reaction product of WbpZ. The donor and acceptor substrate specificities of these two enzymes differ significantly. While WbpX and WbpY transferred one D-Rha residue to the WbpZ product, the combination of WbpX and WbpY resulted in the synthesis of a D-Rhamnan polymer of at least 8 D-Rha residues. Additionally, WbpX was observed to transfer several D-Rha residues to the WbpY product, as well as to a less natural acceptor substrate. The biochemical characterization of WbpX and WbpY allows for the in vitro synthesis of CPA which may serve as the basis for vaccine development. These D-RhaTs are attractive therapeutic targets for the treatment of infections caused by PA and several other pathogens through inhibition of O antigen biosynthesis.
Materials and methods
Materials
Materials were purchased from Sigma-Aldrich (Oakville ON, Canada), unless otherwise stated. GDP-D-[3H]Man and other radiolabelled nucleotide sugars were obtained from American Radiolabeled Chemicals (St. Louis, MO). The enzymatic synthesis of GDP-D-[3H]Rha from GDP-D-[3H]Man using GMD and RMD enzymes, and the chemical synthesis of GDP-D-Rha were described previously [37, 38]. GDP-D-Rha was purified via a C18 Sep-Pak cartridge to remove GMP-morpholidate remaining from the synthesis, followed by reverse-phase HPLC to separate GDP-D-Rha from GMP as well as other contaminants. HPLC was conducted using a 10 µm 100 Å, 250 × 4.6 mm C18 column (Phenomenex, Torrance, CA) and 5 mM triethylammonium acetate (pH 6) as the mobile phase. Nucleotide sugars were detected by absorbance or radioactivity of collected fractions. The syntheses of D-Rhaα1-3-D-GlcNAcα-PP-PhU and D-Galβ1-3-D-GalNAcα-PP-PhU [38, 39] and the synthesis of D-rhamnopyranosideα-O-benzyl (D-Rhaα-Bn) [38], D-GlcNAcα-PP-PhU and D-GalNAcα-PP-PhU [41] were reported previously. The compounds D-Manα1-3-D-Manα-5-benzamidopentyl and L-Rhaα1-6-D-Glcα-O-(CH2)2-phenyl were synthesized by Todd Lowary (University of Alberta, Edmonton, Canada). D-Mannose disaccharides were from V-labs (USA). D-Rha was purchased from Carbosynth (Compton, UK).
Bioinformatics
Homology of proteins was determined by BLAST and ALIGN searches. Characterized α-D-Man-transferases (D-ManTs) were aligned using Clustal Omega. Protein structures of WbpX and WbpY were simulated using Phyre2, and GDP-D-Rha was modelled into the protein according to crystal structure of D-ManT PimA from Mycobacterium smegmatis. [42].
Expression and purification of WbpX and WbpY
Plasmids WbpX-pET-30a( +) and WbpY-pET-30a( +) were generously provided by Joseph Lam (University of Guelph, Guelph, ON, Canada). Enzyme sequences included a His6 tag, thrombin site, and S-tag at the N-termini of WbpX and WbpY. WbpX and WbpY were subcloned from pET-30a( +) into pGEX-6P-1 vectors by BioBasic (Markham, ON, Canada), at the SmaI restriction enzyme site for the expression of glutathione-S-transferase (GST)-tagged fusion proteins. GST fusion proteins also retained original tags downstream of N-terminal GST tag and HRV 3C cleavage site in order to detect enzymes by anti-His6 Western blot following cleavage. Plasmids were transformed into E. coli BL21(DE3), pLysS(DE3), Lemo(DE3) (New England Biolabs) and ArcticExpress(DE3) strains (Agilent Technologies, Santa Clara, CA). Positive transformants were selected from lysogeny broth-Lennox (LB) agar plates containing 20 µg/mL gentamycin, with 50 µg/mL kanamycin (for pET-30a( +) vectors) or 100 µg/mL ampicillin (for pGEX-6P-1 vectors). His6- and GST-tagged enzymes were routinely expressed in 500 mL LB bacterial expression cultures, grown at 37ºC to optical density of 0.6 at 600 nm, with 200 revolutions per min (RPM) shaking with appropriate antibiotics. Cultures were then cooled on ice, and expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside at 10 ºC, 200 RPM for 48 h. Bacteria were harvested via centrifugation and resuspended with phosphate-buffered saline (PBS) containing 10% glycerol.
Soluble and insoluble fractions were prepared by sonication in 50 mM sucrose and centrifugation (8400 × g), yielding the soluble fraction in the supernatant, and insoluble fraction in the pellet. Confirmation of expression was accomplished via SDS-PAGE where uninduced control samples, induced cell lysates, soluble and insoluble fractions were analyzed as described previously [41]. Purification of His6-tagged enzymes was carried out using HisPur Ni–NTA resin (ThermoFisher Scientific, Waltham, MA) as described previously [43]. Purification of GST-WbpX and GST-WbpY was achieved using GST-Bind glutathione-agarose resin (Novagen, Madison, WI). Pelleted cells were sonicated in lysis/binding buffer (PBS pH 7.4) and centrifuged at 8400 × g for 15 min. Supernatant was loaded on 1 mL of equilibrated resin in a sealable chromatography column, and was incubated for 1 h at 4 ºC with end-over-end rotation. Flow-through fractions were collected, and the procedure was repeated to wash column with binding buffer 3x (PBS pH 7.4) for 10 min. Three 20 min elution fractions were collected using a solution of 50 mM Tris, pH 8, and 10 mM reduced L-Glutathione. Purification was assessed by SDS-PAGE and enzyme activity assays using elution fractions.
Assays for D-Rha-transferases and D-Man-transferases
D-RhaT activity was assessed via the transfer of radiolabeled D-[3H]Rha from GDP-D-[3H]Rha to acceptor compounds. For standard assays, bacterial suspensions were centrifuged, and pellets were sonicated in 50 mM sucrose via 3 sonication cycles of 15 s (Fisher Scientific sonicator, setting 3) with 2 min cooling on ice between cycles. Lysate was centrifuged 8400 × g for 15 min to separate supernatant fraction from insoluble enzyme and bacterial debris in the pellet. Total protein content was determined by Bio-Rad Protein Assay reagent (Bio-Rad, Mississauga, ON) Bradford assay, with bovine serum albumin as standard. Standard D-RhaT assays were conducted in a total volume of 40 µL: 10 µL WbpX or WbpY supernatant (0.08 mg total protein), 0.08 mM GDP-D-[3H]Rha (2900 cpm/nmol), 0.25 mM acceptor (D-Rhaα1-3-D-GlcNAcα-PP-PhU), 0.125 M Tris (pH 9) and 5 mM MnCl2. Assay mixtures were incubated at 37 ºC for 10 min, and reactions were quenched by freezing. Reaction product was isolated via C18 Sep-Pak cartridge (Waters, Milford, MA, USA). Cartridges were washed with 4 mL H2O, and hydrophobic product was eluted with 3 mL MeOH. Radioactivity of eluted fractions was measured by scintillation counting. Hydrophilic acceptors lacking a hydrophobic aglycone group were assayed by the Dowex 1 × 8 method [41]. Control assays lacking acceptor were conducted in parallel to normalize for background radioactivity and possible D-[3H]Rha transfer to endogenous acceptors. Conditions of standard enzyme assays were changed to characterize various parameters of WbpX and WbpY activity. D-ManT activities were assayed under similar conditions as D-RhaT assays but using donor substrate GDP-D-[3H]Man. Donor and acceptor specificities were examined under similar conditions replacing GDP-D-[3H]Rha with other radiolabelled nucleotide sugars or replacing D-Rhaα1-3-D-GlcNAcα-PP-PhU with other glycoconjugates as potential acceptors, respectively. Inhibition was studied in standard assays in the presence of potential inhibitors as described [44]. The kinetic parameters were established using GraphPad Prism 9.
Production of non-radioactive reaction products for mass spectrometry
To analyze reaction products by mass spectrometry (MS) nonradioactive nucleotide sugars (GDP-D-Rha or GDP-D-Man) were used in the standard assay as described above. For assays containing both WbpX and WbpY, 5 µL of each enzyme supernatant were added to the assay mixture containing 0.17 mM GDP-D-Rha or GDP-D-Man, 0.25 mM acceptor, 0.125 M Tris (pH 9), and 5 mM MnCl2. Assays were incubated for 24 h at 37 ºC and then frozen. Product was isolated via C18 Sep-Pak cartridge and eluted with MeOH. Samples were dried via rotary evaporation and lyophilization. Reconstituted samples containing diphosphate-lipid moieties were analyzed by electrospray ionization mass spectrometry (ESI–MS) using an Orbitrap Velos Pro spectrometer (Thermo Scientific) in the negative ion mode at Queen’s University. Samples were also analyzed by John Klassen, Edmonton, Alberta, Canada, by ESI–MS. Measurements were carried out in negative ion mode using a Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with the Nanospray Flex™ ion source. The nanoflow ESI (nanoESI) was performed using tips pulled from borosilicate capillaries by a micropipette puller (P-1000, Sutter Instruments, Novato, CA). A voltage of –0.7 kV was applied to a platinum wire inserted into the nanoESI tip and in contact with the sample solution. Data acquisition and processing were performed using Xcalibur (Thermo Fisher Scientific, version 4.4).
Large scale syntheses for NMR
Large scale reactions (1000 × standard assay using non-radioactive GDP-D-Rha) were conducted for linkage analysis of WbpX and WbpY products by 700 MHz NMR. GST-WbpX supernatant was used in reactions with GDP-D-Rha (0.1 mM) and D-Rhaα-Bn (0.125 mM) to form product for NMR analysis in 40 mL reaction volume. After incubation for 20 min at 37 ºC, acceptor and products were separated from GDP-D-Rha by C18 Sep-Pak cartridges. MeOH elution fractions were dried by rotary evaporation and lyophilization. Product was separated from acceptor via reverse phase C18 HPLC using a mobile phase of 10% acetonitrile/90% water. Fractions containing product were pooled and lyophilized. Dried product was exchanged twice with 99.96% D2O by lyophilization prior to NMR experiments. GST-WbpX was also used with GDP-D-Rha (0.1 mM) and D-Rha α1-3-D-GlcNAcα-PP-PhU (0.1 mM) to prepare large scale reaction product for NMR. A large scale WbpY reaction product was prepared with purified GST-WbpY, GDP-D-Rha (0.1 mM) and D-Rhaα1-3-D-GlcNAcα-PP-PhU (0.1 mM) for NMR analysis. 1D and 2D NMR spectroscopy experiments were conducted using a Bruker NEO-700 MHz, performed by Françoise Sauriol (Department of Chemistry, Queen’s University).
Results
Bioinformatics
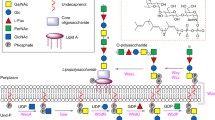
The α1,3-D-RhaT WbpZ and putative D-RhaTs WbpX and WbpY from P. aeruginosa PAO1 have been classified by the CAZy databank as retaining GT4 family enzymes possessing a GT-B fold. They share homology with several GT4 D-ManTs, including WbdC, WbdA and WbdB that are found in several bacterial strains including E. coli O8, O9 and O9a and Klebsiella pneumoniae and synthesize the α1-2 and α1-3 linkages in their O-antigenic D-Man-oligomers, due to multiple GT domains in the proteins [45, 46]. In E. coli O8, WbdA also synthesizes β1-2 linkages. These D-Man-containing polymers are assembled via the ABC transporter-dependent pathway (Fig. 1).

adapted from Melamed and Brockhausen [17])
Proposed mechanism of P. aeruginosa CPA biosynthesis. WbpL transfers a D-GlcNAc-phosphate (or D-GalNAc-phosphate) residue to membrane-bound undecaprenyl phosphate which initiates O antigen synthesis. The α1,3-D-Rhamnosyltransferase WbpZ adds the first D-Rha residue in an α1-3 linkage, as an adapter. Polymerization of the O antigen is directed by WbpX and WbpY up to ~ 70 residues in length (n repeat units = 20–25). The ATP binding cassette (ABC) transporter translocates the assembled CPA D-rhamnan polymer to the periplasmic face of the inner membrane leaflet, where it is transferred and ligated to the core-lipid A molecule by WaaL. The completed LPS molecule is transported to the outer membrane by LPT complex enzymes. (Figure
WbpZ has been shown to possess both D-Rha- and D-ManT activity [38]. It is thus possible that WbpX and WbpY also have dual donor specificity. WbdC from E. coli O9 has 52% sequence identity with WbpZ from PA, and both are thought to synthesize the adapter structure for polymerization. WbdC has 2 GT domains and synthesizes D-Manα1-3-D-GlcNAc-PP-Und as the anchor for O antigen synthesis, while WbpZ transfers D-Man or D-Rha to both D-GlcNAcα-PP-PhU and D-GalNAcα-PP-PhU. WbpZ has essential DxD and Ex7E motifs that are also found in WbdC [38, 45, 46].
WbdB has been shown to be the next D-ManT in the pathway of E. coli O9a synthesis and has 27.5% sequence identity to WbpY and 14.8% identity with WbpX from PA. WbdB from E. coli O9 has 38% identity with WbpY. WbdB sequentially transfers two D-Manα1-3 residues but has only one GT domain (amino acids 207–351) while two GT domains have been assigned to WbpY (amino acids 16–171 and 198 – 335). However, because of the high homology it is expected that WbpY is also the D-RhaT that follows WbpZ [13].
WbdA from E. coli O9a is the polymerizing D-ManT that has 13.4% sequence identity with WbpX from PA but only 10% identity with WbpY. However, WbdA from E. coli O9 has 34% identity with WbpX. In E. coli O9a, WbdA is bifunctional and transfers two D-Manα1-2 and two D-Manα1-3 residues. In E. coli O8, WbdA is trifunctional and synthesizes D-Manα1-2, D-Manα1-3 and D-Manβ1-2 linkages [45, 46]. However, WbpX from PA appears to have only one GT domain (amino acids 268–415). Nevertheless, based on the homology WbpX may be the next D-RhaT in the CPA pathways that follows WbpY action.
Alignment of GT4 ManTs with WbpX, WbpY and WbpZ revealed two highly conserved sequences within the proposed catalytic domains of WbpX and WbpY (Fig. 2). Both WbpX and WbpY have a central PRK sequence (282PRK in WbpX and 203PRK in WbpY) which is absent from WbpZ and from WbdC. Ex7E sequences are frequently found in retaining GT4 enzymes but their role is still uncertain. Several Ex7E sequences are found in WbpZ, WbpX and WbpY and all three enzymes have at least one Ex7E in the assigned GT domain. In addition, there is a conserved FG sequence within the Ex7E sequence in the GT domain (ExFGxxxxE) of all three enzymes. WbpZ and WbpY, but not WbpX, have DxD sequences in the GT domain, which were shown to be essential for activity in WbpZ [38]. WbpX and WbpY lack the QxxRW sequence found in processive enzymes [47] and therefore may act in a non-processive or partially processive mechanism.

Partial sequence alignments of WbpX and WbpY with other GT4 α-D-mannosyltransferases. Alignments performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). Characteristic Ex7E and PRK motifs are shown in black boxes, with identical amino acid residues in yellow, and similar residues in blue. WbpX,Y,Z from P. aeruginosa PAO1 (UniProtKB accession O84908, O84909, Q9HTC0), WbdA (GT domain 1 and 2), WbdB,C from E. coli O9a (UniProtKB accession M4QN28[225–379], M4QN28[663–812], A0A0A8J412, O70052), PimA from Mycobacterium smegmatis (UniProtKB accession A0QWG6), PimB from Mycobacterium tuberculosis H37Rv (UniProtKB accession P9WMZ3), PimC from Mycobacterium tuberculosis 1551 (UniProtKB accession P0CF99), LpcC from Rhizobium leguminosarum bv. viciae (UniProtKB accession O68547), MggA from Petrotoga mobilis (UniProtKB accession A9BHJ0), MtfB,C from E. coli O9 (UniProtKB accession Q47594, Q47595), and WejK from E. coli O99 (UniProtKB accession C8YZ36)
Kyte & Doolittle hydrophobicity plots show more regions of higher hydrophobicity scores in WbpX than WbpY (Fig. S1). Therefore, WbpX may be membrane-associated, leading to challenges in purifying the recombinant enzyme. Models of WbpX and WbpY generated via Phyre2 were docked with GDP-D-Rha using crystallized D-ManT PimA for alignment of the donor substrate within the proposed homologous catalytic domain. Simulated modelling showed that the conserved ExFGxxxxE and PRK sequences are potentially involved in donor substrate binding (Fig. 3). The modeled structures clearly show the GT-B fold characteristic of GT4 family enzymes where the central donor substrate binding site and the putative catalytic domain is between two major folds.

Simulated models of (A) WbpX and (B) WbpY with GDP-D-Rha. Models were generated by Phyre2 with > 90% confidence. N- and C-terminal domains represented in blue and orange, respectively. GDP-D-Rha position and orientation was determined via alignment of PimA (PDB code: 2GEJ) bound to GDP-D-Man
Based on homology of the D-RhaTs with D-ManTs that synthesize O antigens, it is hypothesized that WbpX and WbpY polymerize a D-rhamnan homopolymer possibly up to 70 residues [6, 10, 38]. However, biochemical analyses are necessary to confirm this hypothesis. We outlined our proposed mechanism of CPA assembly in Fig. 1.
Expression and purification
Initial expression tests of His6-tagged WbpX and WbpY in BL21(DE3) bacteria and BL21(DE3)pLysS showed a low yield of protein of interest in the supernatant fraction, with correspondingly low activity in both supernatants and cell lysates (data not shown). Expression was induced in BL21(DE3) at 37 ºC, 23 ºC, and 16 ºC; however, solubility did not increase by lowering induction temperatures. Expression of WbpX and WbpY with a high degree of solubility was achieved in ArcticExpress(DE3), confirmed by SDS-PAGE and Western blot using antibody to the His6-tag (Fig. S2). Solubility was determined by the proportion of enzyme in the soluble fraction after sonication and centrifugation compared to the pellet, as determined by SDS-PAGE, showing that solubility of WbpX and WbpY was approximately 90% and 50%, respectively, while the activity between supernatants was comparable. The presence of soluble enzyme in the supernatant fraction was confirmed by Western blot, also showing a small proportion of degradation products from WbpX. In order to improve purification, wbpX and wbpY were subcloned into pGEX-6-P-1 vectors to express GST fusion proteins. GST-WbpX and GST-WbpY were expressed in ArcticExpress(DE3), induced at 10 ºC. SDS-PAGE showed high expression of fusion proteins of interest at the predicted size (GST-WbpX: 83.7 kDa; GST-WbpY: 73.4 kDa). Purification of GST-WbpX did not yield active enzyme in the elution fractions. GST-WbpY was purified as active enzyme in elution fractions 1 and 2, with low activity in elution 3. However, large amounts of the ArcticExpress chaparonin Cpn60 remained in the elution fractions, suggesting its association with WbpY (Fig S3).
Characterization of D-Rha-transferase activities of WbpX and WbpY
Enzymes were assayed in standard assays conducted at 10 min incubation time which achieved high activity. The pH optima were established using various buffers with pH 5–10 in standard assays. D-RhaT activity was maximal at pH 9 for both His6-WbpX and His6-WbpY (Fig S4). The effect of detergent on D-RhaT activity was assessed using various concentrations of Triton X-100 between 0 and 0.625% and showed that the activity of His6-WbpX was negatively correlated to Triton X-100 concentration, resulting in a 4.6-fold decrease in activity at 0.625% (Fig. 4) from 77.3 to 16.7 nmol·h−1·mg−1. However, Triton X-100 had little effect on His6-WbpY activity. The addition of 5 mM MnCl2, MgCl2 or Mn(CH3CO2)2 led to a substantial increase in His6-WbpX activity, while these ions had no discernable impact on His6-WbpY activity. Assays conducted with 5 and 10 mM ethylenediaminetetraacetic acid (EDTA) had no effect on either His6-WbpX or His6-WbpY (Fig. 5). The addition of 5 mM Zn(CH3CO2)2 resulted in complete loss of activity for both enzymes. This shows that there are significant differences in properties of WbpX and WbpY proteins although both enzymes show properties of retaining GT4 family enzymes.

The effect of Triton X-100 on D-RhaT activity of His6-WbpX and His6-WbpY. Increasing concentrations of the non-ionic detergent led to a decrease in WbpX activity while WbpY seemed to be unaffected. Error bars represent standard error between at least 2 replicates

The effect of various metal ions and EDTA on His6-WbpX and His6-WbpY D-RhaT activity. As compared to assays conducted in the absence of additional metal ions, WbpX activity was enhanced via the addition of 5 mM MgCl2, MnCl2 and Mn(CH3CO2)2, while EDTA had no effect. Addition of 5 mM MgCl2, MnCl2, Mn(CH3CO2)2 and EDTA to assays had very little effect on WbpY activity. Zn(CH3CO2)2 abolished D-RhaT activity in both enzymes. Error bars represent standard error between at least 2 replicates
Donor specificities
The donor specificities of His6-WbpX and His6-WbpY were assessed using [3H]-labelled nucleotide sugars as donors at 0.75 mM in standard assays. Transferase activities using GDP-L-[3H]Fuc, GDP-D-[3H]Man, UDP-D-[3H]Glc, UDP-D-[3H]GlcNAc, UDP-D-[3H]Gal or UDP-D-[3H]GalNAc donors were compared to the activity in the presence of GDP-D-[3H]Rha. WbpX showed no transferase activity using any of the nucleotide sugars other than GDP-D-[3H]Rha, indicating that it is exclusively a D-RhaT. In contrast, His6-WbpY had also significant D-ManT activity using GDP-[3H]Man which was 70% of the D-RhaT activity (Table 1). WbpY is therefore a dual D-RhaT and D-ManT, similar to WbpZ. Inhibition of D-RhaT activity was tested using the enzyme product guanosine diphosphate (GDP) at 1 mM concentration, as well as 1 mM guanosine monophosphate (GMP) and 1 mM GMP-morpholidate, none of which showed significant effects on activity. Thus, possible contaminants in the GDP-D-Rha preparation did not affect D-RhaT activities.
Acceptor specificities
The acceptor specificities of D-RhaT were determined with a variety of synthetic glycans having terminal D-Rha, L-Rha, D-Man, D-Gal, D-GlcNAc, or D-GalNAc residues, in comparison to the standard acceptor D-Rhaα1-3-D-GlcNAcα-PP-PhU. With the exception of mono- and disaccharides, all acceptors possessed a hydrophobic group and were assayed using the C18 Sep-Pak method. His6-WbpX showed broad acceptor specificity, with > 98% D-RhaT activity using D-Rhaα-Bn, D-Manα1-3-D-Manα-5-benzamidopentyl and D-Manα-O-p-nitrophenyl substrates, compared to the standard acceptor D-Rhaα1-3-D-GlcNAcα-PP-PhU (Table 2). Other PP-PhU derivatives having terminal D-GlcNAc, D-GalNAc and D-Galβ1-3-D-GalNAc structures did not yield any activity. This showed that terminal D-Rha (or D-Man) is critical for WbpX activity, but that the enzyme did not require the diphosphate-lipid moiety of the natural acceptor analog. Free D-Rha and D-Man disaccharides were assayed by the Dowex 1 × 8 method and showed no activity at 0.25 mM concentration. However, D-Manα1-2-D-Man and D-Manα1-3-D-Man disaccharides demonstrated 40% and 87% activity at 2.5 mM acceptor concentrations. The preference of WbpX in favour of D-Manα1-3-D-Man over D-Manα1-2-D-Man lends further evidence that WbpX acts on D-Rhaα1-3-terminating acceptor substrates. It also indicates the benefit of having a hydrophobic aglycone group in the acceptor. While D-Manα-p-nitrophenyl was an excellent substrate for His6-WbpX, L-Rhaα-p-nitrophenyl was not active. This indicates the need for the (6-deoxy-)-D-Manα configuration of the terminal sugar.
Thus, WbpX is capable of transferring D-Rha to compounds having D-Rha- or D-Man at the non-reducing terminus, but not to free D-Rha or L-Rha-terminating compounds.
In contrast, His6-WbpY showed a strict acceptor specificity, with D-Rhaα1-3-D-GlcNAcα-PP-PhU being the sole acceptor (Table 2). [D-Rhaα1-2/3]n-D-Rhaα-Bn, the reaction products of WbpX using D-Rhaα-Bn substrate at 2.5 mM concentration, was not an acceptor for WbpY. This indicates that a terminal D-Rhaα residue is not sufficient and WbpY requires a disaccharide or the presence of the diphosphate-lipid moiety in its acceptor. These studies suggest that WbpY is the enzyme that follows WbpZ in the pathway to CPA.
Enzyme kinetics for His6-WbpX in the supernatant fraction showed an apparent KM = 0.15 mM and Vmax = 211 nmol·h−1·mg−1 for GDP-D-[3H]Rha using D-Rhaα1-3-D-GlcNAcα-PP-PhU acceptor, and an apparent KM = 0.22 mM and Vmax = 186 nmol·h−1·mg−1 for D-Rhaα1-3-D-GlcNAcα-PP-PhU acceptor (Fig. 6A, B). Kinetic data were also obtained for His6-WbpX using D-Rhaα-Bn as the acceptor substrate, with apparent KM = 0.02 mM and Vmax = 183 nmol·h−1·mg−1 (Fig. 6C).

His6-WbpX enzyme kinetics. (A) Apparent KM and Vmax for donor substrate GDP-D-[3H]Rha donor substrate were 0.145 mM 211.4 nmol·h−1·mg−1, respectively. (B) Apparent KM and Vmax of acceptor substrate D-Rhaα1-3-D-GlcNAc-PP-PhU were 0.22 mM and 186.4 nmol·h−1·mg−1, and (C) acceptor substrate D-Rhaα-O-benzyl were 0.021 mM and 182.9 nmol·h−1·mg−1, respectively
His6-WbpY in supernatant fraction showed enzyme kinetics for GDP-D-[3H]Rha of apparent KM = 0.73 mM and Vmax = 988 nmol·h−1·mg−1, and for D-Rhaα1-3-D-GlcNAcα-PP-PhU apparent KM = 0.10 mM and Vmax = 384 nmol·h−1·mg−1, respectively (Fig. 7A, B). Kinetic efficiencies expressed as Vmax /KM with respect to GDP-D-[3H]Rha were similar for WbpX (1407) and for WbpY (1353). However, for the acceptor substrate D-Rhaα1-3-D-GlcNAcα-PP-PhU efficiencies were 845 for WbpX and 3840 for WbpY, showing that WbpY was more active towards D-Rhaα1-3-D-GlcNAcα-PP-PhU.

His6-WbpY enzyme kinetics. (A) Apparent KM and Vmax for donor substrate GDP-D-[3H]Rha donor substrate were 0.728 mM and 988.3 nmol·h−1·mg−1, respectively. (B) Apparent KM and Vmax of acceptor substrate D-Rhaα1-3-D-GlcNAc-PP-PhU were 0.097 mM and 384.1 nmol·h−1·mg−1
Mass spectrometry
His6-WbpX and His6-WbpY transferred one D-Rha residue (146 Da) to D-Rhaα1-3-D-GlcNAcα-PP-PhU, forming a product with m/z 918 after 1 or 24 h incubation time (Fig. 8A, B). No Rha-containing compounds with higher mass were detected. However, co-incubation of His6-WbpX and His6-WbpY with D-Rhaα1-3-D-GlcNAcα-PP-PhU acceptor for 24 h showed 8 additional m/z peaks separated by 146 Da in the scanned region (Fig. 8C). In a 1 h incubation, 5 products were seen (data not shown). Although mass spectrometry is considered only to be a “semi-quantitative” means of analysis, relative peak intensities between masses showed that the most intense peak (100%) was the acceptor substrate D-Rhaα1-3-D-GlcNAcα-PP-PhU (m/z 772.287), followed by the product after the addition of one D-Rha (m/z 918.348) at 38% and other multimeric D-Rha products in sequentially decreasing intensity with the highest mass having the lowest intensity. Thus, polymerization of up to 8 D-Rha residues indicated that WbpX and WbpY cooperate to extend the CPA O antigen non-processively. WbpY also transferred a single D-Man residue (162 Da) to D-Rhaα1-3-D-GlcNAcα-PP-PhU, forming D-Man-D-Rhaα1-3GlcNAcα-PP-PhU product (m/z 934) (Fig. 8D).

ESI–MS (negative ion mode) spectra of D-RhaT or ManT assays incubated for 24 h containing (A) His6-WbpX or (B) His6-WbpY. Acceptor compound D-Rhaα1-3-D-GlcNAcα-PP-PhU at m/z 772 is indicated by a star and reaction product at m/z 918 is indicated by an arrow. (C) Both His6-WbpX and His6-WbpY were present in D-RhaT assay. The MS spectrum shows various D-RhaT products with up to 8 D-Rha residues. This shows that the mixture of the two enzymes was able to synthesize D-Rha polymers based on D-Rhaα1-3-D-GlcNAcα-PP-PhU. (D) D-Mannosyltransferase activity of His6-WbpY with GDP-D-Man donor substrate. Acceptor compound D-Rhaα1-3-D-GlcNAcα-PO3-PO3-(CH2)11-O-phenyl (m/z 772) is indicated by a star and D-Man product (m/z 934) is indicated by an arrow
Using D-Rhaα-Bn as acceptor, up to 11 D-Rha residues were transferred by GST-WbpX after 20 min incubation (Fig. 9). HPLC purification of reaction products showed a broad product peak. ESI–MS analysis showed that relative to the most abundant product D-Rha3-D-Rhaα-Bn (m/z 691.28) (100%), the acceptor substrate D-Rhaα-Bn (m/z 253.11) was observed at 19%, likely due to incomplete separation of product from acceptor by HPLC. The distribution of products followed an interesting pattern with m/z peaks from shorter oligosaccharides or from longer oligosaccharides having less intensity than D-Rha3-D-Rhaα-Bn. The large oligosaccharide D-Rha7-D-Rhaα-Bn (m/z 1275.50) had an intensity of 13% compared to that of D-Rha3-D-Rhaα-Bn and subsequent products showed very low intensity. This confirms that the reaction was non-processive but that there is a control of the optimal product length that differs from the reaction using D-Rhaα1-3-D-GlcNAcα-PP-PhU acceptor. Clearly, the polymerizing activity of WbpX seems to be high if the D-GlcNAcα-PP-PhU moiety of the acceptor is replaced by a benzyl group.

ESI–MS (negative ion mode) spectrum of GST-WbpX adding multiple D-Rha residues to acceptor compound D-Rhaα-O-benzyl. Products were purified by HPLC, however multiple products co-eluted. Acceptor D-Rhaα-O-benzyl at m/z 253.11 is indicated by a star and 11 reaction products are indicated with arrows
To further examine the sequence of D-Rha transfer to the natural analog D-Rhaα1-3-D-GlcNAcα-PP-PhU, non-radioactive WbpX-product using D-Rhaα1-3-D-GlcNAcα-PP-PhU acceptor was isolated (D-Rha-D-Rhaα1-3-D-GlcNAcα-PP-PhU) and used as acceptor substrate for GST-WbpY with GDP-D-Rha donor substrate in 1 h incubation. Thus, two acceptors for WbpY were present in the assay (WbpX-product (m/z 918.3) and D-Rhaα1-3-D-GlcNAcα-PP-PhU (m/z 772.3). After 1 h incubation time GST-WbpY added one additional D-Rha residue to the WbpX-product indicated by m/z 1064.4 (Fig. 10A). Conversely, WbpY product D-Rhaα1-3-D-Rhaα1-3-D-GlcNAcα-PP-PhU (m/z 918.3) was isolated and used as acceptor for GST-WbpX in a 1 h incubation. Four different products resulted, showing the sequential addition of 4 D-Rha residues (Fig. 10B, Table 3). This suggests that WbpY transfers one D-Rha residue to D-Rhaα1-3-D-GlcNAcα-PP-PhU. Subsequently, WbpX transfers additional D-Rha residues.

ESI MS of WbpY/WbpX reaction products using WbpX/WbpY products D-Rha-D-Rhaα1-3-D-GlcNAcα-PP-PhU (m/z = 918.3). A. WbpX product with D-Rhaα1-3-D-GlcNAcα-PP-Ph acceptor was produced and isolated, and then used as a substrate for WbpY. The addition of one D-Rha residue is seen as m/z 1064.4. B. WbpY product with D-Rhaα1-3-D-GlcNAcα-PP-Ph acceptor was produced and isolated, and then used as a substrate for WbpX. Several D-Rha residues were sequentially added
Linkage analysis by NMR
A large scale (1000 x) synthesis of WbpX producּt was carried out using D-Rhaα-Bn as the acceptor substrate. GST-WbpX supernatant was reacted with D-Rhaα-Bn and produced 1.41 μmol product, based on parallel radioactive assays used for quantification. Acceptor and products were isolated from assay mixtures using C18 Sep-Pak cartridges, eluted with MeOH and dried. Product was separated from acceptor substrate via reverse-phase HPLC, with a mobile phase of 10% acetonitrile/90% H2O. Acceptor D-Rhaα-Bn eluted in 28–32 min, and products unexpectedly eluted later, in a broad peak between 48–76 min, possibly due to the hydrophobicity of the 6-methyl group of D-Rha. Fractions containing product were pooled, lyophilized, and exchanged with D2O. 1H NMR experiments revealed a complex spectrum with 14 distinct H-1 D-Rhaα proton signals, indicating that WbpX added multiple residues to D-Rhaα-Bn acceptor substrate. Heteronuclear single quantum coherence (HSQC) experiments were useful to assign carbon shifts. Correlation spectroscopy (COSY), total correlation spectroscopy (TOCSY), HSQC and heteronuclear single quantum coherence (HMBC) experiments were used to assign chemical shifts of protons involved in glycosidic linkages (Table S1). There were several different equatorial H-1 signals and extensive overlap of signals making it difficult to assign shifts. At least 3 different H-1 signals of D-Rha-2 were detected linked to benzyl. The nuclear Overhauser effect spectroscopy (NOESY) spectrum showed NOEs between D-Rha-1-H1 (5.13 ppm) and D-Rha-2-H2 (4.00 ppm) indicating a D-Rhaα1-2 linkage synthesized by WbpX. In addition, NOEs were detected between D-Rha-1-H1 (5.13 ppm) and D-Rha-2-H3 (3.68 ppm). This indicated that WbpX has α1,2-D-RhaT and α1,3-D-RhaT activities (Fig. 11).

2D-NOESY NMR spectrum of WbpX products using D-Rhaα-O-benzyl acceptor. There is extensive overlap of signals making it difficult to assign shifts. The NOESY spectrum showed NOEs between D-Rha-1 H1 (5.13 ppm) and D-Rha-2 H2 (4.00 ppm) proving a D-Rhaα1-2D-Rhaα linkage synthesized by WbpX. In addition, NOEs were detected between D-Rha-1 H1 (5.13 ppm) and D-Rha-2 H3 (3.68 ppm) indicating D-Rhaα1-3-D-Rhaα linkages
Product was also synthesized with WbpX and D-Rhaα1-3-D-GlcNAcα-PP-PhU acceptor. However, the 2D NMR analysis showed that a complex mixture of compounds was present and the structures could not be resolved (data not shown).
To analyze the linkage(s) synthesized by WbpY, purified GST-WbpY elution fractions were pooled and used in 1000 × standard assays, using D-Rhaα1-3-D-GlcNAcα-PP-PhU acceptor substrate. Parallel radioactive control assays indicated that 1.36 μmol product was synthesized. Linkage analysis of the D-RhaT product of WbpY was determined using 1D and 2D NMR experiments (Table 4). The acceptor dissolved in D2O was also analyzed by NMR for comparison. The chemical shifts of all D-Rha and D-GlcNAc residues and most of the 13C carbon shifts were determined by COSY and TOCSY experiments. The chemical shift of H-1 D-Rha-1 at 4.93 ppm indicated the α-linkage. A small amount of the original acceptor substrate was also seen. NOE signals were observed in selective rotating-frame nuclear Overhauser effect correlation spectroscopy (ROESY) experiments between D-Rha-1 H-1 and D-Rha-2 H-3. The spectra indicated that WbpY transferred a single D-Rha residue in α1-3 linkage to form D-Rhaα1-3-D-Rhaα1-3-D-GlcNAcα-PP-PhU (Fig. 12). Thus WbpY is a monofunctional α1,3-D-RhaT.

2D-ROESY NMR spectrum of GST-WbpY product D-Rhaα1-3-D-Rhaα1-3-D-GlcNAcα-PO3-PO3 (CH2)11O-phenyl. NOE detected between Rha-1 H1 (4.93 ppm) and Rha-2 H3 (3.67 ppm) proves D-Rhaα1-3 linkage synthesized by WbpY
Discussion
Three enzymes, WbpZ, WbpX and WbpY were the first D-RhaT to be characterized. In this study, we provided biochemical proof that WbpX and WbpY from PAO1 are D-RhaTs involved in the extension of the D-Rha polymer. D-Rha is also found in polysaccharides of other bacterial species. Therefore, this research has applications in the understanding of the syntheses of additional O antigens. Importantly, D-Rha is not found in human glycoconjugates, and can be exploited in vaccine design.
NMR analysis revealed that WbpX synthesizes both α1-2 and α1-3 D-Rha linkages while WbpY synthesizes a single D-Rhaα1-3 linkage. Co-incubation of these enzymes with GDP-D-Rha nucleotide donor and synthetic D-Rhaα1-3-D-GlcNAcα-PP-PhU acceptor achieved the in vitro synthesis of a D-Rha polymer with at least 8 D-Rha residues which is a close analog of the natural CPA synthesis intermediate [D-Rha]n-D-GlcNAcα-PP-Und. It remains to be established if the D-Rha polymer synthesized in vitro has the [D-Rhaα1-2-D-Rhaα1-3-D-Rhaɑ1-3] repeating unit structure of CPA and if it has immunoreactivity as CPA. We conclude that WbpZ synthesizes the D-Rhaα1-3-D-GlcNAc-PP-Und primer which is a substrate for WbpY to synthesize an adapter or anchor structure upon which the D-Rha polymer is built. It is interesting that the polymerization by WbpX was clearly facilitated by replacing the D-GlcNAcα-PP-Und moiety of the acceptor with a benzyl group that appeared to enhance the acceptor affinity.
Genetic studies by Rocchetta et al. [13] first proposed that WbpZ forms the adapter D-Rha-D-GlcNAcα-PP-Und which we confirmed by biochemical characterization of WbpZ [38]. It was also hypothesized that WbpY transfers two α1-3 linked D-Rha residues, followed by WbpX that synthesizes the D-Rha α1-2 linkage. We partially confirmed these hypotheses. However we were unable to confirm that WbpY by itself adds a second D-Rhaα1-3 linkage although it has low homology to WbdB that transfers 2 D-Man residues. WbpX clearly was able to synthesize multiple α1-2 and α1-3 D-Rha linkages and therefore acts as a bifunctional D-RhaT albeit having only one proposed GT domain. This sequence of WbpZ → WbpY → WbpX action may not be required when using a non-natural synthetic D-Rhaα-Bn acceptor.
WbpX has a broad acceptor specificity and also transfers D-Rha to several other acceptors that terminate in D-Rha or D-Man. However, it is not a D-ManT, thus D-Man-polymers or mixed D-Man-D-Rha- polymers are unlikely to be synthesized. WbpY could transfer a single D-Man but due its restricted acceptor specificity it is not likely to build a D-Man polymer. This supports the idea that WbpX is the main polymerase enzyme. CPA from various PA strains is essentially a D-rhamnan but has been shown to contain a small amount of D-Man as well as other monosaccharides including xylose and ribose [6, 9, 48, 49]. The relatively high purity of CPA is clearly due to the polymerizing activity of WbpX that exclusively utilizes GDP-D-Rha as donor. GTs possessing bifunctional activities have been identified, including WbdA from E. coli O9a, capable of transferring D-Man residues in α1-3 and α1-2 linkages during O antigen biosynthesis [17, 45]. WbdA has 2 or 3 GT domains while WbpX has only one predicted GT domain that appears to add several D-Rha residues in sequence and also alternate between creating α1-2 and α1-3 linkages. Thus, WbpX may have a flexible and dynamic catalytic site while WbpY only synthesizes the α1-3 linkage although it has 2 proposed GT domains.
Characterization of WbpX and WbpY by varying conditions of the standard assay found some interesting properties of these enzymes. Optimal enzyme activity was observed at high pH in both WbpX and Y. This is despite the abnormally acidic pH present in the cystic fibrosis lung, ~ 6.4, due to defective ion transport and established measurements of cytosolic Pseudomonas pH of 7.63–7.87 across multiple species and strains [50]. Furthermore, the previously characterized D-RhaT WbpZ involved in the initiation of CPA biosynthesis had a pH optimum of 7.5 [38]. The addition of Triton X-100 to assays led to a significant reduction in WbpX activity, while WbpY activity was unaffected. This suggests that WbpX may be associated with the inner membrane of PA and requires hydrophobic membrane components or anchoring to catalyze its D-RhaT reactions. Alternatively, Triton X-100 may slightly alter the tertiary structure of WbpX. In contrast, WbpZ showed 2.5-fold increase in activity at 0.25% Triton X-100, suggesting that the detergent may be involved in the stabilization of the enzyme [38]. The mechanism remains to be shown by which WbpX can continue to transfer D-Rha residues at high speed to form CPA while associated with the membrane.
WbpX and WbpY are retaining GTs having a GT-B fold and do not require metal ions for function. D-RhaT activity of WbpY was unchanged in the presence or absence of Mn2+ and Mg2+ salts or EDTA. However, the addition of Mn2+ and Mg2+ salts enhanced the activity of WbpX although they were not required for D-RhaT activity. These results showed that there are significant differences in the properties of these two enzymes although they both are inhibited by Zn2+. The low sequence identity of 19.5% between WbpX and WbpY reflects significant differences in properties and function in the assembly of CPA. However, both enzymes have α3-D-RhaT activities, utilize the same GDP-D-Rha and D-Rhaα1-3GlcNAcα-PP-PhU substrates and the structural models are remarkably similar. Because of the broad acceptor specificity of WbpX it is able to extend the polymer without the proximity to PP-Und. In contrast, the narrow acceptor specificity of WbpY seems to dictate that it has to act near the PP-Und moiety. The synthesis of additional acceptor substrates containing D-Rha in both α1-2 and α1-3 linkages would clarify these possibilities.
Mutagenesis of the PRK sequences in WbpX and WbpY may reveal its role in protein structure. The role of the Glu residues of the Ex7E motif and the conserved FG residues within this motif may also be clarified by mutagenesis. Studies of crystal structures and protein complexes of these D-RhaTs, as well as additional synthetic acceptors with D-Rhaα1-2 and α1-3 termini may further elucidate the mechanisms of polymer synthesis and the cooperative interactions between the two enzymes. Since the QxxRW sequence [47] is not present in WbpX and the biosynthetic intermediates are clearly released from the enzyme, the polymerization may be only partially processive. The observed in vitro cooperation between WbpX and WbpY lends insight into the mechanism of CPA biosynthesis and establishes the technology for in vitro chemo-enzymatic CPA synthesis and antibacterial vaccines.
Data availability
All data will be made available upon request (brockhau@queensu.ca). Supplementary files are attached.
Abbreviations
- CF:
-
Cystic fibrosis
- COSY:
-
Correlation spectroscopy
- CPA:
-
Common polysaccharide antigen
- EDTA:
-
Ethylenediaminetetraacetic acid
- ESI:
-
Electrospray ionization
- GST:
-
Glutathione S-transferase
- HMBC:
-
Heteronuclear multiple bond correlation
- HSQC:
-
Heteronuclear single quantum coherence
- LPS:
-
Lipopolysaccharide
- Man:
-
Mannose
- ManT:
-
Man-transferase
- MS:
-
Mass spectrometry
- NMR:
-
Nuclear magnetic resonance spectroscopy
- OSA:
-
O-specific antigen
- PA:
-
Pseudomonas aeruginosa
- PBS:
-
Phosphate-buffered saline
- PP-PhU:
-
Diphosphate-phenylundecyl
- Rha:
-
Rhamnose
- RhaT:
-
Rha-transferase
- RPM:
-
Rotations per min
- TOCSY:
-
Total correlation spectroscopy
References
Knirel, Y.A.: Polysaccharide antigens of Pseudomonas aeruginosa. Crit. Rev. Microbiol. 17, 273–304 (1990)
Middleton, M.A., Layeghifard, M., Klingel, M., Stanojevic, S., Yau, Y.C.W., Zlosnik, J.E.A., Coriati, A., Ratjen, F.A., Tullis, E.D., Stephenson, A., Wilcox, P., Freitag, A., Chilvers, M., McKinney, M., Lavoie, A., Wang, P.W., Guttman, D.S., Waters, V.J.: Epidemiology of clonal Pseudomonas aeruginosa infection in a Canadian cystic fibrosis population. Ann. Am. Thorac. Soc. 15, 827–836 (2018)
Hauser, A.R., Jain, M., Bar-Meir, M., McColley, S.A.: Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin. Microbiol. Rev. 24, 29–70 (2011)
Ciofu, O., Tolker-Nielsen, T.: Tolerance and resistance of Pseudomonas aeruginosa biofilms to antimicrobial agents–How P. aeruginosa can escape antibiotics. Front. Microbiol. 10, 913 (2019)
Johnson, L., Mulcahy, H., Kanevets, U., Shi, Y., Lewenza, S.: Surface-localized spermidine protects the Pseudomonas aeruginosa outer membrane from antibiotictreatment and oxidative stress. J. Bacteriol. 194, 813–826 (2012)
King, J.D., Kocíncová, D., Westman, E.L., Lam, J.S.: Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 15, 261–312 (2009)
Rocchetta, H.L., Burrows, L.L., Lam, J.S.: Genetics of O-antigen biosynthesis in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 63, 523–553 (1999)
Murphy, K., Park, A.J., Hao, Y., Brewer, D., Lam, J.S., Khursigaraa, C.M.: Influence of O polysaccharides on biofilm development and outer membrane vesicle biogenesis in Pseudomonas aeruginosa PAO1. J. Bacteriol. 196, 1306–1317 (2014)
Arsenault, T.L., Hughes, D.W., MacLean, D.B., Szarek, W.A., Kropinski, A.M.B., Lam, J.S.: Structural studies on the polysaccharide portion of “A-band” lipopolysaccharide from a mutant (AK1401) of Pseudomonas aeruginosa strain PAO1. Can. J. Chem. 69, 1273–1280 (1991)
Hao, Y., King, J.D., Huszczynski, S., Kocíncová, D., Lam, J.S.: Five new genes are important for common polysaccharide antigen biosynthesis in Pseudomonas aeruginosa. MBio 4, e00631-e712 (2013)
Rivera, M., Chivers, T.R., Lam, J.S., McGroarty, E.J.: Common antigen lipopolysaccharide from Pseudomonas aeruginosa AK1401 as a receptor for bacteriophage A7. J. Bacteriol. 174, 2407–11 (1992)
Lam, M.Y.C., McGroarty, E.J., Kropinski, A.M., MacDonald, L.A., Pedersen, S.S., Hoiby, N., Lam, J.S.: Occurrence of a common lipopolysaccharide antigen in standard and clinical strains of Pseudomonas aeruginosa. J. Clin. Microbiol. 27, 962–967 (1989)
Rocchetta, H.L., Burrows, L.L., Pacan, J.C., Lam, J.S.: Three rhamnosyltransferases responsible for assembly of the A-band D-rhamnan polysaccharide in Pseudomonas aeruginosa: a fourth transferase, WbpL, is required for the initiation of both A-band and B-band lipopolysaccharide synthesis. Mol. Microbiol. 28, 1103–1119 (1998)
Whitfield, C., Trent, M.S.: Biosynthesis and export of bacterial lipopolysaccharides. Annu. Rev. Biochem. 83, 99–128 (2014)
Islam, S.T., Lam, J.S.: Synthesis of bacterial polysaccharides via the Wzx/Wzy-dependent pathway. Can. J. Microbiol. 60, 697–716 (2014)
Huszczynski, S.M., Coumoundouros, C., Pham, P., Lam, J.S., Khursigara, C.M.: Unique Regions of the Polysaccharide Copolymerase Wzz2 from Pseudomonas aeruginosa Are Essential for O-Specific Antigen Chain Length Control. J. Bacteriol. 201, e00165-e219 (2019)
Melamed, J., Brockhausen, I.: Biosynthesis of bacterial polysaccharides. Manuscript code: 00072. Comprehensive Glycoscience 2nd edition, edited by Joe Barchi (2021). Vol.3pp 143–178 (2021)
Abeyrathne, P.D., Daniels, C., Poon, K.K., Matewish, M.J., Lam, J.S.: Functional characterization of WaaL, a ligase associated with linking O-antigen polysaccharide to the core of Pseudomonas aeruginosa lipopolysaccharide. J. Bacteriol. 187, 3002–3012 (2005)
Poon, K.K., Westman, E.L., Vinogradov, E., Jin, S., Lam, J.S.: Functional characterization of MigA and WapR: putative rhamnosyltransferases involved in outer core oligosaccharide biosynthesis of Pseudomonas aeruginosa. J. Bacteriol. 190, 1857–1865 (2008)
Blankenfeldt, W., Giraud, M.F., Leonard, G., et al.: The purification, crystallization and preliminary structural characterization of glucose-1-phosphate thymidylyltransferase (RmlA), the first enzyme of the dTDP-L-rhamnose synthesis pathway from Pseudomonas aeruginosa. Acta Crystallogr. D. Biol. Crystallogr. 56, 1501–1504 (2000)
Owens, T.W., Taylor, R.J., Pahil, K.S., et al.: Structural basis of unidirectional export of lipopolysaccharide to the cell surface. Nature 567, 550–553 (2019)
King, J.D., Poon, K.K.H., Webb, N.A., et al.: The structural basis for catalytic function of GMD and RMD, two closely related enzymes from the GDP-D-rhamnose biosynthesis pathway. FEBS J. 276, 2686–2700 (2009)
Smith, A.R.W., Zamze. S.E., Munro, S.M., Carter, K.J., Hignett, R.C.: Structure of the sidechain of lipopolysaccharide from Pseudomonas syringae pv. morsprunorum C28 Eur. J. Biochem. 149, 73–78 (1985)
Ovod, V., Rudolph, K., Knirel, Y., Krohn, K.: Immunochemical characterization of O polysaccharides composing the α- D-rhamnose backbone of lipopolysaccharide of Pseudomonas syringae and classification of bacteria into serogroups O1 and O2 with monoclonal antibodies. J. Bacteriol. 178, 6459–6465 (1996)
Senchenkova, S.N., Shashkov, A.S., Knirel, Y.A., McGovern, J.J., Moran, A.P.: The O specific polysaccharide chain of Campylobacter fetus serotype B lipopolysaccharide is a D-rhamnan terminated with 3-O-methyl-D-rhamnose (D-acofriose). Eur. J. Biochem. 239, 434–438 (1996)
Hickman, J., Ashwell, G.: Isolation of a bacterial lipopolysaccharide from Xanthomonas campestris containing 3-acetamido-3,6-dideoxy-D-galactose and D-rhamnose. J. Biol. Chem. 241, 1424–1428 (1966)
Kocharova, N.A., Knirel, Y.A., Widmalm, G., Jansson, P.E., Moran, A.P.: Structure of an atypical O-antigen polysaccharide of Helicobacter pylori containing a novel monosaccharide 3-C-methyl-D-mannose. Biochemistry 39, 4755–4760 (2000)
Winn, A.M., Wilkinson. S.G.: The O7 antigen of Stenotrophomonas maltophilia is a linear D-rhamnan with a trisaccharide repeating unit that is also present in polymers for some Pseudomonas and Burkholderia species. FEMS Microbiol. Lett. 166, 57–61 (1998)
Perepelov, A.V., Li, D., Liu, B., Senchenkova, S.N., Guo, D., Shevelev, S.D., Shashkov, A.S., Guo, X., Feng, L., Knirel, Y.A., Wang, L.: Structural and genetic characterization of Escherichia coli O99 antigen. FEMS Immunol. Med. Microbiol. 57, 80–87 (2009)
Ortega, X., Hunt, T.A., Loutet, S., Vinion-Dubiel, A.D., Datta, A., Choudhury, B., Goldberg, J.B., Carlson, R., Valvano, M.A.: Reconstitution of O-specific lipopolysaccharide expression in Burkholderia cenocepacia strain J2315, which is associated with transmissible infections in patients with cystic fibrosis. J. Bacteriol. 187, 1324–1333 (2005)
Cérantola, S., Montrozier, H.: Structural elucidation of two polysaccharides present in the lipopolysaccharide of a clinical isolate of Burkholderia cepacia. Eur. J. Biochem. 246, 360–366 (1997)
Tonetti, M., Zanardi, D., Gurnon, J.R., Fruscione, F., Armirotti, A., Damonte, G., Sturla, L., De Flora, A., Van Etten, J.L.: Paramecium bursaria Chlorella virus 1 encodes two enzymes involved in the biosynthesis of GDP-L-fucose and GDP-D-rhamnose. J. Biol. Chem. 278, 21559–21565 (2003)
Van Etten, J.L., Agarkova, I., Dunigan, D.D., Tonetti, M., De Castro, C., Duncan, G.A.: Chloroviruses Have a Sweet Tooth. Viruses. 9, 88 (2017)
McCaughey, L.C., Grinter, R., Jost, I., Roszak, A.W., Waløen, K.I., Cogdell, R.J., Milner, J., Evans, T., Kelly, S., Tucker, N.P., Byron, O., Smith, B., Walker, D.: Lectin-like bacteriocins from Pseudomonas spp. utilise D-rhamnose containing lipopolysaccharide as a cellular receptor. PLoS Pathog. 10, e1003898 (2014)
Ghequire, M.G.K., Swings, T., Michiels, J., Buchanan, S.K., De Mot, R.: Hitting with a BAM: Selective Killing by Lectin-Like Bacteriocins. mBio. 9, e02138–17 (2018)
Chen, W.X., Cheng, L., Pan, M., Qian, Q., Zhu, Y.L., Xu, L.Y., Ding, Q.: D Rhamnose β Hederin against human breast cancer by reducing tumor-derived exosomes. Oncol. Lett. 16, 5172–5178 (2018)
Wang, S., Tanaka, H., Hindsgaul, O., Lam, J.S., Brockhausen, I.: A convenient synthesis of GDP-D-rhamnose: The donor substrate for D-rhamnosyltransferase WbpZ from Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 23, 3491–3495 (2013)
Wang, S., Hao, Y., Lam, J.S., Vlahakis, J.Z., Szarek, W.A., Vinnikova, A., Veselovsky, V.V., Brockhausen, I.: Biosynthesis of the common polysaccharide antigen of Pseudomonas aeruginosa PAO1: Characterization and role of GDP-D-Rhamnose: GlcNAc/GalNAc-diphosphate-lipid α1,3-D-rhamnosyltransferase WbpZ. J. Bacteriol. 197, 2012–2019 (2015)
Torgov, V., Danilov, L., Utkina, N., Veselovsky, V., Brockhausen, I.: Synthesis of P1-(11-phenoxyundecyl)-P2-(2-acetamido-2-deoxy-3-O-α-D-rhamnopyranosyl-a-D-glucopyranosyl) diphosphate and P1-(11- phenoxyundecyl)-P2-(2-acetamido-2-deoxy-3-O-β-D- galactopyranosyl-α-D-galactopyranosyl) diphosphate. Carbohydr. Res. 453, 19–25 (2017)
Hao, Y., Murphy, K., Lo, R.Y., Khursigara, C.M., Lam, J.S.: Single-nucleotide polymorphisms found in the migA and wbpX glycosyltransferase genes account for the intrinsic lipopolysaccharide defects exhibited by Pseudomonas aeruginosa PA14. J. Bacteriol. 197, 2780–2791 (2015)
Wang, S., Czuchry, D., Liu, B., Vinnikova, A., Gao, Y., Vlahakis, J.Z., Szarek, W.A., Feng, L., Wang, L., Brockhausen, I.: Characterization of two UDP-Gal: GalNAc-diphosphate-lipid β1,3-Galactosyltransferases WbwC from Escherichia coli Serotypes O104 and O5. J. Bacteriol. 196, 3122–3133 (2014)
Guerin, M.E., Kordulakova, J., Schaeffer, F., et al.: Molecular recognition and interfacial catalysis by the essential phosphatidylinositol mannosyltransferase PimA from mycobacteria. J. Biol. Chem. 282, 20705–20714 (2007)
Czuchry, D., Szarek, W.A., Brockhausen, I.: Identification and biochemical characterization of WbwB, a novel UDP-Gal: Neu5Ac-R α1,4-galactosyltransferase from the intestinal pathogen Escherichia coli serotype O104. Glycoconj. J. 35, 65–76 (2018)
Gao, Y., Vlahakis, J.Z., Szarek, W.A., Brockhausen, I.: Selective inhibition of glycosyltransferases by bivalent imidazolium salts. Bioorg. Med. Chem. 21, 1305–1311 (2013)
Greenfield, L.K., Richards, M.R., Li, J., Wakarchuk, W.W., Lowary, T.L., Whitfield, C.: Biosynthesis of the polymannose lipopolysaccharide O-antigens from Escherichia coli serotypes O8 and O9a requires a unique combination of single- and multiple-active site mannosyltransferases. J. Biol. Chem. 287, 35078–35091 (2012)
Greenfield, L.K., Richards, M.R., Vinogradov, E., Wakarchuk, W.W., Lowary, T.L., Whitfield, C.: Domain organization of the polymerizing mannosyltransferases involved in synthesis of the Escherichia coli O8 and O9a lipopolysaccharide O-antigens. J. Biol. Chem. 287, 38135–38149 (2012)
Yakovlieva, L., Walvoort, M.T.C.: Processivity in Bacterial Glycosyltransferases. ACS Chem. Biol. 15, 3–16 (2020)
Choudhury, B., Carlson, R.W., Goldberg, J.B.: The structure of the lipopolysaccharide from a galU mutant of Pseudomonas aeruginosa serogroup-O11. Carbohydr. Res. 340, 2761–2772 (2005)
Yokota, S., Kaya, S., Sawada, S., Kawamura, T., Araki, Y., Ito, E.: Characterization of a polysaccharide component of lipopolysaccharide from Pseudomonas aeruginosa IID 1008 (ATCC 27584) as D-rhamnan. Eur. J. Biochem. 167, 203–209 (1987)
Arce-Rodríguez, A., Volke, D.C., Bense, S., Häussler, S., Nikel, P.I.: Non-invasive, ratiometric determination of intracellular pH in Pseudomonas species using a novel genetically encoded indicator. Microb. Biotechnol. 12, 799–813 (2019)
Acknowledgements
The authors are grateful the late Leonid Danilov, Zelinsky Institute, Academy of Sciences, Moscow, Russia, for his continuing essential contribution to the work reported here. We thank Joseph Lam (University of Guelph) for the plasmids encoding WbpXY and John Klassen (University of Alberta) for mass spectrometry analyses. Jiaxi Wang (Queen’s University) carried out mass spectrometry and Françoise Sauriol (Queen’s University) recorded NMR spectra. For the synthesis of GDP-D-Rha and acceptor substrates D-Rhaα-Bn and GalNAc/GlcNAcα-PP-PhU, we thank Jason Vlahakis (Department of Chemistry, Queen’s University) and the late Walter A. Szarek, to whom we are indebted for his long-time collaboration and support. We thank Cassidy Philips for preliminary enzyme assays. This work was funded by a Discovery grant from the Natural Sciences and Engineering Research Council (to I.B).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Melamed, J., Kocev, A., Torgov, V. et al. Biosynthesis of the Pseudomonas aeruginosa common polysaccharide antigen by D-Rhamnosyltransferases WbpX and WbpY. Glycoconj J 39, 393–411 (2022). https://doi.org/10.1007/s10719-022-10040-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-022-10040-4