
Abstract
Recently, bats have gained attention as potential reservoir hosts for emerging zoonotic single-stranded (ssRNA) viruses that may prove fatal for humans and other mammals. It has been hypothesized that some features of their innate immune system may enable bats to trigger an efficient early immune response. Toll-like receptors (TLRs) represent a first line defense within the innate immune system and lie directly at the host–pathogen interface in targeting specific microbe-molecular patterns. However, the direction and strength of selection acting on TLRs are largely unknown for bats. Here, we studied the selection on viral ssRNA sensing TLR8 based on sequence data of 21 bat species. The major part (63 %) of the TLR8 gene evolved under purifying selection, likely due to functional constraints. We also found evidence for persistent positive selection acting on specific amino acid sites (7 %), especially when compared to viral TLR evolution of other mammals. All of these putatively positively selected codons were located in the ligand-binding ectodomain, some coincidenced or were in close proximity to functional sites, as suggested by the crystallographic structure of the human TLR8. This might contribute to the inter-species variation in the ability to recognize molecular patterns of viruses. TLR8 evolution within bats revealed that branches leading to ancestral and recent lineages evolved under episodic positive selection, indicating selective selection pressures in restricted bat lineages. Altogether, we found that the TLR8 displays extensive sequence variation within bats and that unique features separate them from humans and other mammals.
Similar content being viewed by others


Introduction
Zoonotic viruses that emerge from wildlife pose a serious threat for humans and domestic livestock. Over the past years, bats have attracted growing attention as a potential reservoir of emerging zoonotic viruses that may have a high impact on human health such as the severe acute respiratory syndrome-like (SARS) and Middle East respiratory syndrome-like (MERS) coronaviruses, Ebola and Marburg filoviruses, Hendra, Nipah, and other newly recognized paramyxoviruses, hepaciviruses, pegiviruses, and hantavirus, as well as new lineage of influenza A virus (reviewed in Calisher 2015). In addition, bats have long been known as vectors for lyssaviruses causing rabies. Apparently, all aforementioned viruses are single-stranded RNA (ssRNA) viruses and, except for lyssaviruses, cause little or no pathology in bats (e.g., Wynne and Wang 2013; Brook and Dobson 2015). This raises the question why certain viruses can infect and persist in apparently healthy bats whereas they may cause severe symptoms and even death in other vertebrate species (Wang et al. 2011; Bean et al. 2013; Plowright et al. 2015).
Typically, mechanisms of infection and resistance involve direct interactions of the host and the pathogen at a molecular level. In general, viruses are first recognized by the innate immune system via pattern recognition receptors (PRR), including the Toll-like receptor (TLR) family, which sense microbe-associated molecular patterns (MAMPs; Takeda et al. 2003; Kawai and Akira 2010). Resistance to virus infection can evolve by selection of mutations that alter amino acids in the binding region of receptor molecules. This sort of positively selected residues point to specific molecular recognition interfaces between host and viral components that have adapted and counter adapted in a long series of classical Red Queen conflicts and can be used to infer the history of the host–virus “arms race” during their co-evolution (Daugherty and Malik 2012; Hancks et al. 2015). Thus, PRRs may rapidly evolve to evade pathogen antagonism and to ensure activation of a robust immune response. Although bats appear to share the immune cell and gene repertoire with other mammals, it has been repeatedly hypothesized that the long-term co-existence of bats and viruses must have imposed strong selective pressures on the bat genome and that those genes related to the first line of antiviral defense within the innate immune system, e.g., TLRs, may reflect this co-evolution (Wibbelt et al. 2010; Cowled et al. 2011; Baker et al. 2013; Wynne and Wang 2013; Zhang et al. 2013).
Binding of MAMPs to TLRs triggers a series of events leading to increased expression of proinflammatory genes (Kawai and Akira 2006). Within TLRs, the extracellular part, consisting of multiple leucine rich repeats (LRR), is involved in pathogen recognition and evolves faster than the highly conserved intercellular Toll/interleukin-1 receptor (TIR) domain which is responsible for intracellular signalling (Mikami et al. 2012). Most mammals have 10–12 different TLRs, each recognizing different ligands, hereby nucleic acids of viruses are recognized by endosomal TLRs (3, 7, 8, and 9; Alexopoulou et al. 2001; Lund et al. 2004; Xagorari and Chlichlia 2008). Single nucleotide polymorphisms (SNPs) in all TLRs have been correlated with disease susceptibility demonstrating that single amino acid alterations may have an effect on immune response mechanisms (humans: Schroder and Schumann 2005; Wong et al. 2010; Taylor et al. 2012; Uciechowski et al. 2011; Wang et al. 2014; rodents: Tschirren et al. 2013).
Studies of the evolutionary dynamics of TLRs have revealed that selective pressures vary between TLRs and taxa. In general, a high degree of purifying selection is found for all TLRs and is particularly reported for nucleic acid-sensing TLRs due to functional constraints (Roach et al. 2005; Barreiro et al. 2009; Mukherjee et al. 2009). However, an increasing number of studies detected specific residues within the LRR binding region to be under positive selection in almost all TLRs (mammals: Areal et al. 2011; apes: Wlasiuk and Nachman 2010; rodents: Tschirren et al. 2011; bats: Zhang et al. 2013, Escalera-Zamudio et al. 2015; birds: Alcaide and Edward 2011; Grueber et al. 2012; amphibians: Babik et al. 2014) indicating that the ligand-binding domain is subject to classical Red Queen conflicts as mentioned above (Daugherty and Malik 2012; Hancks et al. 2015). Indeed, a recent study highlighted that nucleic acid sensing TLRs in bats exhibited unique mutations fixed in ligand-binding sites (Escalera-Zamudio et al. 2015). They found further evidence for episodic positive selection acting specifically upon the bat lineage (based on eight bat species belonging to three families) suggesting potential functional differences between bat and other mammalian TLRs. Further, several ancestor genes involved in innate immunity (e.g., TLR7, NLRP3, and MAP3K7) were found to be under stronger positive selection in the genomes of the Australian black flying fox (Pteropus alecto) and David’s Myotis (Myotis davidii) than their orthologues in seven other mammalian species (Zhang et al. 2013). Interestingly, TLR7, which is besides TLR8 and RIG-1 (an intracellular PRR of the retinoic acid-inducible gene I family, Jensen and Thomsen 2012) responsible for the detection of ssRNA viruses, was one of those being under positive selection. This is remarkable because TLR7 has been suggested to be under strong purifying selection in other mammalian taxa (Barreiro et al. 2009; Georgel et al. 2009; but see Areal et al. 2011). Here, we investigated traces of selection acting upon the viral ssRNA sensing TLR8 gene to shed further light on evolutionary dynamics of the viral sensing TLRs within different bat orders and their potential implications in ligand-binding affinities. In addition to the aforementioned studies, we expanded the number of investigated bat taxa to 21 bat species, belonging to seven families, to increase resolution. We included three species of the Rhinolophoidae, which are known as a main reservoir for beta-coronaviruses. Specifically, we studied the molecular variation, the strength and direction of selection, focussing on the presence of putatively positively selected amino acids in the LRR-binding region. Finally, we discuss the altered amino acid residues and their potential implications in ligand-binding affinities in detail and in comparison to new data on two ssRNA ligand-binding sites of the human TLR8 molecule (Tanji et al. 2015).
Materials and methods
Tissue collection, DNA and RNA extraction
Tissue samples from 15 species where obtained both from free-ranging bats and also from museum specimen (Table S1, Supplementary Material). Wild bats were captured at the following locations: Bulgaria (Rhinolophus euryale, R. hipposiderus, R. ferrumequinum, and Myotis myotis, permission number permit of the Ministerstvo na Okolnata Sreda i Vodita, Sofia and RIOSV Ruse; No. 57/18.04.2006, No. 100/04.07.2007), Costa Rica (Saccopteryx bilineata, Pteronotus parnelli, Carollia perspicilatta, Vampyressa pusilla, Vampyressa bidens, Ministerio del Ambiente y Energia; # 036-2009-SINAC: No. 137–2010–SINAC and 168–2011–SINAC) and from Panama (Noctilio albiventris, Noctilio leporinus, Ministerio de Agricultura, Ganaderia, Acuacultura y Pesca #04508). We captured wild bats using mist-nets or harptraps, depending on the specific efficacy, when bats emerged from their roosts. A 4-mm wing-tissue sample was collected using a sterile biopsy punch and the sample immediately stored in 96 % ethanol until DNA isolation. Liver samples (stored in RNAlater at −20 °C) of M. myotis and N. albiventris were used for RNA analyses. Animals were euthanized in accordance with appropriate guidelines (Gannon and Sikes 2007) and under the license of national authorities. In addition, we received frozen tissue material from a museum specimen from Eidolon helvum (Natural History Museum Berlin). In Germany, we collected tissue material from four bat species that were obtained from central depositories where bats killed at wind turbines are deposited. Samples were stored at −80 °C after collection (Nyctalus noctula, Nyctalus leisleri, Vespertilio murinus, and Pipistrellus nathusii, Landesumweltamt Brandenburg, Vogelschutzwarte Buckow). Total DNA was extracted from the biopsies using standard protocols (DNA extraction Kit, Genial). RNA of M. myotis and N. albiventris was extracted using RNA extraction-kit (RNeasyKit, Quiagen). Possibly remaining genomic DNA was removed by digestion and the cDNA-library established by reverse transcriptase (RevertAid H Minus First cDNA Synthesis Kit, Fermentas).
Available TLR8 sequences of other bat species were included in our analyses (Pterpous alecto GenBank accession number XM_006906028, Eptesicus fuscus XM_008156576, Myotis davidii XM_006763795, M. brandtii XM_005880947, M. lucifugus XM_006088606) and compared to the equine (XM_014728737) and human (XM_005274543) orthologous.
Primer design
We first aligned the published bat TLR8 sequences from the Australian black flying fox (P. alecto) and three species of mouse-eared bats (M. davidii, M. brandtii, and M. lucifugus) to identify highly conserved regions. We then designed a diverse array of primer sets to amplify the whole TLR8 coding region (3,056 bp without primers out of 3,123 bp of the human TLR8-coding sequence) in all selected bat species (Supplementary Table S2). Established primers spaced overlapping fragments between 700 and 800 bp in length.
TLR8 amplification and sequencing
We amplified the TLR8 coding region in two to three overlapping amplicons using a proofreading, long-range polymerase (Myfi-Polymerase, Thermoscintific). PCR reactions were conducted in a total volume of 25 μL consisting of 10–50 ng genomic DNA, 10 μmol/L of primers and 5 × MyFi reaction buffer with 1 mmol/L dNTPs, 3 mmol/L MgCl2, and 2 U/μL MyFi DNA Polymerase. PCR conditions were as follows: initial denaturation at 95 °C for 3 min followed by 36 cycles of denaturation at 95 °C for 15 s, annealing between 50 and 58 °C for 15 s and elongation at 72 °C for 4–5 min depending on fragment length and a final elongation step by 72 °C for another 10 min. PCR products were electrophoresed in a 1 % agarose gel. Positive amplicons were purified (Exonuclease I, FastAP thermosensitive alkaline phosphatase, Thermoscientific) and bi-directly sequenced using Big-Dye Terminator technology and an ABI PRISM 310 Automated Genetic Analyser (Applied Biosystems, Foster City CA, USA). Bat TLR8 sequences have been deposited in GenBank under the following accession numbers KU163591-KU163606.
Statistical analysis
Nucleotide sequences were edited and aligned manually using Chromas Lite 2.1.1 (Technelysium Pty Ltd 1998–2012) and MEGA 6.0 (Tamura et al. 2013). Sequence percent identities compared to other mammalian species (human and horse) were obtained by homology Blast search (http://blast.ncbi.nlm.nih.gov) and calculated by MEGA. To estimate evolutionary divergence of the TLR8 gene sequences within bats, we calculated besides sequence percent identities the nucleotide diversity π by estimating the mean number of base differences per site using a bootstrap procedure with 500 replicates with Mega 6.0.
Under neutrality, coding sequences are expected to present a ratio (ω) of non-synonymous (dN) over synonymous (dS) base pair substitutions that does not significantly deviate from 1 (ω = dN/dS = 1), while significant deviations may be interpreted as either the result of positive (ω > 1) or purifying (ω < 1) selection (Ellegren 2008). We estimated the relative rates of dN and dS according to the Nei and Gojobori (1986) method, applying the Jukes-Cantor correction for multiple hits in 500 replicates (Jukes and Cantor 1969) implemented in MEGA. Evidence for persistent positive selection in the past was assessed through the program CODEML (included in the software package Paml 3.15, Yang 2007), which estimates ω among sites applying different models of codon evolution (neutral, purifying, or positive). First we tested whether ω differs among sites by comparing model M0, which assumes a constant ω across all sites to model M3 (discrete), which allows ω to vary among sites, estimating the proportion of conserved, neutral and positive selected codons from the data. To test for the presence of sites evolving under positive selection we used the nested model pair M7, allowing codons only to evolve neutrally or under purifying selection and M8, which adds a class of sites under positive selection assuming a beta distribution for ω. The two nested models pairs were compared using the likelihood ratio test (LRT, α = 95 %) with two degrees of freedom (Yang et al. 2005). For a better comparison to number and position of positively selected sites detected in TLRs of other taxa we applied a set of methods to detect candidate-codons under persistent positive selection. Based on the methodology adopted by other authors and in previous studies (Wlasiuk and Nachman 2010; Areal et al. 2011; Matos et al. 2013; Castel et al. 2014; Darfour-Oduro et al. 2015), only codons identified by three or more methods were considered to be putatively under positive selection. It has been suggested that the application of several selection assessment methods, followed by the classification of sites detected in consensus as positively selected can minimize false positives (e.g., Kosakovsky Pond and Frost 2005, Maldonado et al. 2014). We applied Model M8 implemented in the PAML package and the SLAC (single likelihood ancestor counting), FEL (fixed-effect likelihood), REL (random effect likelihood), and FUBAR (Fast, Unconstrained AppRoximation analyses) methods implemented in the Hyphypackage (www.datamomkey.org; Kosakovsky Pond et al. 2005; Delport et al. 2010; Murell et al. 2013). Codons with p values <0.1 SLAC and FEL, with Bayes Factor >50 for REL, and with posterior probabilities >0.9 for M8 and FUBAR were considered as candidates under positive selection. We further tested the degree of dissimilarity of amino acid substitutions according to their physiochemical properties using PRIME (Property Informed Model of Evolution), with the amino acid property set according to Atchley et al. (2005), also available in the Datamonkey web server. A change on this properties was considered significant if the posterior probabilities exceeded >0.9. The evolutionary conservation of amino acid positions was then predicted using the ConSurf tool (Ashkenazy et al. 2010), with the assumption that positively selected residues were least conserved.
Finally, to test for episodic positive selection Branch-Site REL and MEME (mixed effects model of evolution) implemented in the Hyphypackage were used. The Branch-Site REL model estimates proportion of sites under selection along tree branches and allows evolutionary rate to simultaneously vary along phylogenetic branches and sites (Kosakovsky Pond et al. 2011). The MEME method identifies lineage-specific events of positive selection at specific sites, even though the same site is under purifying or neutral selection in other lineages (Murell et al. 2012). Codon sites were considered to be under episodic positive selection at a significant level p < 0.05. A Neighbour Joining tree was used as working topology in all analyses.
The domain architecture for TLR8 was determined by using a Simple Modular Architecture Research Tool (SMART, Schultz et al. 1998), which combines homology searches with predictive algorithms to identify putative protein domains. TM domains were additionally identified by TMPRED (Hofmann and Stoffel 1993), which estimates transmembrane regions on the basis of hydrophobicity of predicted TM-helices. The LRRfinder tool (www.lrrfinder.com, Offord et al. 2009) was used to calculate, in addition to the domain architecture, the surface accessibility (buried or exposed) and the predicted secondary structure (helix, coil or strand) for each amino acid, which we used to discuss the potential functionality of amino acids that have been predicted to have evolved under persistent positive selection. Tertiary protein structure predictions were generated using Swiss Model (Biasini et al. 2014) based on the known crystallised human (hTLR8) structure. Positions of sites of persistent positive selection affecting particular amino acid positions in the protein were visualized by PyMol 1.2.8 (De Lano 2002). Amino acid positions and LRR numbering were assigned according to the human h TLR8 molecule throughout the article (Tanji et al. 2013, 2015).
Results
Domain architecture and diversity
Similar to other mammalian TLRs, TLR8 of all investigated bat species showed the typical domain architecture with a signal peptide followed by a LRR ectodomain for ligand binding, a transmembrane domain (TM), and a cytoplasmatic TIR domain for signal transduction. Number and locations of LRRs within the ectodomain varied substantially among species (15–19 LRR, Supplementary Fig. S1). We could not detect TM domains in the Noctilio and Rhinolophus species by SMART analyses. However, based on TMPRED analyses, we identified the TM domain in the appropriate region for species of these two genera (Supplementary Fig. S2). Expression was confirmed by RT-PCR in N. albiventris and M. myotis, in spleen and liver.
The open-reading frame of the amplified single large TLR8 exon displayed no stop codons. We detected several indels (1–3 amino acids), which always concerned three contiguous nucleotides or a multiple of it, thus frame shifts never occurred (Supplementary Fig. S3). We obtained 1,018 amino acids (aa) out of the 1,024 aa that were previously described for hTLR8. However, the obtained lengths of amino acid sequences varied between 1,015 aa (Vespertilionidae) and 1,020 aa (Phyllostomidae), as a result of the presence of indels. Sequence length variations were all located in the LRR ectodomain.
The nucleotide sequences showed 79–85 and 76–81 % sequence identity to their equine and human orthologues. The adopted TLR8 protein sequences showed less similarity with 74–76 % (equine) and 70–72 % (human) amino acid sequence identity, respectively, with higher similarities in the TM and TIR-domain compared to the LRR ectodomain (Supplementary Table S3a, b, c). Mean nucleotide (nt) and amino acid (aa) similarities within bats reached 84 % (nt) and 80 % (aa), and ranged from 78 % (nt) and 72 % (aa) between M. myotis and R. hipposideros to 99 % (nt and aa) identity between E. helvum and P. alecto, displaying a remarkable intra-order diversity. In all comparisons of the LRR ectodomain, amino acid sequences showed a lower level of similarity between taxa than the corresponding nucleotide sequences. The opposite was true for the TM and TIR domain, were amino acid sequences showed higher percent sequence identities between all taxas than the nucleotide sequences (Supplementary Table S3a, b, c).
Evidence for persistent positive selection
Non-synonymous substitutions did not exceed those of synonymous substitutions when the whole sequence was considered or when only the LRR-region was analyzed with highest values for ω in the LRR ectodomain (Table 1). However, we detected significant ω heterogeneity within the TLR8 sequences (M0 vs M3, p < 0.001). According to model M3 the large majority of codons evolved under purifying selection (63 %, ω = 0.06), 30 % of sites evolved neutrally (ω = 0.83) and 7 % of all sites were detected to evolve under positive selection (ω = 2.34; Table 2). When comparing models that estimate ω with a beta distribution also model M8 allowing for positive selection fitted the data significantly better than model M7 that considered only neutral sites (p < 0.001). To identify specific sites that might have evolved under persistent positive selection, we used different analytical approaches (see method section, Supplementary Table S4). Estimated PSS occurred clustered in certain regions within the amino acid sequence alignment and were all located in the ligand-binding LRR ectodomain (Fig. 1, Supplementary Fig. S3). This clustering was even pronounced in the 3D structure of the molecule (see below). 24 specific codon sites were estimated as positive selected sites (PSS) by at least three model analyses and were thus elected as putatively PSS in bats (Table 3, Supplementary Table S4). Amino acid character alterations were present in all 24 PSS including non-polar and polar as well as basic polar and acidic polar side chain polarities (Table 3). This was underlined by the amino acid conservation scores of the ConSurf analyses, which indicated all PSS to be least conserved (scores >1.5, Table 3).

Predicted protein domain architecture of the TLR8 gene of Rhinolophus ferrumequinum and putatively positive selected sites in bats. TM domains are shown in dark blue; other domains are labeled accordingly. Protruding loops are marked in orange. Predicted regions involved in ligand binding (red rectangles) and dimerization (circles: orange inactivated, red activated dimer) according to the human TLR8 molecule (Tanji et al. 2013, 2015) are marked below the gen cartoon. Amino acid sites with statistically significant ω values in bats are indicated by triangles (M8: posterior probabilities shaded triangles >95 %, blanc triangles >90 %; FEL, PRIME: p values shaded triangles <0.05, blanc triangles <0.1; REL: Bayes factor shaded triangles >50; SLAC: p values shaded triangles <0.1; FUBAR: posterior probabilities shaded triangles >90 %, blanc triangles >85 %). Putatively positive selected sites (PSS) in bats estimated by more than three models are shown as red x. The four clustered PSS regions are marked by red rectangles. PSS in mammals according to Areal et al. (2011) in Suidae (Darfour-Oduro et al. 2015) and in primates (Wlasiuk and Nachman 2010) are mapped as green triangles
The eight PSS detected by Escalera-Zamudio et al. (according to their M8 analyses) matched with the estimated PSS of our M8 model analyses, except position 506, which was identified to have undergone episodic positive selection by our analyses (see below and Supplementary Fig. S3). Four PSS (position 236, 338, 481, and 712) matched with predicted positive selected codons in mammals (Areal et al. 2011) and others were situated in close proximity (43, 238, 351, 354, 420, 421, 420, 447, and 608). Of the three PSS described in TLR8 of other taxa (491 in primates (Wlasiuk and Nachman 2010), 178, 388 in suidae (Darfour-Oduro et al. 2015)) one (388) showed amino acid alterations and the other two were conserved in bats (Fig. 1, Supplementary Table S4, Fig. S3).
Evidence for episodic positive selection
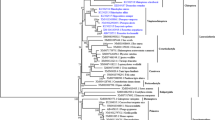
We performed branch-site REL and MEME analyses to detect signatures of episodic positive selection in specific bat lineages within TLR8 evolution. We found evidence for episodic positive selection in the ancestral lineages of different higher taxonomic clades and families, including the ancestors of Yinchiroptera, Rhinolophoidae, Noctilionoidae, and Pteropodidae with strongest support for the Rhinolophoid lineage (Table 4, Fig. 2). Analyses also indicated that terminal branches of single species have been under positive selection pressure, including M. brandtii, R. hipposideros, S. bilineata, and V. murinus. The branch leading to S. bilineata might concern the whole family of Emballonuridae, because only a single species of this family was included in our analyses. The MEME analyses revealed 34 sites of episodic positive selection along branches (Fig. 2, Table 4, Supplementary Table S4). Besides lineage-specific codons, some of the identified sites coincided with codons detected to evolve as well under persistent positive selection. Position 351 involved in ligand binding and dimerization was positively selected in the ancestral branches leading to Yinchiroptera, Rhinolophoidae and within the Myotis branch. Position 539 which has been described as a ligand-binding-site mutation in bats by Escalera-Zamudio et al. (2015) was identified as a site underlying episodic positive selection in our analyses (Table 4, Supplementary Table S4, Fig. S3).

Phylogenetic tree for Chiropteran TLR8. Neighbor-joining tree was estimated based on a distance matrix of Kimuras two-parameter evolutional model using the human TLR8 sequence (XM_005274543) as outgroup to root the tree. Bootstrap percent probabilities were based on 500 replicates. The scale bar represents a genetic distance of 0.02. Estimated lineages evolving under episodic positive selection by the branch-site REL analyses are shown in red and mean ω values are represented. Amino acid sites under episodic positive selection detected singularly by MEME analyses are mapped above branches (arrows); see also Table 4
Predicted molecule structure and location of PSS according to human TLR8
Analyses of surface accessibility confirmed that all PSS, except three (aa position 104, 420, and 447), should prepossess exposed positions in the three-dimensional molecule structure (Table 3). However, position 104 was located in a protruding loop within LRR2 and 447 was located within the Z-loop and might thus be incorrectly defined by the LRR-finder-tool.
Projection of PSS onto the 3D protein model of R. euryale revealed that the sites under selection localized to approximately four distinct regions within the LRR ectodomain (Fig. 3). The main region concerned several PSS located in a section between LRR10-18 including the Z-loop, which has been found to be involved in dimerization and in binding uridine-rich degraded ssRNA at two distinct binding sites (Tanji et al. 2013, 2015). Five PSS (312, 338, 368, 420, 421, and 481) were situated in a concavity surrounded region of LRR 10, 11, and 14 and parts of the Z-loop, which are considered to bind ssRNA oligonucleotides (Tanji et al. 2015). Here, position 338 had the strongest support by convergent findings in all our analyses and was also indicated as PSS in mammals (Areal et al. 2011, Supplementary Table S4). Out of the 21 amino acids predicted to be directly involved in RNA-binding at that site, 12 exhibited amino acid mutations and nine were conserved in bats (Supplementary Table S4). Three PSS (352, 354, and 356) were located within or next to another binding site predicted to bind the base uridine and small chemical ligands, also known to be responsible for TLR8 dimerization (Figs. 3 and 4). Here, PSS 352 has been found to interact directly with the base uridine, as does position 351, which was found to be under episodic positive selection in bats. Some of the predicted amino acids representing this uridine binding site in hTLR8 were also conserved in bats (11 out of 16 aa) and five positions showed variable amino acid substitutions (Supplementary Table S4). Interestingly, PSS 447 was located in the Z-loop within a part (position 442–457) predicted to be cleaved in hTLR8 (Tanji et al. 2013).

Predicted tertiary molecule structure of a chiropteran TLR ectodomain (R. euryale) and sites of persistent positive selection. Tertiary structure was generated by Swiss-Model based on known human-crystallized TLR8 structure, Z-loop is shown in orange, cleaved part in black, ligand-binding site predicted to be involved in binding ssRNA oligonucleotids are marked in green, and the site predicted to be involved in dimerization and binding the base uridine are indicated in yellow (Tanji et al. 2015). Positions of sites under persistent positive selection in bats are shown in pink; purple indicate positions that coincide with amino acids of human binding sites. a Front, b side, and c top view

Predicted tertiary molecule structure of a chiropteran TLR8-dimer (R. euryale) and sites of persistent positive selection at the dimerization interface. Tertiary structure was generated by Swiss-Model based on known human-crystallized TLR8-dimer, ligand-binding site predicted to be involved in dimerization and binding the base uridine (Tanji et al. 2015) are indicated in light (α-chain) and dark green (ß-chain). Positions of sites under persistent positive selection in bats are shown in light (α-chain) and dark (ß-chain) blue; light (α-chain) and dark (ß-chain) purple indicate positions that coincide with amino acids of the human binding sites. a Front, b side, and c top view
Besides the PSS found within the ligand-binding region, we found several PSS clustering in three additional regions of the protein (Fig. 3). One region concerned LLR1 (43) and a protruding loop within LRR2 (104, 109). A second region concerned three amino acids located next to each other within LRR7 and 8 (236, 238, 277) at the protein surface. Of those, position 236 was also described as PSS in mammals (Areal et al. 2011). A last region showed five PSS (691, 677, 685, 712, and 732) in close proximity within LRR23 and 24, also one (677) with strong support (Supplementary Table S4). Position 677, 699, and 723 were in close neighborhood to each other located at the surface towards the dimerization interface. Position 712 was also identified as PSS in mammals (Areal et al. 2011).
Discussion
Similar to TLR8 of other mammal species, the TLR8 gene of bats is highly conserved concerning gene and domain architecture, suggesting an analogous function. In all investigated bat species, the coding sequence of TLR8 is encoded within a single large exon, which is in agreement with the gene structure of other mammals. However, indels lead to structural length differences in the LRR ectodomain between different bat families. Also, the number and location of LRRs within the ectodomain varied substantially between species, suggesting alterations in molecule structures which might have an effect on ligand-binding affinities. The observed percent sequence identities at the amino acid level were highest in the TM and TIR domain when compared to that of other mammals and within bat species, indicating the importance of a conserved function in these domains for triggering the intracellular immune cascade. These contrasts to finding in the LRR ectodomain were less similarity was found at the amino acid level indicating selection mechanisms towards amino acid mutations. Altogether intraorder variability between bat taxa was high especially in the ligand-binding LRR ectodomain, and reached percent sequence identity values as different as to that to other mammal taxa, like human and horse. This finding might point to a long history of species-specific adaptation processes in the co-evolution with viral pathogens within the species rich order of Chiroptera.
As a result of the constraints imposed by the receptor protein function, purifying selection has been argued as the dominant selective force in the evolution of all TLRs, particularly for nucleic acid sensing TLRs (e.g., Georgel et al. 2009; Wlasiuk and Nachman 2010; Fornuskova et al. 2013; but see Areal et al. 2011). In bats, we found an excess of synonymous over non-synonymous substitutions when considering the whole TLR8 gene and the LRR ectodomain exclusively, which is consistent with processes of purifying selection. However, the ratio of non-synonymous over synonymous base pair substitutions reached a relatively high value of 0.517 for omega, a value that is much higher than those observed in most other proteins in mammals (range of ω = 0.1–0.25, Ellegren 2008). This fact was underlined in our analytical approach of codon evolution, where maximum likelihood models that allowed for positive selection fitted the data significantly better than models that considered only neutral sites indicating besides purifying selection also the occurrence of a positive selection pressure in the history of the TLR8 gene evolution. Overall, the large majority of codons evolved under neutrality or purifying selection, but 7 % of all sites were detected to evolve under persistent positive selection, a high value compared, e.g., to the TLR8 evolution in primates, where only 3 % evolved under positive selection (Wlasiuk and Nachman 2010) and might reflect the longer evolutionary history of bats compared to that of primates (first radiation of bats in the early Eocene compared to the Oligocene in primates).
All positively selected sites were located in the LRR ectodomain several within or next to predicted sites involved in ligand binding, which is consistent with findings in TLRs of other taxa (e.g., Areal et al. 2011; Wlasiuk and Nachman 2010; Tschirren et al. 2011, Escalera-Zamudio et al. 2015; Alcaide and Edward 2011; Grueber et al. 2012; Babik et al. 2014, Vinkler et al. 2014). As the recognition of viral RNA is essential for host defense, only such mutations that are advantageous in the recognition of co-evolving viruses should have become fixed and reflected as differences between species (Morozumi and Uenishi 2009). This was supported by the observation that the amino acid alterations within PSS led to physio-chemical changes. Physio-chemical alterations are thought to be important in defining the molecular structure and are therefore expected when natural selection promotes a functional change in the structure of proteins (Smith 2003, Vinkler et al. 2014; Darfour-Oduro et al. 2015). Our result indicate a similar or even higher level of persistent positive selection acting in TLR8 as described for non-viral TLRs in other mammalian orders (e.g., Tschirren et al. 2011; Areal et al. 2011; Smith et al. 2012, Darfour-Oduro et al. 2015) and contrast the picture of a more constrained evolution for viral TLRs as reported for other mammalian orders (humans: Barreiro et al. 2009; primates: Wlasiuk and Nachman 2010; rodents: Fornuskova et al. 2013; Suidae: Morozumi and Uenishi 2009; Darfour-Oduro et al. 2015, but see mammals: Areal et al. 2011). However, we lack information about the diversity of non-viral TLRs in bats, thus an intra-order comparison of viral and non-viral TLR evolution is still needed for the order of Chiroptea.
To get a closer insight in the strength of selection acting on TLR8 evolution within bats, we examined whether episodic positive selection has contributed to the diversity pattern. We found evidence that the branches leading to higher taxonomic clades have undergone episodic positive selection at several amino acids sites, indicating that the ancestral species within these lineages, especially those of Rhinolophoidae and Pteropodidae, had to undergo adaptive changes at these sites in response to their pathogen environment. Additionally, we detected episodic positive selection in terminal branches leading to single species (M. brandtii, R. hipposiderus, M. brandtii, and V. murinus). Our results indicate that repeated episodes of positive selection, interspersed with purifying selection shaped the observed TLR8 diversity pattern throughout the evolution of bats, and we assume that this adaptive process is currently still in operation.
The arrangement of sites under positive selection can predict locations of binding interactions between host and pathogen proteins (Werling et al. 2009; Daugherty and Malik 2012). Projection of persistent PSS onto the predicted 3D protein model revealed that the sites under selection localized to approximately four distinct regions within the LRR ectodomain (Fig. 3). Three of them concerned parts of the protein that have not been described before to be involved in ligand binding in humans (Tanji et al. 2015). However, some of them coincidence with PSS detected also in mammals (236 and 712), or were in close proximity (43 and 608) and might thus be of functional relevance. Some others were located in close neighbourhood in a protruding loop (104 and 109) or in the dimerization interface (677, 699, and 723) and might have functional relevance for structural stability, because protruding loops and the dimerization interface are thought to be involved in dimerization or stabilisation of the dimeric structure (Gautam et al. 2006; Tanji et al. 2013).
The fourth and main region with 12 PSS coincided with the predicted ligand-binding region of LLR10-18 and the Z-loop in humans (Fig. 3). Crystallographic analyses of the human TLR8 revealed that this region binds degradation products of ssRNA at two distinct sites (Tanji et al. 2015). One site within the concave surface of LRR10-14 interacts with ssRNA oligomers of different length and different sequence motifs, but seems to prefer sequences containing purine-pyrimidine dinucleotides. The other site is known to interact with small chemical ligands (e.g., imiquimod, R484) and to bind non-phosphorylated uridine mononucleoside and is furthermore important for the correct dimerization of the TLR8 dimer. These two sites have a synergistic immunostimulatory effect, because uridine-binding has to be stimulated by ligand-binding of RNA-oligonucleotides, but only uridine binding provokes reorganization of the dimer into the active conformation (Geyer et al. 2015). Thereby, it is conceivable that species-specific pyrimidine rich RNAs determine whether TLR activation is initiated or not. Some PSS (312, 338, 368, 420, 421, and 481) where located in next proximity of this binding site and three of them (236, 338, and 481), matched with PSS in mammals which did not include any chiropteran sequence (Areal et al. 2011). Here, we found strongest support for PSS at position 338. Whether these PSS might be involved in ligand binding of RNA oligomers and thus responsible for shaping a more effective immune response is highly speculative. It might also be possible that they provoke conformational differences in affecting the relation among predicted amino acids directly involved in ligand binding with the result of species specific differences in ligand-binding affinities.
Even more interesting was the location of five PSS in the site predicted to bind uridine and to be involved in dimerization and conformational activation of TLR8. Here, PSS 352 is known to directly interact with uridine. Others (354 and 356) were again in close proximity to amino acid sites predicted to interact with uridine and/or involved in dimerization. Interestingly, position 351, also predicted to interact with uridine was detected to be positive selected within the branch of Rhinolophoidae and Pteropodidae, both known as main reservoirs of ssRNA viruses. Another PSS (447) was situated within a part of the Z-loop (442–457) known to be cleaved in humans (Tanji et al. 2013), which was also defined as episodically selected in Rhinolophoidae and Pteropodidae. According to the 3D localization, a likely involvement in dimerization seems to be possible (Fig. 4). The amino acids at the cleavage site in the Z-loop (441/442) showed variable amino acid alterations in bats including a deletion of three amino acids in Vespertilioninae (corresponding to human SYA) and coincided with a deletion of five amino acids in mouse and rat (human: RQSYA). The deletion of this motif has been stated to be responsible for the dysfunction of TLR8 in rodents and has been suggested as an important determinant for distinct ligand recognition between rodent and non-rodent TLR8s (Liu et al. 2010; Govindaraj et al. 2011).
Whether and how the interspecific variation of TLR8 detected in bats play a role in TLR8 activation after binding to the RNAs from different viral pathogens is a complex question. However, it is well known that non-synonymous amino acid alterations at sites which are not directly involved in pathogen binding might indirectly influence the pathogen-binding ability of a receptor by, e.g., changing the conformation of the dimer structure in both the inactivated and activated form (Gautam et al. 2006; Tschirren et al. 2011; Geyer et al. 2015). This could also account for the three other detected PSS rich regions located outside the defined ligand-binding region of the hTLR8. There is also accumulating evidence that in distinct TLRs different species recognizes either a different ligand or that a particular TLR ligand interaction does not induce the same response within different hosts, as a result of structural differences between the proteins (e.g., TLR4: Lien et al. 2000, Lizundia et al. 2008; TLR5: Anderson-Nissen et al. 2007; TLR8: Govindaraj et al. 2011). Concerning TLR8, for instance imiquimod failed to activate hTLR8, but activated bovine and porcine TLR8 (Zhu et al. 2008, 2009); mouse TLR8 failed to respond to ligand stimulation in the absence of polyT-oligodeoxynucleotides, most probably because of the missing RQSYA motif (Gordon et al. 2006, Liu et al. 2010); and the synthetical ligand R848 induced human and sheep TLR8 activation 10 to 15-fold, whereas bovine and cat TLR8 were induced less than 5-fold (Liu et al. 2010). These variable results indicate complex mechanisms involved in TLR8 dimerization and activation which vary between different species and might concern also TLR8 function within bats. Differences in either ligand–recognition and/or receptor–ligand interactions as well as modifications in the dimer conformation are thought to reflect co-evolution of a host TLR with species-specific microbes, allowing for the detection of species-specific pathogens or discrimination between pathogenic agents and tolerated commensals (Werling et al. 2009; Govindaraj et al. 2011). Within the co-evolution of bats and their viruses, it could thus be speculated that viruses may inactivate TLR8 simply by inhibiting correct dimerization or conformation of ligand-binding sites.
Further studies are urgently needed to investigate whether TLR8 polymorphism of bats provoke any functional differences in immune response compared to humans and other mammals, which would improve our understanding of bats being potential reservoirs of certain viruses. Our findings do not necessarily elucidate how bats can remain asymptotic to viral infections, but they indicate that the TLR8 gene is subject to diversifying as well as purifying selection pressures. In addition, our results provide important insights into which codons might be candidates for influencing species-specific ligand-binding affinities of TLR8 in bats, which in turn may influence vulnerability or tolerance to specific viruses.
References
Alcaide M, Edward SV (2011) Molecular evolution of the toll-like receptor multigene family in birds. Mol Biol Evol 28:1703–1715
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double-stranded RNA and activation of NF-kB by toll like receptor 3. Nature 413:732–738
Anderson-Nissen E, Smith KD, Bonneau R, Strong RK, Aderem A (2007) A conserved surface on toll-like receptor 5 recognizes bacterial flagellin. J Exp Med 204:393–403
Areal H, Abrantes J, Esteves PJ (2011) Signatures of positife selection in toll-like receptor genes in mammals. BMC Evol Biol 11:368
Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N (2010) ConSurf 2010: calculation evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38:W529–W533
Atchley WR, Zhao JP, Fernandes AD, Druke T (2005) Solving the protein sequence metric problem. Proc Natl Acad Sci USA 102:6395–6400
Babik W, Dudek K, Fijarczyk A, Pabijan M, Stuglik M, Szkotak R, Zielinsky P (2014) Constraint and adaptation in newt toll-like receptor genes. Genome Biol Evol 7(1):81–95
Baker ML, Schountz T, Wang LF (2013) Antiviral immune response of bats: a review. Zoonoses Public Health 60:104–116
Barreiro LB, Ben-Ali M, Quach H, Laval G, Patin E, Pickrell JK et al (2009) Evolutionary dynamics of human toll-like receptors and their different contributions to host defense. PLoS Genet 5:e1000562
Bean AGD, Baker ML, Stewart CR, Cowled C, Deffrasnes C, Wang LF, Lowenthal JW (2013) Studying immunity to zoonotic diseases in the natural host—keeping it real. Nature Rev Immunol 13:815–861
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G et al (2014) SWISS-MODEL:modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acid Res 42:W252–W258
Brook CE, Dobson AP (2015) Bats as ‘special’ reservpors for emerging zoonotic pathogens. Trends in Microbiol 23:172–180
Calisher CH (2015) Viruses in bats: a historic review. In: Wang L-F, Cowled C (eds) Bats and viruses: a new frontier of emerging infectious diseases. Wiley, New York, p. 23–47.
Castel G, Razzauti M, Jousselin E, Kergoat GJ, Cosson J-F. Changes in Diversification Patterns and Signatures of Selection during the Evolution of Murinae-Associated Hantaviruses. Viruses. 2014;6(3):1112-1134. doi:10.3390/v6031112
Cowled C, Baker M, Tachedjian M, Zhou P, Bulach D, Wang LF (2011) Molecular characterization of toll-like receptors in the black flying fox. Pteropus alecto Dev Comp Immunol 35:7–18
Darfour-Oduro KA, Megens HJ, Roca AL, Groenen M, Schook LB (2015) Adaptive evolution of toll-like receptors (TLRs) in the family Suidae. PLoS ONE 10(4):e0124069
Daugherty MD, Malik HS (2012) Rules of engagement: molecular insights from host-virus arms-races. Ann Rev Genet 46:677–700
De Lano WL (2002) The PyMol Molecular Graphics System Version 1.5.0.4. Schrödinger, LLC, San Carlos, http://www.pymol.org
Delport W, Poon AF, Frost SDW, Kosakovsky Pond SL (2010) Datamonkey 2010: a suite of phylogenetic analyses tools for evolutionary biology. Bioinformatics 26(19):2455–2457
Ellegren H (2008) Comparative genomics and the study of evolution by natural selection. Mol Ecol 17:4586–4596
Escalera-Zamudio M, Zepeda-Mendoza ML, Loza-Rubio E, Rojas-Anaya E, Mendez-Ojeda ML, Arias CF, Greenwood AD (2015) The evolution of bat nucleic acid-sensing toll-like receptors. Molecula Ecology 24:5899–5909
Fornuskova A, Vinkler M, Pages M, Galan M, Jousselin E et al (2013) Contrasted evolutionary histories of two toll-like receptors (Tlr4 and Tlr7) in wild rodents (MURINAE). BMC Evol Biol 13:194
Gannon WL, Sikes (2007) Guidelines of the American Society of Mammalogists for the use of wild mammals in research. J Mammal 88:809
Gautam JK, Ashish CLD, Krueger JK, Smith MF (2006) Structural and functional evidence for the role of the TLR2 DD loop in TLR1/TLR2 heterodimerization and signalling. J Bio Chem 281:30132–30142
Georgel P, Macquin C, Bahram S (2009) The heterogeneous allelic repertoire of human toll-like receptor (TLR) genes. PLoS ONE 4(11):e7803. doi:10.13717/journal.pone.0007803
Geyer M, Pelka K, Latz E (2015) Synergistic activation of toll-like receptor 8 by two RNA degradation products. Nat Struct Mol Biol 22(2):99–101
Gordon KK, Qiu XX, Binsfeld CC, Vasilakos JP, Alkan SS (2006) Cutting edge: activation of murinr TLR8 by a combination of imidazoquinoline immune response modifiers and polyT oligodeoxynucleotides. J Immunol 177:6584–6587
Govindaraj EG, Manavalan B, Basith S, Choi S (2011) Comparative analysis of species-specific ligand recognition in toll-like receptor 8 signaling: a hypothesis. PLoS ONE 6(9):e25118
Grueber CE, Wallis GP, King TM, Jamieson IG (2012) Variation at innate immunity toll-like receptor genes in a bottlenecked population of a New Zealand robin. PLoS One 7:e45011
Hancks DC, Hartley MK, Hagen C, Clark NL, Elde NC (2015) Overlapping patterns of rapid evolution in the nucleic acid sensors cGAS and OAS1 suggest a common mechanism of pathogen antagonism and escape. PLoS Genet 11(5):e1005203
Hofmann K, Stoffel W (1993) TMbase—a database of membrane spanning proteins segments. Biol Chem Hoppe-Seyler 374:166
Jensen S, Thomsen AR (2012) Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 86(6):2900–2910
Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munroe HN (ed) Mammalian protein metabolism. Academic, New York, pp 21–132
Kawai T, Akira S (2006) TLR signaling. Cell Death Differ 13:861–825
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol 11:373–384
Kosakovsky Pond SL, Frost SDW (2005) Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1209–1222
Kosakovsky Pond SL, Frost SDW, Muse SV (2005) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21(5):676–679
Kosakovsky Pond SL, Mirrell B, Fourment M, Frost SDW, Delport W, Scheffler K (2011) A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol 28:3033–3043
Lemos de Matos A, McFadden G, Esteves PJ (2013) Positive Evolutionary Selection On the RIG-ILike Receptor Genes in Mammals. PLoS ONE 8(11): e81864. doi:10.1371/journal.pone.0081864
Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S et al (2000) Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharids. J Clin Invest 105:497–504
Liu J, Xu C, Hsu L-C, Luo Y, Xiang R, Chuang T-H (2010) A five-amino-acid motif in the undifined region of the TLR8 ectodomain is required for species-specific ligand recognition. Mol Immunol 47(5):1083
Lizundia R, Sauter KS, Taylor G, Werlin D (2008) Host species-specific usage of the TLR4-LPS receptor complex. Innate Immun 14:223–231
Lund JM, Alexopoulou L, Sato A (2004) Recognition of single-stranded RNA viruses by toll-like receptor 7. P Nat Acad Sci 101:5598–5603
Maldonado E, Sunagar K, Almeida D, Vasconcelos V, Antunes A (2014) IMPACT_S: intergrated multiprogram platform to analyse and combine tests of selection. PLoS ONE 9(10), e96243
Mikami T, Miashita H, Takatsuka S, Kuroki Y, Matsushima N (2012) Molecular evolution of vertebrate toll-like receptors: evolutionary rate differences between their leucin-rich repeats and their TIR domains. Genes 503:235–243
Morozumi T, Uenishi H (2009) Polymorphism distribution and structural conservation in RNA-sensing toll-like receptors 3, 7, and 8 in pigs. Biochim Biophys Acta 1790:267–274
Mukherjee S, Sarkar-Roy N, Wagener DK, Majumder PP (2009) Signatures of natural selektion are not uniform across genes of innate immune system, but purifying selection is the dominant signature. PNAS 106(17):7073–7078
Murell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Pond SLK (2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8(7), e1002764
Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K (2013) FUBAR: a fast, unconstrained Bayesian AppRoximation for inferring selection. Mol Biol Evol 30(5):1196–1205
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nuclotide substitutions. Mol Biol Evol 3:418–426
Offord V, Coffey TJ, Werlin D (2009) LRRfinder: a web application for the identification of leucine-rich repeats and an integrative toll-like receptor database. Dev Comp Immunol 34:1035–1041
Plowright RK, Eby P, Hudson PJ, Smith IL, Westcott D et al (2015) Ecological dynamics of emerging bat viruses spillover. Proc R Soc B 282:20142124
Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A (2005) The evolution of vertebrate toll-like receptors. Proc Natl Acad Sci U S A 102:9577–9582
Schroder NW, Schumann RR (2005) Single nucleotide polymorphisms of toll-like receptors and susceptibility to infectious disease. Lancet Infect Dis 5:156–164
Schultz J, Milpetz F, Bork P, Pontig CP (1998) SMART, a simple modular architecture research tool: identification of signalling domains. PNAS 95:5857–5864
Smith NGC (2003) Are radical and conservative substitution rates useful statistics in molecular evolution? J Mol Evol 57:467–478
Smith SA, Jann OC, Haig D, Russell GC, Werling D, Glass EJ, Emes RD (2012) Adaptive evolution of toll-like receptor 5 in domesticated mammals. BMC Evol Biol 12:122
Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Re Immunol 21:335–376
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tanji H, Ohto U, Shibata T, Miyaka K, Shimizu T (2013) Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 339:1426–1429
Tanji H, Ohto U, Shibata T, Miyaka K, Taoka M, Yamauchi Y, Shimizu T et al (2015) Toll-like receptor 8 sense degradation products of single stranded RNA. Nat Struct Mol Biol 22(2):109–115
Taylor BD, Darville T, Ferrell RE, Kammerer CM, Ness RB, Haggerty CL (2012) variants in toll-like receptor 1 and 4 genes are associated with Chlamydia trachomatis among women with pelvic inflammatory disease. J Inect Dis 205:603–609
Technelysium Pty Ltd. (1998–2012) Chromas Lite 2.1.1. http://www.technelysium.com.au
Tschirren B, Raberg L, Westerdahl H (2011) Signatures of selection acting on the innate immunity gene toll-like receptor 2 (TLR2) during the evolutionary history of rodents. J Evol Biol 24(6):1232–1240
Tschirren B, Andersson M, Scherman K, Westerdahl H, Mittl PRE, Raberg L (2013) Polymorphisms at the innate immune receptor TLR2 are associated with Borrelia infection in a wild rodent population. Proc R Soc B 280:20130364
Uciechowski P, Imhoff H, Lange C, Meyer CG, Browne EN, Kirsten DK et al (2011) Susceptibility to tuberculosis is associated with TLR1 polymorphisms resulting in a lack of TLR1 cell surface expression. J Leucocyte Biol 90:377–388
Vinkler M, Bainova H, Bryja J (2014) Protein evolution of toll-like receptors 4, 5 and 7 within Galloanserae birds. Genet Sel Evol 46:72
Wang LF, Walker PJ, Poon LLM (2011) Mass extinctions, biodiversity and mitochondrial function: are bats ‘special’ as reservoirs for emerging viruses? Curr Op Virol 1:649–657
Wang CM, Chang SW, Wu YJJ, Lin JC, Ho HH, Chou TC et al (2014) Genetic variations in toll-like receptors (TLRs3/7/8) are associated with systemic lupus erythematosus in a Taiwanese population. Scientific Reports 4:3792
Werling D, Jann OC, Offord V, Glass EJ, Coffey TJ (2009) Variation matters: TLR structure and species-specific pathogen recognition. Trend Immunol 30(3):124–130
Wibbelt G, Moore MS, Schountz T, Voigt CC (2010) Emerging diseases in Chiroptera: why bats? Biological Letters 6:438–440
Wlasiuk G, Nachman MW (2010) Adaptation and constraint at toll-like receptors in primates. Mol Biol Evol 27(9):2172–2186
Wong SH, Gochhait S, Malhotra D, Petterson FH, Teo YY et al (2010) Leprosy and the adaptation of human toll-like receptor 1. PLoS Pathog 6(7), e1000979. doi:10.13717/journal.ppat.1000979
Wynne JM, Wang LF (2013) Bats and viruses: friend or foe? PLoS Pathog 9(10), e1003651
Xagorari A, Chlichlia K (2008) Toll-like receptors and viruses: induction of innate antiviral responses. Open Microbiol J 2:49–59
Yang ZH (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang ZH, Wong WSW, Nielsen R (2005) Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly KA et al (2013) Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science 339:456–460
Zhu J, Lai K, Brownile R, Babiuk LA, Mutwiri GK (2008) Porcine TLR8 and TLR7 are both activated by selective TLR7 ligand imiquimod. Mol Immunol 45:3238–3243
Zhu J, Lai K, Brownile R, Liu Q, Babiuk LA, Potter A et al (2009) Characterization of bovine toll-like receptor 8: ligand specificity, signaling essential sites and dimerization. Mol Immunol 46:978–990
Acknowledgments
We thank several co-workers for the help in capturing bats in the field. This work was supported by the Deutsche Forschungsgemeinschaft (priority program SPP1596 (Vo890/23) to CCV).
Author contributions
JS and CV designed the study and wrote the manuscript. JS designed laboratory protocols and analyzed samples. CV collected field samples.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Table S1 Investigated bat species, sample accession numbers due to the IZW biobank and geographic origin. Table S2 Designed primers to amplify the chiropteran TLR8 gene. Table S3 Nucleotide/amino acid sequence percent identities of the whole (a), the LRR ectodomain (b) and the TM and TIR domain (c) of the TLR8 sequences between investigated bat taxa and between the equine and human orthologous. Table S4 Positive selected sites identified by different model analyses for the Chiropteran TLR8 gene in relation to PSS of other taxa and to relevant binding an dimerization sites of the human TLR8 (hTLR8) molecule according to Tanji et al. (2013, 2015). (PDF 3309 kb)
ESM 2
Figure S1 Predicted protein domain architecture of bat TLR8 genes determined by SMART analyses (Schultz et al. 1998). Figure S2 Estimated transmembrane domains within the TLR8 gene. Noctilio leporinus (A) and Rhinolophus euryale (B) identified by TMPRED. Figure S3 Amino acid sequence variability of the TLR8 gene between 21 bat species and predicted positive selected sites. (PDF 111 kb)
Rights and permissions
About this article

Cite this article
Schad, J., Voigt, C.C. Adaptive evolution of virus-sensing toll-like receptor 8 in bats. Immunogenetics 68, 783–795 (2016). https://doi.org/10.1007/s00251-016-0940-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-016-0940-z