Standardization of Plant Microbiome Studies: Which Proportion of the Microbiota is Really Harvested?

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microbiota Recovery
2.2. Cultivable Microbiota Enumeration
2.3. Bacterial Microbiota Composition Analysis by 16S rRNA Gene
3. Results
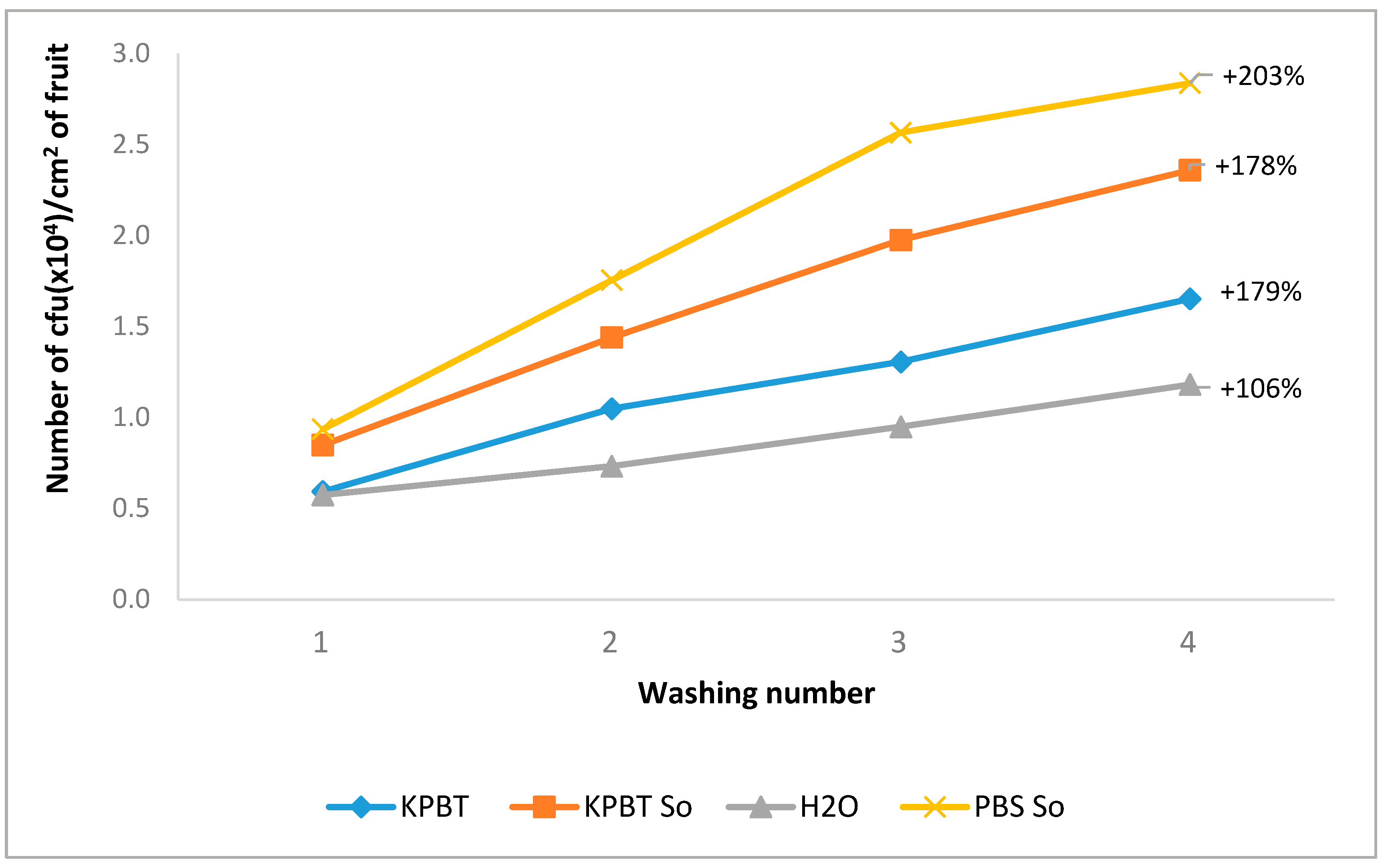
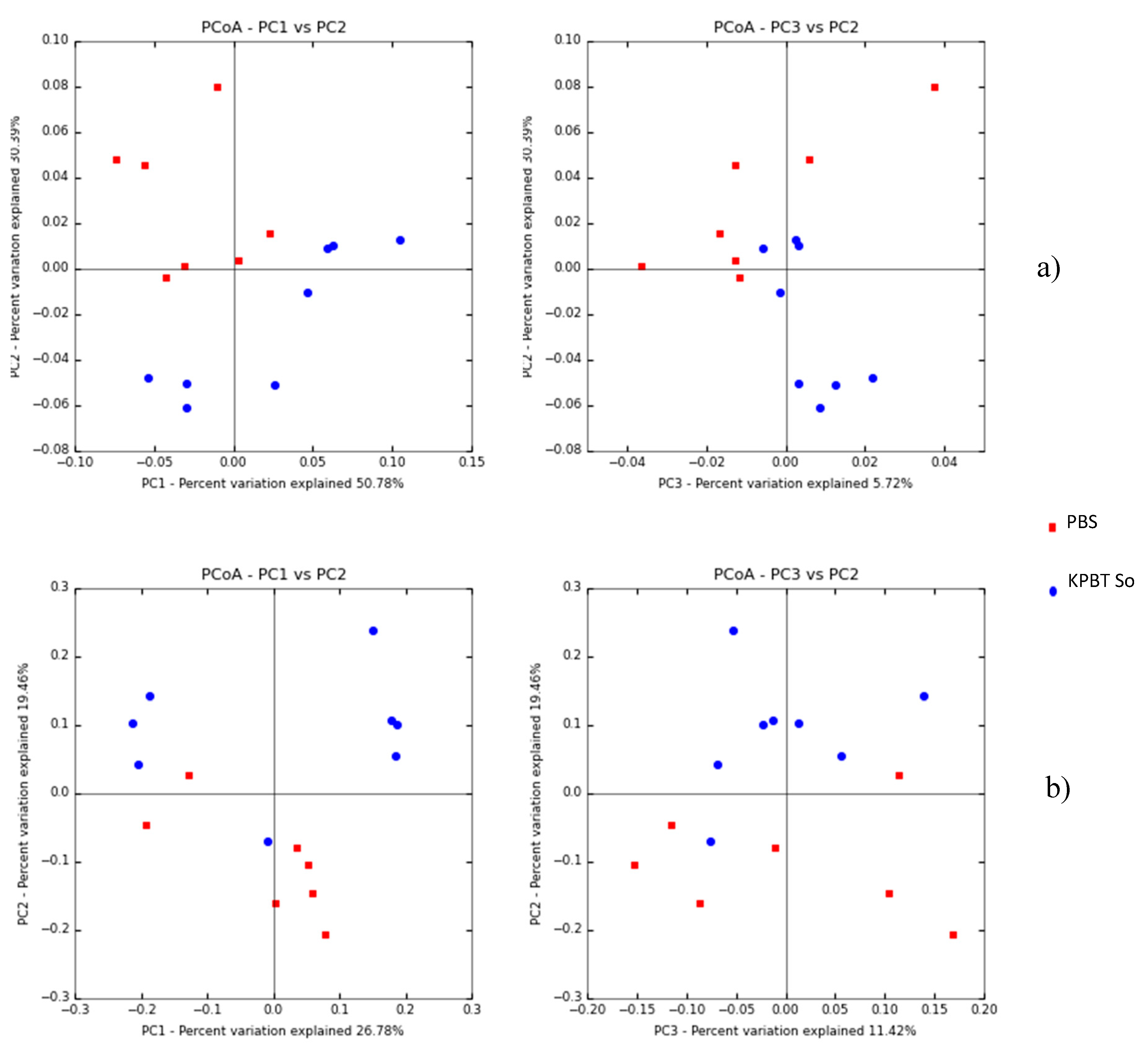
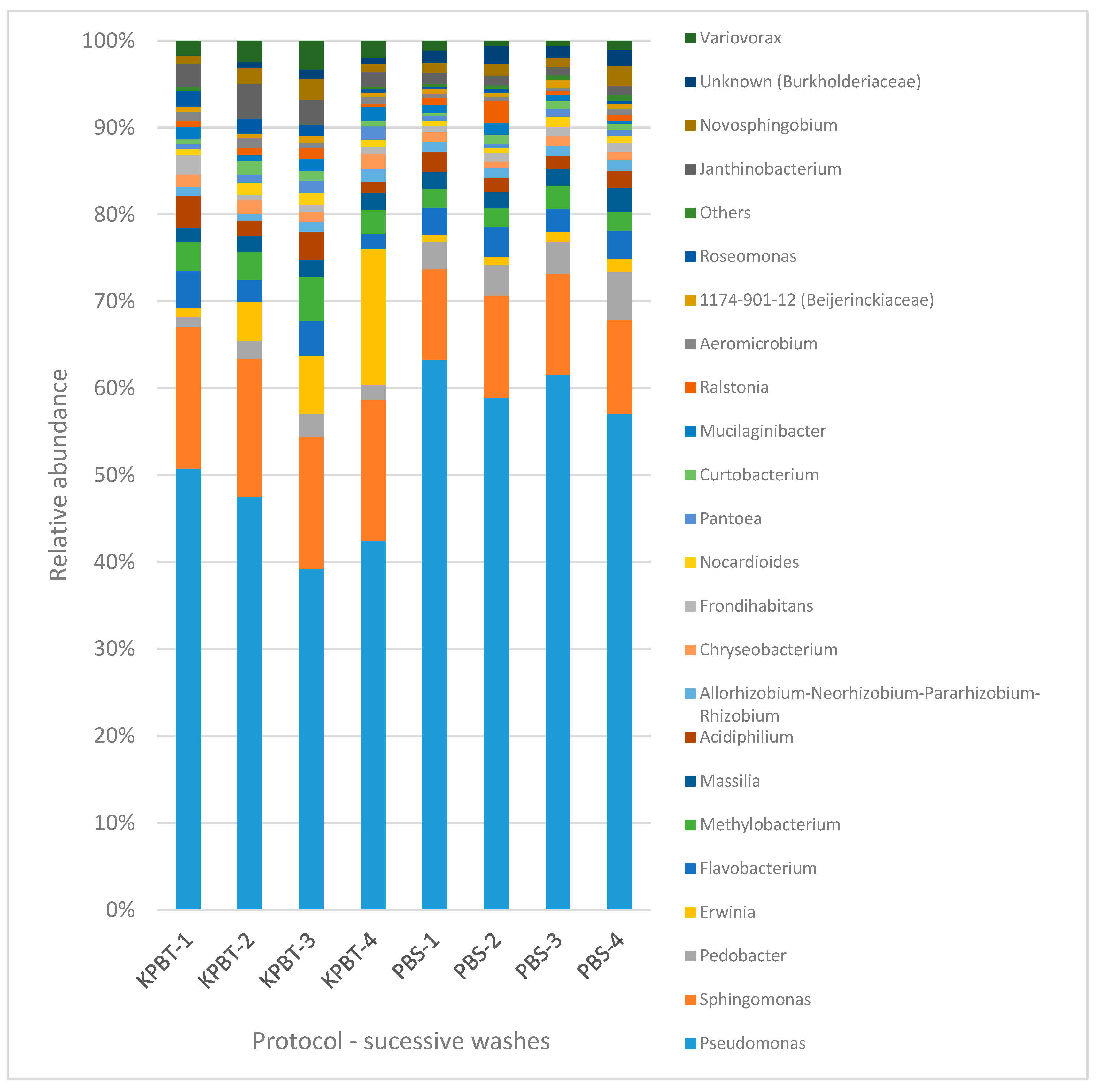
3.1. Apple Carposphere Microbiota
3.1.1. Cultivable Microbiota Enumeration
3.1.2. Bacterial Diversity Analysis by 16S rRNA Gene
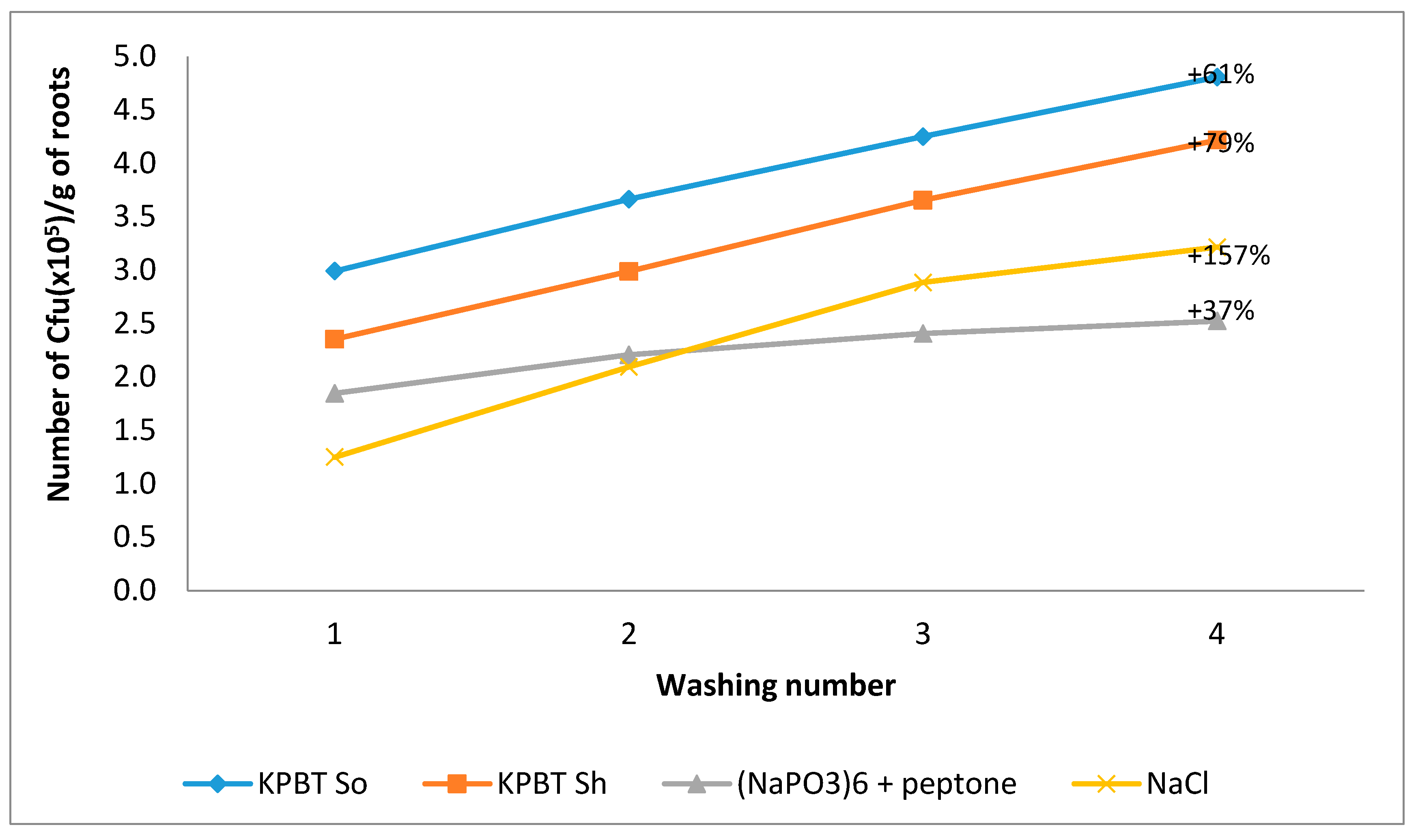
3.2. Lettuce Rhizoplane Microbiota
3.2.1. Cultivable Microbiota Enumeration
3.2.2. Bacterial Diversity Analysis by 16S rRNA Gene
4. Discussion
4.1. Impact of the Protocol on the Quantity and Diversity of Harvested Microorganisms
4.2. Impact of Successive Washes on the Quantity and Diversity of Harvested Microorganisms
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Reinhold-Hurek, B.; Bünger, W.; Burbano, C.S.; Sabale, M.; Hurek, T. Roots Shaping Their Microbiome: Global Hotspots for Microbial Activity. Annu. Rev. Phytopathol. 2015, 53, 403–424. [Google Scholar] [CrossRef] [PubMed]
- Lugtenberg, B.; Kamilova, F. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compant, S.; Clément, C.; Sessitsch, A. Plant growth-promoting bacteria in the rhizo- and endosphere of plants: Their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 2010, 42, 669–678. [Google Scholar] [CrossRef] [Green Version]
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Bringel, F.; Couée, I. Pivotal roles of phyllosphere microorganisms at the interface between plant functioning and atmospheric trace gas dynamics. Front. Microbiol. 2015, 6, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glassner, H.; Zchori-Fein, E.; Compant, S.; Sessitsch, A.; Katzir, N.; Portnoy, V.; Yaron, S. Characterization of endophytic bacteria from cucurbit fruits with potential benefits to agriculture in melons (Cucumis melo L.). FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramey, B.E.; Koutsoudis, M.; Bodman, S.B. Von; Fuqua, C. Biofilm formation in plant-microbe associations. Curr. Opin. Microbiol. 2004, 7, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Abdelfattah, A.; Wisniewski, M.; Droby, S.; Schena, L. Spatial and compositional variation in the fungal communities of organic and conventionally grown apple fruit at the consumer point-of-purchase. Nature. Publ. Gr. 2016, 347. [Google Scholar] [CrossRef] [Green Version]
- Massart, S.; Martinez-Medina, M.; Jijakli, M.H. Biological control in the microbiome era: Challenges and opportunities. Biol. Control 2015, 89, 98–108. [Google Scholar] [CrossRef]
- Izard, J. Steps in Metagenomics: Let’s Avoid Garbage in and Garbage Out. In Metagenomics for Microbiology; Elsevier Inc.: Amsterdam, The Netherlands, 2014; ISBN 9780124105089. [Google Scholar]
- Pollock, J.; Glendinning, L.; Wisedchanwet, T.; Watson, M. The Madness of Microbiome: Attempting To Find Consensus “Best Practice” for 16S Microbiome Studies. Appl. Environ. Microbiol. 2018, 84, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Boers, S.A.; Jansen, R.; Hays, J.P. Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barillot, C.D.C.; Sarde, C.-O.; Bert, V.; Tarnaud, E.; Cochet, N. A standardized method for the sampling of rhizosphere and rhizoplan soil bacteria associated to a herbaceous root system. Ann. Microbiol. 2013, 63, 471–476. [Google Scholar] [CrossRef]
- Knief, C. Analysis of plant microbe interactions in the era of next generation sequencing technologies. Front. Plant Sci. 2014, 5, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Shi, Y.; Geng, L.; Chu, H.; Zhang, J.; Song, F.; Duan, J.; Shu, C. Template Preparation Affects 16S rRNA High-Throughput Sequencing Analysis of Phyllosphere Microbial Communities. Front. Plant Sci. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Schlatter, D.; Gohl, D.M.; Kinkel, L.L. Run-to-run sequencing variation can introduce taxon-specific bias in the evaluation of fungal microbiomes. Phytobiomes J. 2018, 2, 165–170. [Google Scholar] [CrossRef]
- Kloepper, J.W.; Beauchamp, C.J. A review of issues related to measuring colonization of plant roots by bacteria.pdf. Can. J. Microbiol. 1992, 38, 1219–1232. [Google Scholar] [CrossRef]
- Donegan, K.; Matyac, C.; Seidler, R.; Porteous, A. Evaluation of methods for sampling, recovery, and enumeration of bacteria applied to the phylloplane. Appl. Environ. Microbiol. 1991, 57, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Richter-heitmann, T.; Eickhorst, T.; Knauth, S.; Friedrich, M.W. Evaluation of strategies to separate root-associated microbial communities: A crucial choice in rhizobiome research. Front. Microbiol. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Vadkertiová, R.; Molnárová, J.; Vránová, D.; Sláviková, E. Yeasts and yeast-like organisms associated with fruits and blossoms of different fruit trees. Can. J. Microbiol. 2012, 58, 1344–1352. [Google Scholar] [CrossRef]
- Wilson, C.L.; Wisniewski, M.E.; Droby, S.; Chalutz, E. A selection strategy for microbial antagonists to control postharvest diseases of fruits and vegetables. Sci. Hortic. (Amsterdam). 1993, 53, 183–189. [Google Scholar] [CrossRef]
- Fan, Q.; Tian, S. Postharvest biological control of grey mold and blue mold on apple by Cryptococcus albidus (Saito) Skinner. Postharvest Biol. Technol. 2001, 21, 341–350. [Google Scholar] [CrossRef]
- Viñas, I.; Usall, J.; Teixidó, N.; Sanchis, V. Biological control of major postharvest pathogens on apple with Candida sake. Int. J. Food Microbiol. 1998, 40, 9–16. [Google Scholar] [CrossRef]
- Qin, G.; Tian, S.; Xu, Y. Biocontrol of postharvest diseases on sweet cherries by four antagonistic yeasts in different storage conditions. Postharvest Biol. Technol. 2004, 31, 51–58. [Google Scholar] [CrossRef]
- Glenn, D.M.; Bassett, C.; Dowd, S.E. Effect of pest management system on ‘Empire’ apple leaf phyllosphere populations. Sci. Hortic. (Amsterdam). 2015, 183, 58–65. [Google Scholar] [CrossRef]
- Ottesen, A.R.; White, J.R.; Skaltsas, D.N.; Newell, M.J.; Walsh, C.S. Impact of Organic and Conventional Management on the Phyllosphere Microbial Ecology of an Apple Crop - ProQuest. J. Food Prot. 2009, 72, 2321–2325. [Google Scholar] [CrossRef] [PubMed]
- Teixidó, N.; Usall, J.; Gutierrez, O.; Viñas, I. Effect of the antagonist Candida sake on apple surface microflora during cold and ambient (shelf life) storage. Eur. J. Plant Pathol. 1998, 104, 387–398. [Google Scholar] [CrossRef]
- Lahlali, R.; Massart, S.; De Clercq, D.; Serrhini, M.N.; Jijakli, M.H. Assessment of Pichia anomala (strain K) efficacy against blue mould of apples when applied pre- or post-harvest under laboratory conditions and in orchard trials - Springer. Eur. J. Plant Pathol. 2009, 123, 37–45. [Google Scholar] [CrossRef]
- Manso, T.; Nunes, C. Metschnikowia andauensis as a new biocontrol agent of fruit postharvest diseases. Postharvest Biol. Technol. 2011, 61, 64–71. [Google Scholar] [CrossRef]
- Shen, Y.; Nie, J.; Li, Z.; Li, H.; Wu, Y.; Dong, Y.; Zhang, J. Differentiated surface fungal communities at point of harvest on apple fruits from rural and peri-urban orchards. Sci. Rep. 2018, 8, 2165. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Nie, J.; Dong, Y.; Kuang, L.; Li, Y.; Zhang, J. Compositional shifts in the surface fungal communities of apple fruits during cold storage. Postharvest Biol. Technol. 2018, 144, 55–62. [Google Scholar] [CrossRef]
- Vepštaitė-Monstavičė, I.; Lukša, J.; Stanevičienė, R.; Strazdaitė-Žielienė, Ž.; Yurchenko, V.; Serva, S.; Servienė, E. Distribution of apple and blackcurrant microbiota in Lithuania and the Czech Republic. Microbiol. Res. 2018, 206, 1–8. [Google Scholar] [CrossRef]
- Grosch, R.; Dealtry, S.; Schreiter, S.; Berg, G.; Mendonça-Hagler, L.; Smalla, K. Biocontrol of Rhizoctonia solani: Complex interaction of biocontrol strains, pathogen and indigenous microbial community in the rhizosphere of lettuce shown by molecular methods. Plant Soil 2012, 361, 343–357. [Google Scholar] [CrossRef]
- Bonaldi, M.; Chen, X.; Kunova, A.; Pizzatti, C.; Saracchi, M.; Cortesi, P. Colonization of lettuce rhizosphere and roots by tagged Streptomyces. Front. Microbiol. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Schmautz, Z.; Graber, A.; Jaenicke, S.; Goesmann, A.; Junge, R.; Smits, T.H.M. Microbial diversity in different compartments of an aquaponics system. Arch. Microbiol. 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mangmang, J.S.; Deaker, R.; Rogers, G. Response of lettuce seedlings fertilized with fish effluent to Azospirillum brasilense inoculation. Biol. Agric. Hortic. 2014, 31, 61–71. [Google Scholar] [CrossRef]
- Wilson, C.L.; Chalutz, E. Postharvest biological control of penicillium rots of citrus with antagonistic yeasts and bacteria. Sci. Hortic. 1989, 40, 105–112. [Google Scholar] [CrossRef]
- Delaide, B.; Delhaye, G.; Dermience, M.; Gott, J.; Soyeurt, H.; Jijakli, M.H. Plant and fish production performance, nutrient mass balances, energy and water use of the PAFF Box, a small-scale aquaponic system. Aquac. Eng. 2017, 78, 130–139. [Google Scholar] [CrossRef]
- Chave, M.; Dabert, P.; Brun, R.; Godon, J.J.; Poncet, C. Dynamics of rhizoplane bacterial communities subjected to physicochemical treatments in hydroponic crops. Crop Prot. 2008, 27, 418–426. [Google Scholar] [CrossRef]
- Rosberg, A.K.; Gruyer, N.; Hultberg, M.; Wohanka, W.; Alsanius, B.W. Monitoring rhizosphere microbial communities in healthy and Pythium ultimum inoculated tomato plants in soilless growing systems. Sci. Hortic. (Amsterdam). 2014, 173, 106–113. [Google Scholar] [CrossRef]
- Clayton, M.; Amos, N.D.; Banks, N.H.; Morton, R.H. Estimation of apple fruit surface area. New Zeal. J. Crop Hortic. Sci. 1995, 23, 345–349. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Eck, M.; Sare, A.R.; Massart, S.; Schmautz, Z.; Junge, R.; Smits, T.H.M.; Jijakli, M.H. Exploring Bacterial Communities in Aquaponic Systems. Water 2019, 11, 260. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Ram Rideout, J.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Heal. Dis. 2015, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanshew, A.S.; Mason, C.J.; Raffa, K.F.; Currie, C.R. Minimization of chloroplast contamination in 16S rRNA gene pyrosequencing of insect herbivore bacterial communities. J. Microbiol. Methods 2013, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, S.C.; Song, Z.; Gutknecht, J.L.M.; Rosen, C.J.; Kinkel, L.L. DNA Template Dilution Impacts Amplicon Sequencing-Based Estimates of Soil Fungal Diversity. Phytobiomes J. 2018, 2, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.-I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.M.; Kim, M.; Lee-Cruz, L.; Lai-Hoe, A.; Go, R.; Ainuddin, N.; Rahim, R.A.; Shukor, N.; Adams, J.M. Distinctive bacterial communities in the rhizoplane of four tropical tree species. Microb. Ecol. 2012, 64, 1018–1027. [Google Scholar] [CrossRef]
- Kloepper, J.W.; Mahaffee, W.F.; McInroy, J.A.; Backman, P.A. Comparative analysis of five methods for recovering rhizobacteria from cotton roots. Can. J. Microbiol. 1991, 37, 953–957. [Google Scholar] [CrossRef]
- Rose, L.; Jensen, B.; Peterson, A.; Banerjee, S.N.; Arduino, M.J. Swab materials and Bacillus anthracis spore recovery from nonporous surfaces. Emerg. Infect. Dis. 2004, 10, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
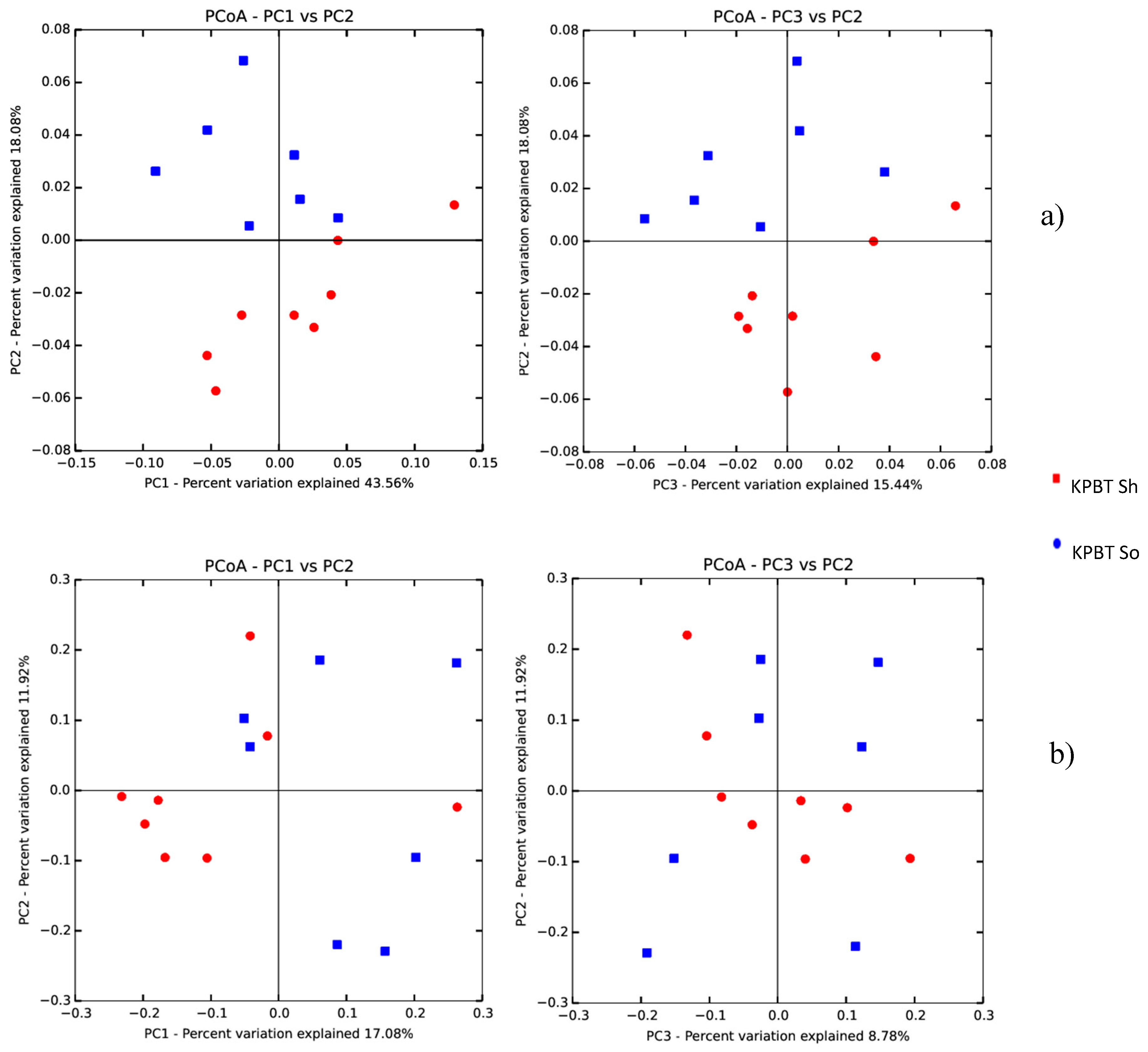{kind=link}
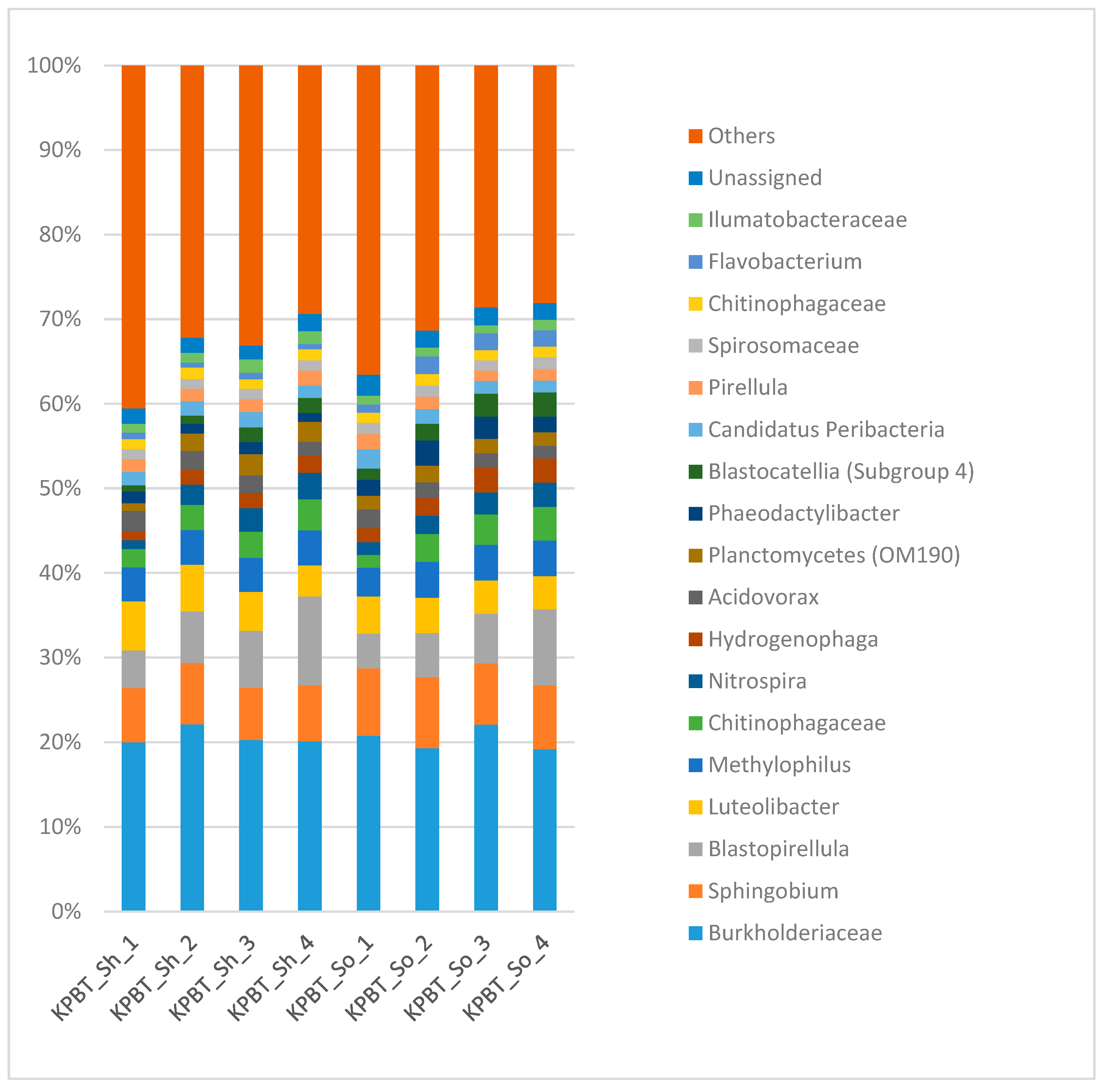{kind=link}
| Host | Buffer | Technic Use | Purpose | Reference |
|---|---|---|---|---|
| Apple | Malt extract + glucose + antibiotic | Culture of fruit slice shaken at 100 rpm (3–10 days) | Plating | [20] |
| Sterile deionized water | Two washes (by dipping) | Biological assay | [21] | |
| Phosphate buffer, pH 6.5 | Use of second wash; 30 s sonication + 100 rpm shaking for 10 min (the first washing with no sonication is thrown) | Plating | [22] | |
| Phosphate buffer, pH 6.5 | Use of second wash; 10 min sonication + 150rpm shaking for 10 min (the first wash with no sonication is thrown) | Plating | [23] | |
| Sterile water | 100 rpm shaking for 10 min | Plating | [24] | |
| Double deionized water (DDI) | 2 min mixing the sample + 1 min sonication on each side | 454-amplicon sequencing | [25] | |
| Deionized sterile water | 5 min sonication | Amplicon sequencing | [26] | |
| Phosphate buffer, pH = 7 | Fruit dissection + shaking for 20 min at 150 rpm + 10 min sonication | Plating | [27] | |
| Phosphate buffer, pH = 6.5 | Rinsing by shaking at 120 rpm for 20 min | Plating | [28] | |
| Water and phosphate buffer, pH 6.5 | Rinsing first time in water; and once with sonication bath with phosphate buffer | Plating | [29] | |
| No buffer | Wiping the fruit surface with moistened cotton swab | Amplicon sequencing | [30,31] | |
| Phosphate buffer, pH 6.8 | Rinsing by shaking at 120 rpm for 2 h | Amplicon sequencing | [32] | |
| Lettuce in soil | Sterile saline water | Shaking by rotary shaker at 307 rpm with glass bead for 1 h | PCR-DGGE, | [33] |
| Sterile 0.9% NaCl + 0.02% Silwet L-77 solution | Vortexing 2 times for 15s | Plating | [34] | |
| Lettuce in aquaponics | Sterile ultrapure water | Vortexing for 2 min followed by 5 min ultrasonic bath | Illumina sequencing | [35] |
| Sterile peptone phosphate buffer (1g peptone + 1.21 g K2HPO4 + 0.34 g KH2PO4 + 1 L deionized water | Crushing with a tissue lyser | T-RFLP | [36] |
| Treatment Label | Material | Solution | Treatment 1 | Duration 1 | Intensity 1 | Treatment 2 | Duration 2 | Intensity 2 | Reference |
|---|---|---|---|---|---|---|---|---|---|
| KPBT | 4 apples | 1 L of a sterile 0.05M potassium phosphate buffer + 0.1% tween80 pH 6.5 | Shaking | 20 min | 120 rpm | - | - | - | [28] |
| KPBT So | 4 apples | 1 L of a sterile 0.05 M potassium phosphate buffer + 0.1% tween80 pH 6.5 | Sonication in ultra-bath | 10 min | - | Shaking | 20 min | 120 rpm | Modified from [28] |
| PBS So | 4 apples | 1 L of sterile Phosphate Buffer Saline, pH 7.4 | Sonication in ultra-bath | 20 min | - | Shaking | 20 min | 120 rpm | Modified from [23] |
| H2O | 4 apples | 1 L of sterile distilled water | Shaking | 10 min | 120 rpm | - | - | - | [24,37] |
| A | |||||||||
| Treatment Label | Material | Solution | Treatment 1 | Duration 1 | Intensity 1 | Reference | |||
| (NaPO3)6 + peptone | 2 g of roots | 30 mL of a 2 g/L (NaPO3)6 + 1 g/L peptone sterile solution | Shaking | 20 min | 200 rpm | [40] | |||
| NaCl | 2 g of roots | 5 mL of isotonic sterile water (0.85% NaCl) | Shaking | 10 min | 150 rpm | [39] | |||
| KPBT Sh | 2 g of roots | 30 mL of a sterile 0.05 M potassium phosphate buffer + 0.05% tween80 pH 6.5 | Shaking | 20 min | 150 rpm | Intern protocol | |||
| KPBT So | 2 g of roots | 30 mL of a sterile 0.05 M potassium phosphate buffer + 0.05% tween80 pH 6.5 | Ultra-bath sonication | 10 min | - | Intern protocol | |||
| B | |||||||||
| Faith_pd | Pielou_Eveness | Observed_Otus | Shannon | |
|---|---|---|---|---|
| KPBT So | 5.13 | 0.85 | 184 | 6.36 |
| PBS So | 5.14 | 0.78 | 172 | 5.76 |
| Comparison between KPBT So and PBS So | ns | 0.005 | Ns | 0.015 |
| First wash | 5.37 | 0.80 | 193 | 6.06 |
| Second wash | 5.15 | 0.82 | 180 | 6.13 |
| Third wash | 5.13 | 0.82 | 175 | 6.11 |
| Fourth wash | 4.89 | 0.81 | 165 | 5.92 |
| Pairwise comparisons (six) between the successive washes | ns for all | ns for all | ns for all | ns for all |
| (a) Comparison of non-pooled washes (rarefied at 6524 sequences); | ||||
| Faith_pd | Pielou_Eveness | Observed_Otus | Shannon | |
| KPBT So | 6.29 | 0.83 | 251 | 6.59 |
| PBS So | 5.92 | 0.75 | 229 | 5.82 |
| Comparison between KPBT So and PBS So | ns | ns | ns | ns |
| First wash | 5.24 | 0.78 | 189 | 5.91 |
| Pooled of the four washes | 6.71 | 0.78 | 276 | 6.33 |
| Comparisons between the first wash and the pooled of the four successive washes | 0.033 | ns | 0.032 | ns |
| (b) Comparison of pooled washes (rarefied at 7500 sequences). | ||||
| Faith_PD | Pielou_Eveness | Observed_Otus | Shannon | |
|---|---|---|---|---|
| KPBT Sh | 106 | 0.73 | 764 | 6.99 |
| KPBT So | 85 | 0.73 | 622 | 6.75 |
| Pairwise comparison between KPBT Sh and KPBT So | 0.037 | ns | 0.028 | ns |
| First wash | 100 | 0.76 | 750 | 7.19 |
| Second wash | 98 | 0.73 | 780 | 6.96 |
| Third wash | 88 | 0.73 | 604 | 6.77 |
| Fourth wash | 100 | 0.71 | 670 | 6.65 |
| Pairwise comparisons (six) between the successive washes | ns for all | ns for all | ns for all | ns for all |
| (a) Comparison of non-pooled washes (rarefied at 17,463 sequences); all the successive washes were pairwise compared | ||||
| Faith_PD | Pielou_Eveness | Observed_Otus | Shannon | |
| KPBT Sh | 120 | 0.73 | 925 | 7.2 |
| KPBT So | 80 | 0.72 | 609 | 6.95 |
| Pairwise comparison between KPBT Sh and KPBT So | ns | ns | ns | ns |
| First wash | 98 | 0.76 | 755 | 7.2 |
| Pooled of the four washes | 116 | 0.72 | 885 | 7.00 |
| Comparisons between the first wash and the pooled of the four successive washes | ns | ns | ns | ns |
| (b) Comparison of pooled washes (rarefied at 17,463 sequences) | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sare, A.R.; Stouvenakers, G.; Eck, M.; Lampens, A.; Goormachtig, S.; Jijakli, M.H.; Massart, S. Standardization of Plant Microbiome Studies: Which Proportion of the Microbiota is Really Harvested? Microorganisms 2020, 8, 342. https://doi.org/10.3390/microorganisms8030342
Sare AR, Stouvenakers G, Eck M, Lampens A, Goormachtig S, Jijakli MH, Massart S. Standardization of Plant Microbiome Studies: Which Proportion of the Microbiota is Really Harvested? Microorganisms. 2020; 8(3):342. https://doi.org/10.3390/microorganisms8030342
Chicago/Turabian StyleSare, Abdoul Razack, Gilles Stouvenakers, Mathilde Eck, Amber Lampens, Sofie Goormachtig, M. Haïssam Jijakli, and Sebastien Massart. 2020. "Standardization of Plant Microbiome Studies: Which Proportion of the Microbiota is Really Harvested?" Microorganisms 8, no. 3: 342. https://doi.org/10.3390/microorganisms8030342