
Abstract
Aims/hypothesis
Human α1-antitrypsin (hAAT) gene therapy prevents type 1 diabetes in a NOD mouse model of diabetes. However, repeated i.p. injections of hAAT into NOD mice leads to fatal anaphylaxis. The aim of the study was to determine if an alternative route of administration avoids anaphylaxis and allows evaluation of hAAT’s potential for diabetes prevention and reversal. We also sought to determine if the addition of granulocyte colony-stimulating factor (G-CSF), augments hAAT’s capacity to prevent or reverse disease in the NOD mice.
Methods
To evaluate hAAT pharmacokinetics, serum hAAT levels were monitored in NOD mice receiving a single dose (2 mg) of hAAT by i.p., s.c. or i.d. injection. For studies of type 1 diabetes prevention and reversal, mice received i.d. hAAT (2 mg/mouse/3 days) for 8 or 10 weeks or hAAT and G-CSF (i.p., 6 μg/day) for 6 weeks. Blood glucose determinations, glucose tolerance testing and insulin tolerance tests were performed.
Results
Both i.p. and s.c. injections resulted in fatal anaphylaxis. The i.d. injection avoided anaphylaxis and i.d. injection of hAAT into 11-week-old NOD mice prevented disease (p = 0.005, AAT vs PBS at 40 weeks of age). Treatment of diabetic NOD mice with hAAT or hAAT plus G-CSF provided long-term (at least 100 days) reversal of diabetes in 50% of treated animals. G-CSF did not enhance the reversal rates of hAAT. Glucose tolerance and insulin levels were normalised in mice with hAAT prevention and reversal.
Conclusions/interpretation
Intradermal hAAT prevents and reverses disease in a NOD mouse model of type 1 diabetes without inducing anaphylaxis.
Similar content being viewed by others


Introduction
Type 1 diabetes is an autoimmune disease that results from an imbalanced and over-reactive immune response, resulting in the destruction of insulin-producing pancreatic beta cells. Due to the pathogenic complexity of this disease, development of an effective method for late prevention or reversal post-onset of the disorder has been remarkably challenging [1]. Although recent studies have shown promising results in terms of reversing type 1 diabetes, many of these treatments can lead to detrimental side effects [2–4]. As such, the exploration of therapies with more tolerable adverse event profiles is urgently needed.
The α1-antitrypsin (AAT) is a multifunctional protein with both proteinase inhibitor and anti-inflammatory activities. These facets render it a potential therapeutic candidate for immune disease intervention including in type 1 diabetes. We demonstrated that human AAT (hAAT) gene therapy prevented type 1 diabetes in NOD mice as a model of diabetes [5, 6]. Follow-up investigations showed that AAT protein therapy protected beta cells from apoptosis [7]. Work performed by Lewis et al. demonstrated that AAT therapy induced immune tolerance and prolonged survival of transplanted islets [8, 9] and Koulmanda et al. demonstrated a profound ability for hAAT to reverse type 1 diabetes in a NOD mouse model through a combination of beneficial mechanisms [10]. However, our own attempts, designed to assess the therapeutic effects of AAT protein therapy in NOD mice, demonstrated that repeated i.p. administration of hAAT led to fatal anaphylaxis [11]. The reasons for the discrepancy (in terms of anaphylaxis induction) between ours and the aforementioned studies remain unclear. Here, we report that i.d. administration of hAAT avoids fatal anaphylaxis and show that hAAT has the ability to both prevent as well as reverse disease in a NOD mouse model of type 1 diabetes.
Granulocyte-colony stimulating factor (G-CSF) is another relatively low-risk agent that has the potential for use in type 1 diabetes immunotherapies. G-CSF induces an immunoregulatory shift from a T-helper type 1 to a T-helper type 2 cytokine phenotype, increases tolerogenic dendritic cells and mobilises regulatory T cells [12–14]. G-CSF has successfully prevented both the onset of disease in the NOD mouse and cyclophosphamide-mediated acceleration of diabetes [15, 16]. Parker et al. have recently shown that G-CSF enhances the long-term reversal of diabetes afforded by murine antithymocyte globulin (ATG) [4]. To determine if the combination of G-CSF and hAAT would enhance the protective effect of hAAT alone on type 1 diabetes prevention and reversal, we included G-CSF as a second drug in the present study.
Methods
Animals
Female NOD/LtJ mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and housed in specific-pathogen-free facilities at the University of Florida. The Institutional Animal Care and Use Committee at the University of Florida approved all animal manipulations.
Kinetics studies
Eight-week-old female NOD mice (n = 6) received i.p., s.c. or i.d. injections or s.c. implantation of an osmotic pump (Alzet Osmotic Pumps, Cupertino, CA, USA). All mice received a single administration of clinical-grade hAAT (Prolastin, Bayer, Elkhart, IN, USA) at a dose of 2 mg/mouse. All mice were bled at 0 min, 5 min, 15 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 4 h, 8 h, and daily thereafter until 7 days after hAAT administration. Serum hAAT levels were detected using a hAAT-specific ELISA.
Area under the curve of serum hAAT concentration in the first 7 days (AUC0–7 days) was calculated by WinNonlin 5.2 (Pharsight Inc., Mountain View, CA, USA) using the linear trapezoidal rule.
Prevention studies
Cohorts of 11-week-old female NOD mice were injected i.d. with hAAT (2 mg/mouse/3 days, for 10 weeks), with hAAT (2 mg/mouse/3 days, for 10 weeks) plus G-CSF (Neupogen, i.p., 6 μg daily, for 8 weeks), with G-CSF alone or with saline. Blood glucose was monitored weekly. Serum hAAT levels and anti-hAAT antibodies were analysed biweekly until 20 weeks of age.
Reversal studies
Mice used in this study were monitored three times per week for hyperglycaemia, defined as a blood glucose >13.32 mmol/l, by tail bleed. Animals measuring above this threshold on two consecutive days were considered diabetic. At the onset of diabetes, mice received i.d. injection of hAAT (Prolastin, 2 mg/3 days, for 8 weeks), hAAT plus G-CSF (i.p., 6 μg daily for 8 weeks), G-CSF alone or saline. All mice at the onset of diabetes received insulin treatment with s.c. insulin pellets (LinBit; LinShin Canada, Inc., Toronto, ON, Canada). The dose of insulin was adjusted to achieve a blood glucose level between 5.55 and 11.1 mmol/l in the first 3 days after the treatment. Blood glucose levels in all animals were continually monitored.
Intraperitoneal glucose tolerance test (IPGTT)
Mice were fasted for 6 h by removal to a clean cage without food at the end of their dark (feeding) cycle. A fasting glucose level was obtained from tail venous blood. Mice were weighed and i.p. injected with glucose (1 mg/g bodyweight). Blood glucose values were obtained at 5, 15 30, 60, 120 and 240 min after glucose challenge.
Insulin tolerance test (ITT)
The test was performed on random-fed mice at about 14:00 hours. The mice were i.p. injected with insulin (0.75 U/kg) in approximately 0.1 ml 0.9% NaCl. Blood glucose was detected at 0, 15, 30, 45 and 60 min after the injection of insulin.
Histology and immunohistochemistry
Insulitis was evaluated and scored on pancreatic sections stained with haematoxylin and eosin, as described previously [4–6]. Briefly, the degree of lymphocytic infiltration in each islet was scored according to the following scale: 0, none; 1, peri-islet infiltrates; 2, <50% intra-islet infiltrates; 3, >50% intra-islet infiltrates. Insulin immunohistochemistry was performed as previously described [4–6]. Fractional insulin area was determined from whole digital slide scans using a positive pixel count algorithm (Spectrum, Aperio, Vista, CA, USA).
ELISA for the detection of serum hAAT and BAFF levels and antibodies against hAAT
Detection of hAAT and anti-hAAT antibodies in mouse serum was performed as previously described [17]. Purified hAAT was used as standard (Athens Research & Technology, Athens, GA, USA). Detection of B cell activating factor (BAFF) in serum was performed according to the manufacturer’s instructions (R&D Systems, Inc. Minneapolis, MN, USA).
Results
Slow release of hAAT prevents fatal anaphylaxis in NOD mice
In a pharmacokinetics study, cohorts of 8-week-old NOD mice (n = 6) received 2 mg of hAAT (Prolastin) by an i.p., s.c., or i.d. injection, or by osmotic pump (Alzet Pump). As shown in Fig. 1a, administration of this protein by different routes resulted in distinct kinetics: i.p. administration resulting in the highest levels for the entire 60 min. At 30 min after injection, serum hAAT levels in s.c., i.d. and pump groups were 20%, 5% and 2%, respectively, of that in the i.p. group. The i.p., s.c. and i.d. injections resulted in similar AUC0–7 days, whereas Alzet Pump resulted in a twofold increase of AUC0–7 days (Fig. 1b,c). Because serum hAAT levels in the s.c. injected group were dramatically lower than those in the i.p. injected group, we administered repeated s.c. injections of hAAT (2 mg per 3 days) to NOD mice (4 weeks old). Unfortunately, 90% (9/10) of mice died after the fourth or fifth injection. We next performed repeated i.d. injections of hAAT, which resulted in fourfold lower levels of hAAT at 30 min when compared with s.c. injections (Fig. 1a). Surprisingly, no animal died after up to 15 injections (Table 1, groups 2 and 3). Furthermore, animals that received more than nine i.d. injections also survived seven additional i.p. injections of hAAT (Table 1, group 3), indicating that the animals developed tolerance to hAAT after nine i.d. injections. Hence, the fatal anaphylaxis could be avoided by controlling the release of hAAT in the first 30–40 min of administration. Therefore, we chose to use i.d. injections for the following studies.

Pharmacokinetics of hAAT in NOD mice. Cohorts of 8-week-old NOD mice (n = 6) were injected with 2 mg of hAAT (Prolastin). Serum hAAT levels were detected by hAAT-specific ELISA. Black diamond, intraperitoneal injection (IP); white square, subcutaneous injection (SC); black triangle, intradermal injection (ID); white circle, using an osmotic pump (Alzet Pump). a hAAT levels within 60 min of administration. b Long-term (7 days) hAAT levels after single administration. c AUC0–7 days of hAAT levels
AAT protein therapy prevents development of type 1 diabetes in a NOD mouse model
To determine whether i.d. hAAT protein therapy prevents the development of type 1 diabetes, cohorts of 11-week-old female NOD mice were i.d. injected with hAAT, hAAT plus G-CSF, G-CSF alone or saline. The G-CSF treatment lasted 8 weeks (11 to 18 weeks of age) and hAAT treatment lasted 10 weeks (11 to 20 weeks of age). Injection of hAAT resulted in high serum levels of hAAT and anti-hAAT antibodies (Fig. 2a,b). All mice in the hAAT-treated and hAAT plus G-CSF-treated groups remained diabetes free until 25 weeks of age, whereas 67% of the G-CSF-treated and saline-treated groups developed diabetes at 21 weeks (Fig. 2c). At 40 weeks of age (the end of the experiment), 63% of mice in the AAT group were diabetes free (p = 0.005, AAT vs PBS). These data clearly demonstrated that hAAT protein therapy significantly prevented disease in a NOD mouse model of type 1 diabetes, whereas G-CSF had no enhancing effect.

Therapy with hAAT protein prevents type 1 diabetes development in a NOD mouse model. Cohorts of 11-week-old female NOD mice were i.d. injected with hAAT (2 mg/mouse, every 3 days) for 10 weeks (indicated by a grey bar) and/or G-CSF (6 mg/mouse, daily) for 8 weeks (indicated by a white bar). White diamond, hAAT (n = 8); black diamond, hAAT plus G-CSF (n = 7); black triangle, G-CSF alone (n = 8); white triangle, saline (n = 9). a Serum levels of hAAT. b Levels of anti-hAAT antibodies detected by ELISA. c Life table analysis for diabetes development. At 25 weeks of age, all mice in the hAAT-treated and hAAT plus G-CSF-treated groups remained diabetes free. At 40 weeks of age (the end of the experiment), 63% of mice in AAT group were diabetes free (p = 0.005, AAT vs PBS)
AAT reversed type 1 diabetes in NOD mice
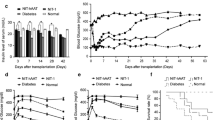
To test the feasibility of hAAT in reversing type 1 diabetes, newly diagnosed diabetic mice were treated with hAAT, hAAT plus G-CSF, G-CSF alone or saline. Both hAAT and G-CSF treatments were continued for 8 weeks. All mice were given insulin pellets, which provided a source of exogenous insulin for 10 to 15 days. Blood glucose levels of all mice given saline or G-CSF alone were greater than 16.65 mmol/l after exhaustion of the insulin pellets (Fig. 3a,b). In the AAT-treated group, more than 50% of mice (4/7) remained diabetes free for at least 100 days (Fig. 3c). In the hAAT plus G-CSF-treated group, 50% of the mice similarly remained diabetes free (Fig. 4a,b). Blood glucose in two hAAT plus G-CSF-treated mice oscillated considerably for 8 weeks, remained below 13.32 mg/dl for 2 more weeks and finally rose to 27.75 mmol/l (Fig. 4b) indicating that the treatment has a partial effect on the intervention of diabetes in these mice. Notably, the starting blood glucose levels in mice whose disease was successfully reversed by AAT plus G-CSF therapy were significantly lower than in mice in which therapy failed (Fig. 4c). A similar trend could be observed in hAAT-treated mice with the exception of one mouse with a very high starting blood glucose level, in which the disease was successfully reversed. Consistent with the observations in a previous study using ATG and G-CSF for reversal of type 1 diabetes, these results indicate that the severity of disease at the onset or before treatment affects the efficacy of the treatment.

Reversal of type 1 diabetes by hAAT protein therapy. New onset diabetic mice were treated with hAAT (2 mg/mouse), G-CSF (6 mg/mouse) alone or saline. Both hAAT and G-CSF treatments were administered for 8 weeks. Each line represents data from an individual animal. a Blood glucose levels in saline-treated group (n = 5). b Blood glucose levels in G-CSF-treated group (n = 6). c Blood glucose levels in mice treated with hAAT and successfully reversed from type 1 diabetes (n = 4). d Blood glucose levels in mice treated with hAAT that failed to reverse type 1 diabetes (n = 3)

Reversal of type 1 diabetes by hAAT and G-CSF therapy. New onset diabetic mice (n = 6) were treated with hAAT (2 mg/mouse) and G-CSF (6 mg/mouse) for 8 weeks. Each line represents data from an individual animal. a Blood glucose in long-term reversed mice. b Blood glucose in mice who failed to reverse type 1 diabetes. c Effect of disease severity on the efficacy of treatments. Starting blood glucose levels (at the onset of diabetes) in different groups are plotted. † p = 0.0044
AAT treatment enhanced islet function and decreased BAFF levels
In addition to monitoring blood glucose, we performed glucose tolerance tests and insulin resistance tests. As shown in Fig. 5a, mice that survived in the prevention and reversal studies responded to glucose similarly to normal mice, indicating that islet function was retained in these mice. The AAT-treated mice also responded to insulin challenge similarly to normal mice, indicating that no insulin resistance developed in these mice (Fig. 5b). Insulitis was evaluated as previously described. As shown in Fig. 5c, more islets were found in hAAT prevented and reversed mice than in mice with new-onset diabetes. Similarly, larger insulin-positive areas were found in those mice compared with control mice.

The effects of hAAT treatment on islet function, insulin resistance and immune system. a Intraperitoneal glucose tolerance test. White diamond, diabetic mice (n = 4); black square, hAAT-prevented mice (40 weeks of age, n = 4); white triangle, hAAT-reversed mice (euglycaemic for more than 100 days, n = 3); black circle, normal diabetes-free mice (n = 4). b Insulin tolerance test. c The effects of hAAT on insulitis. Insulitis was evaluated in AAT-prevented mice (40 weeks of age, n = 4) and reversed mice (euglycaemic for more than 100 days, n = 3). The new onset diabetic mice served as control (n = 2)
To further understand the effects of AAT on the immune system, we measured BAFF levels in both control and AAT-treated mice. We showed that AAT significantly decreased serum BAFF levels 2 weeks after treatment (Fig. 6) suggesting an effect of hAAT on B cell-mediated autoimmunity.

The effect of hAAT on serum BAFF levels. BAFF levels in hAAT-treated group (n = 8) and saline-treated mice (PBS, n = 10) were detected by ELISA 2 weeks after the treatment. p = 0.023 (AAT vs PBS)
Discussion
Although hAAT has potential in the prevention and reversal of type 1 diabetes [5, 6, 8, 9], repeated administration of hAAT may lead to fatal anaphylaxis in NOD mice [11]. The immune response is not AAT specific, but is instead the result of the over-reactive immune system of the NOD mice. This high death rate has been a major hurdle for the further investigation of AAT for treatment of type 1 diabetes [11]. In the present study, we showed that i.d. injection of hAAT resulted in slow release in NOD mice and effectively avoided fatal anaphylaxis. Given the fact that the serum hAAT levels at 30 min after i.d. injection were 20-fold and fourfold lower than i.p. and s.c. injections, respectively, the most important factor for avoiding fatal anaphylaxis appears to be controlling the serum levels of AAT in the first 30 min following injection [18]. These results suggest that controlled interaction of AAT and AAT-specific IgE on the mast cells is critical to avoid anaphylaxis. Although i.p., s.c. and i.d. injections of AAT displayed distinct kinetics in the first 2 h, the AUC0–7days were similar in these groups. Delivery of AAT by an osmotic pump resulted in a twofold increase of the AUC0–7days suggesting a possible clinical application in large animals and in humans. These results not only revealed the kinetics of AAT administration by various routes, but also enabled us to further investigate the therapeutic effect of AAT.
Previously, we showed that AAT gene therapy prevented disease in a NOD mouse model of type 1 diabetes [5, 6]. AAT inhibited caspase-3 activity and protected against islet cell death [7]. In the present study, we showed that AAT protein therapy partially prevented and reversed type 1 diabetes in NOD mice. Consistent with our observations and those of other groups, these results support our hypothesis that AAT is beneficial for the treatment of type 1 diabetes [8, 9]. However, partial prevention (i.e. delay of diabetes development) and reversal (50%) of type 1 diabetes by hAAT monotherapy indicate that hAAT therapy has the potential for further improvement. Therefore, a combination therapy using drugs targeting different pathways may improve the treatment effect compared with hAAT monotherapy. The rationale of using G-CSF in this study was to employ its immunoregulatory properties, including its ability to mobilise regulatory T cells, induce tolerogenic dendritic cells and shift cytokine phenotype from T-helper type 1 to type 2 [13, 14]. Previously we demonstrated that G-CSF enhanced the efficacy of ATG-mediated reversal of type 1 diabetes in the NOD mouse model, but as in the present study G-CSF monotherapy did not prevent or reverse disease [4]. Unfortunately, in the present study, we did not observe an enhancing effect when G-CSF was used in combination with AAT. In fact, although there was no statistical difference, we noticed that the addition of G-CSF to hAAT decreased the preventive effect of hAAT alone. These results indicated that G-CSF in combination with different drugs could lead to different outcomes. The possible mechanism behind this observation requires further investigation. Future studies will also focus upon the development of combination therapies of AAT with other drugs.
It is interesting to note that blood glucose levels oscillated in some of the AAT and G-CSF-treated animals. Although these mice eventually developed diabetes, they survived for 10 weeks post diabetes onset, during which time they were mostly euglycaemic. The results indicate that the treatment was partially effective in these mice and provides strong evidence of considerable individual differences in NOD mice despite being an inbred strain. Based on this treatment effect, one can classify NOD mice into three groups: reversal, partial-reversal, and non-reversal groups. The partial-reversal and non-reversal groups may require dose optimisation or additional treatments. It is notable that the reversal efficacy of hAAT therapy appears to be inversely correlated with the blood glucose levels at the onset of diabetes. This tendency is consistent and similar with the observations in a previous study using ATG and G-CSF for reversal of type 1 diabetes [4]. These results suggest that the severity or beta cell mass remaining at the onset of diabetes is probably a major determinant of the success of a treatment.
In the prevention studies, we observed that late AAT treatment (at 10 weeks of age) resulted in the complete prevention of type 1 diabetes up to 25 weeks of age. These results, when compared with controls, clearly demonstrated the powerful protective effect of AAT on disease development. On the other hand, some AAT-treated mice developed diabetes after withdrawal of AAT treatment. This delayed diabetes development suggests that the protective effect was AAT-dependent and/or longer-term treatment with AAT may be required to induce long-term prevention. In fact, our previous observation that gene therapy mediated long-term AAT expression and resulted in long-term prevention supports the latter notion [6]. Together, these results imply that AAT gene therapy rather than AAT protein therapy may one day be useful in preventing type 1 diabetes.
AAT is a multifunctional protein. In addition to acting as a serine proteinase inhibitor in the circulation, AAT can inhibit production of major inflammatory cytokines, such as IL-6 and TNF-α, and enhance the production of the anti-inflammatory cytokine IL-10 through increasing cellular cAMP levels [19, 20]. Evidence from previous studies has shown that AAT treatment may enhance pancreatic beta cell function [7, 8]. In the present studies, we showed that AAT-treated mice responded to glucose challenge similarly to the normal mice. Interestingly, AAT treatment also decreased the serum levels of BAFF. These results are consistent with our previous observations that AAT gene therapy reduced insulin autoantibody levels and that AAT protein and gene therapy reduced serum levels of BAFF and autoantibodies in a collagen-induced arthritis mouse model. These results strongly suggest that hAAT reduces autoantibody levels through inhibition of BAFF production. As BAFF is a B cell activator, it is possible that this reduction may contribute to the inhibition of other B cell-mediated immunity. Detailed molecular mechanism(s) underlying the effect of hAAT on B cell immunity remains to be investigated in future studies. Together, these results indicate that AAT may play an important role in controlling B cell-mediated immunity and imply a new function of AAT.
In summary, we have shown that NOD-specific anaphylaxis can be avoided by controlling the drug’s release within 30 min after the injection and that AAT protein therapy effectively prevents and reverses disease in a NOD mouse model of type 1 diabetes. These results are consistent with previous observations, demonstrate the therapeutic effect of AAT and support the possible clinical application of AAT in patients with type 1 diabetes.
Abbreviations
- ATG:
-
Anti-thymocyte globulin
- BAFF:
-
B cell activating factor
- G-CSF:
-
Granulocyte colony-stimulating factor
- hAAT:
-
Human α1-antitrypsin
- IPGTT:
-
Intraperitoneal glucose tolerance test
- ITT:
-
Insulin tolerance test
References
Atkinson MA, Eisenbarth GS (2001) Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 358:221–229
Cernea S, Pozzilli P (2008) New potential treatments for protection of pancreatic B cell function in type 1 diabetes. Diabet Med 25:1259–1267
Pasquali L, Giannoukakis N, Trucco M (2008) Induction of immune tolerance to facilitate beta cell regeneration in type 1 diabetes. Adv Drug Deliv Rev 60:106–113
Parker MJ, Xue S, Alexander JJ et al (2009) Immune depletion with cellular mobilization imparts immunoregulation and reverses autoimmune diabetes in nonobese diabetic mice. Diabetes 58:2277–2284
Song S, Goudy K, Campbell-Thompson M et al (2004) Recombinant adeno-associated virus-mediated alpha-1 antitrypsin gene therapy prevents type I diabetes in NOD mice. Gene Ther 11:181–186
Lu Y, Tang M, Wasserfall C et al (2006) Alpha1-antitrypsin gene therapy modulates cellular immunity and efficiently prevents type 1 diabetes in nonobese diabetic mice. Hum Gene Ther 17:625–634
Zhang B, Lu Y, Campbell-Thompson M et al (2007) Alpha1-antitrypsin protects beta-cells from apoptosis. Diabetes 56:1316–1323
Lewis EC, Shapiro L, Bowers OJ, Dinarello CA (2005) Alpha1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc Natl Acad Sci U S A 102:12153–12158
Lewis EC, Mizrahi M, Toledano M et al (2008) Alpha1-antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci U S A 105:16236–16241
Koulmanda M, Bhasin M, Hoffman L et al (2008) Curative and beta cell regenerative effects of alpha1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc Natl Acad Sci U S A 105:16242–16247
Lu Y, Parker M, Pileggi A et al (2008) Human alpha 1-antitrypsin therapy induces fatal anaphylaxis in non-obese diabetic mice. Clin Exp Immunol 154:15–21
Rutella S (2007) Granulocyte colony-stimulating factor for the induction of T cell tolerance. Transplantation 84:S26–30
Hartung T, Docke WD, Gantner F et al (1995) Effect of granulocyte colony-stimulating factor treatment on ex vivo blood cytokine response in human volunteers. Blood 85:2482–2489
Rutella S, Bonanno G, Pierelli L et al (2004) Granulocyte colony-stimulating factor promotes the generation of regulatory DC through induction of IL-10 and IFN-alpha. Eur J Immunol 34:1291–1302
Kared H, Masson A, Adle-Biassette H, Bach JF, Chatenoud L, Zavala F (2005) Treatment with granulocyte colony-stimulating factor prevents diabetes in NOD mice by recruiting plasmacytoid dendritic cells and functional CD4+ CD25+ regulatory T cells. Diabetes 54:78–84
Hadaya K, Kared H, Masson A, Chatenoud L, Zavala F (2005) G-CSF treatment prevents cyclophosphamide acceleration of autoimmune diabetes in the NOD mouse. J Autoimmun 24:125–134
Lu Y, Song S (2009) Distinct immune responses to transgene products from rAAV1 and rAAV8 vectors. Proc Natl Acad Sci U S A 106:5
Finkelman FD (2007) Anaphylaxis: lessons from mouse models. J Allergy Clin Immunol 120:506–515, quiz 516–507
Janciauskiene SM, Nita IM, Stevens T (2007) Alpha1-antitrypsin, old dog, new tricks. Alpha1-antitrypsin exerts in vitro anti-inflammatory activity in human monocytes by elevating cAMP. J Biol Chem 282:8573–8582
Grimstein C, Choi Y-K, Satoh M et al (2010) Combination of alpha-1 antitrypsin and doxycycline suppresses collagen induced arthritis. J Gene Med 12:35–44
Acknowledgements
This work was supported by grants from NIH (HL079132, DK062652), Juvenile Diabetes Research Foundation and University of Florida Office of Research. H. Ma was supported by China Scholarship Council.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
H. Ma and Y. Lu contributed equally to this work.
Rights and permissions
About this article
Cite this article
Ma, H., Lu, Y., Li, H. et al. Intradermal α1-antitrypsin therapy avoids fatal anaphylaxis, prevents type 1 diabetes and reverses hyperglycaemia in the NOD mouse model of the disease. Diabetologia 53, 2198–2204 (2010). https://doi.org/10.1007/s00125-010-1829-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1829-2