
Abstract
CALHM1 is a cell surface calcium channel expressed in cerebral neurons. CALHM1 function in the brain remains unknown, but recent results showed that neuronal CALHM1 controls intracellular calcium signaling and cell excitability, two mechanisms required for synaptic function. Here, we describe the generation of Calhm1 knockout (Calhm1−/−) mice and investigate CALHM1 role in neuronal and cognitive functions. Structural analysis revealed that Calhm1−/− brains had normal regional and cellular architecture, and showed no evidence of neuronal or synaptic loss, indicating that CALHM1 deficiency does not affect brain development or brain integrity in adulthood. However, Calhm1−/− mice showed a severe impairment in memory flexibility, assessed in the Morris water maze, and a significant disruption of long-term potentiation without alteration of long-term depression, measured in ex vivo hippocampal slices. Importantly, in primary neurons and hippocampal slices, CALHM1 activation facilitated the phosphorylation of NMDA and AMPA receptors by protein kinase A. Furthermore, neuronal CALHM1 activation potentiated the effect of glutamate on the expression of c-Fos and C/EBPβ, two immediate-early gene markers of neuronal activity. Thus, CALHM1 controls synaptic activity in cerebral neurons and is required for the flexible processing of memory in mice. These results shed light on CALHM1 physiology in the mammalian brain.
Similar content being viewed by others

Introduction
Calcium homeostasis modulator protein 1 (CALHM1)1 was identified by a tissue-specific gene expression profiling approach2, which was designed to screen for genes preferentially expressed in the hippocampus and located on susceptibility loci for Alzheimer’s disease (AD). AD is a neurodegenerative brain disorder characterized by an array of cognitive disturbances, one of the earliest and most prominent being an impairment in the flexible use of episodic memory3. CALHM1 does not appear to be associated with the risk of developing AD4, but independent genetic studies have shown that it influences the onset of the disease1,4. At the mechanistic level and in cell culture systems, CALHM1 controls the accumulation of the amyloid-β (Aβ) peptide1,5,6, a main culprit in AD7. Some studies8,9, but not all10, have also reported an association between a CALHM1 variant and Aβ levels in human cerebrospinal fluid. These results support the notion that CALHM1 might be involved in molecular mechanisms relevant to AD pathogenesis and thus warrant further studies aimed at understanding the exact physiology of CALHM1 in the brain.
Despite one report of failed detection of Calhm1 mRNA in the mouse brain11, CALHM1 expression has now been unambiguously demonstrated in human and murine brains, particularly in hippocampal and cortical neurons1,12,13. The physiological function associated with its neuronal expression in the brain, however, remains incompletely understood. CALHM1 is a plasma membrane, voltage-gated calcium-permeable channel that is regulated by extracellular calcium concentration1,13,14. Its expression in different cell systems induces cationic currents and elevates cytoplasmic calcium levels in response to removal of extracellular calcium and its subsequent add-back1,12 (hereafter termed the calcium add-back condition, CaAB). The response of CALHM1 to CaAB might be highly relevant to synaptic function because transient decreases in extracellular calcium levels are known to occur within the synaptic cleft during neuronal electrical activity15. In mouse cortical neurons, CALHM1 responds to CaAB not only by elevating intra-neuronal calcium levels but also by controlling the cell’s conductance and action potential firing13. Studies performed in CALHM1-transfected hippocampal HT-22 cells, as well as Calhm1−/− primary neurons, have further demonstrated that CALHM1 not only controls calcium influx but also intracellular calcium signal transduction via the activation of a kinase signaling cascade involving extracellular signal-regulated kinase-1/2 (ERK1/2)12. Thus, CALHM1 plays an important role in cerebral neuronal calcium homeostasis and signaling, as well as in neuronal excitability. Here, we show that CALHM1 deficiency in mice leads to major cognitive and neuronal deficits, manifested by the presence of impairments in memory flexibility and hippocampal long-term potentiation (LTP). These deficits could not be explained by alterations in brain development or brain integrity in adulthood, but could be associated with impairments in neuronal activity signaling.
Results
Generation of a constitutive Calhm1−/− mouse line
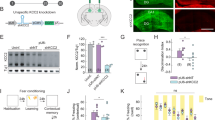
The mouse Calhm1 gene is located on chromosome 19, extends over 3.1 kb, and contains 2 exons separated by one intron (Fig. 1A). Calhm1 overlaps at its 3′ end with the putative promoter region of the uncharacterized homolog gene Calhm21. We chose the strategy of deleting the Calhm1 exon 1 to prevent a potential interference with the Calhm2 promoter. The resulting Calhm1−/− mice (Fig. 1B,C) were viable and fertile. They generated litters of normal size, with normal Mendelian inheritance of the mutant allele, ruling out any essential functions of CALHM1 in mouse embryogenesis.

(A) Schematic representation of the Calhm1 targeting strategy resulting in exon (Ex) 1 deletion. Grey boxes represent Calhm1 coding sequences and solid lines the chromosome sequence. The initiation (ATG) and stop (Stop) codons are indicated. Arrows show primers used for genotyping; neo, neomycin cassette. (B) PCR genotyping of Calhm1−/− mouse lines. PCR was performed on tail genomic DNA of a C57BL/6J mouse control, Calhm1+/+ (+/+), Calhm1+/− (+/−), and Calhm1−/− (−/−) littermates. Arrows point to 4.4-kb and 3.8-kb PCR products corresponding to amplified regions of Calhm1 endogenous locus and recombined locus, respectively (as shown schematically in (A)). (C) Real time PCR analyzing Calhm1 expression levels in whole brains from Calhm1+/+ and Calhm1−/− mice. Calhm1 expression was normalized to the reference genes Hprt1, Tbp, and Polr2a; ND, not detected. (D) Nissl (panels a–f), NeuN (g–l), and GFAP (m–r) staining of sagittal brain sections of a group of old Calhm1+/+ (a–c,g–i,m–o) and Calhm1−/− (d–f,j–l,p–r) littermates. Hippocampal formation (panels a,d,g,j,m,p), cerebral cortex (b,e,h,k,n,q), and cerebellum (c,f,i,l,o,r) are shown. (E–H) Percent area occupied with positive staining for cortical (E) CA1 (F) and cerebellar (G) NeuN expression, and hippocampal (HP) GFAP (H) expression, analyzed by immunohistochemistry as in D (n = 3).
A cohort of adult Calhm1−/− and Calhm1+/+ littermates (see Methods) was sacrificed to examine their brain morphology. Immunohistological analyses of brain sections, using Nissl staining and the neuronal marker protein NeuN, demonstrated that Calhm1−/− brains had normal cerebral architecture when compared to WT controls, and displayed no significant alterations of the neuronal populations of the cortex, hippocampus, and cerebellum (Fig. 1D, panels a-l, and Fig. 1E–G). Also, Calhm1−/− mice displayed normal immunoreactivity for the glial marker, glial fibrillary acidic protein (GFAP), in these brain regions (Fig. 1D, panels m–r, and Fig. 1H). WB analysis of whole brain homogenates showed no differences between Calhm1−/− and Calhm1+/+ mice in the expression of several neuronal markers, such as NeuN, microtubule-associated protein 2 (MAP-2), brain-derived neurotrophic factor (BDNF), tyrosine receptor kinase B (TrkB), and the synaptic marker post-synaptic density protein 95 (PSD-95; Fig. 2A–F). Furthermore, in situ hybridization revealed no alteration in the levels and regional expression of mRNA for the pre-synaptic makers synaptosome-associated protein of 25 kDa (Snap25) and synaptotagmin-1 (Syt1) in the Calhm1−/− hippocampus and cortical brain (Fig. 2G). Thus, Calhm1−/− mice presented grossly normal brain cytoarchitecture and showed no evidence of neuronal or glial degeneration, indicating that CALHM1 deficiency did not affect brain development or brain maintenance in adulthood.

(A) WB analysis of the levels of the indicated proteins in the whole brain from a group of young-adult Calhm1+/+ and Calhm1−/− littermates. (B–F) Densitometric analysis and quantification of the expression levels of the indicated neuronal makers, analyzed by WB as in (A) (n = 3). (G) In situ hybridization of Snap25 (panels a–d) and Syt1 (e–h) in the cerebral cortex (a,b,e,f) and hippocampus (c,d,g,h) of young-adult Calhm1+/+ and Calhm1−/− mice.
Impaired memory flexibility in Calhm1−/− mice
To study whether the deletion of the Calhm1 gene produced phenotypic alterations, Calhm1−/− mice (n = 27) and Calhm1+/+ (n = 29) controls were assessed behaviorally at a young-adult and old age (6-mo-old and 17-mo-old, respectively). The animals belonged to either the C57BL/6J strain (6-mo-old, Calhm1−/−, n = 9, Calhm1+/+, n = 10) or the original hybrid 129 × C57 strain (6-mo-old, Calhm1−/−, n = 8, Calhm1+/+, n = 9; 17-mo-old, Calhm1−/−, n = 10, Calhm1+/+, n = 10). Both genotypes performed similarly in an observational screen (Fig. 3B), the rotarod test (Fig. 3C, but notice that old Calhm1−/− mice acquired the task more slowly), the open field test (Fig. 3D), and fear conditioning (Fig. 3E), indicating that Calhm1−/− mice did not have generalized phenotypic abnormalities. These are important controls because CALHM1 is expressed in the peripheral taste system where it mediates taste perception16. Therefore, CALHM1 could have been involved in another sensory mechanism required for basic behavioral responses during cognitive testing.

(A) Young-adult animals of both genotypes have similar weights, but old Calhm1−/− mice show lower weight than controls, *P < 0.05 (t = 2.89, t test). (B) Both genotypes behave similarly in the observational screen as shown by their scores for the five functions. (C) The rotarod test reveals no differences between genotypes in the time to fall from the rotating drum across the last 2 trials (graph at right), although old Calhm1−/− mice show slow motor learning (trial 2 for BL6 × 129 17-mo mice, *P < 0.05, t = 2.56, t test). (D) Open field test is similar between genotypes. Left, top view of the chamber showing the center and periphery (Per.) zones, as well as heat-maps for representative Calhm1+/+ and Calhm1−/− mice during the test. Middle, no difference in zone occupancy between genotypes. Right, similar time moving during the 20-min test for both genotypes. (E) Fear conditioning is equivalent in both genotypes. Left top, schematic of the task. Left bottom, time courses for the freezing response (% of total time, in 10-sec bins, presented as continuous lines) during the acquisition phase, and the two types of memory testing (context and tone). Right top, both genotypes show similar freezing during the last 5 min of the context memory test. Right bottom, both genotypes freeze equally during the presentation of the tones in the tone memory test. ns, non significant.
Notably, both the young-adult and old Calhm1−/− groups were significantly impaired in a Morris water maze task for memory flexibility17 that required animals to find a hidden platform that was first located in the North quadrant (phase 1) and then in the South quadrant (phase 2) (Fig. 4A). Probe tests were performed at the end of each phase. Mice of both genotypes performed equally on phase 1, in terms of latency (Fig. 4A,B), and swimming speed (Fig. 4E), but showed clear differences in latency during phase 2, whether the mice were in the young-adult or old age group (Fig. 4B, Z = 2.15, P < 0.001, Kolmogorov-Smirnov test). An analysis centered on the change in location of the platform (from North to South quadrants) revealed that Calhm1−/− mice had a significantly higher perseveration ratio (Fig. 4C; Calhm1+/+, n = 29, 0.15 ± 0.03; Calhm1−/−, n = 27, 0.39 ± 0.03; t = 5.38, P < 0.001, t test) and poorer learning score difference (Fig. 4D; Calhm1+/+, n = 29, 0.0217 ± 0.0035; Calhm1−/−, n = 27, −0.0028 ± 0.0035; t = 4.92, P < 0.001, t test), revealing a clear deficit in memory flexibility. Additionally, a probe test at the end of phase 1 revealed no differences between genotypes (Fig. 4F), whereas a second probe test at the end of phase 2 (Fig. 4G) showed that, compared to controls, Calhm1−/− mice had a reduced spatial bias for their trained quadrant, which was evidenced by their significantly lowered spatial memory index (Fig. 4G; Calhm1+/+, n = 29, 0.53 ± 0.03; Calhm1−/−, n = 27, 0.32 ± 0.03; t = 4.67, P < 0.001, t test).

The Morris water maze task was used to assess spatial cognition. (A) Left, the diagrams show phase 1 of training with the platform (yellow circle) in the North location, and phase 2 with the platform in the South location. Right, mice of both genotypes show comparable latencies in phase 1, but Calhm1−/− mice display a clear deficit in phase 2, when the platform is switched to a novel location (left graph, F = 11.9, P < 0.001 ; middle graph, F = 17.8, P < 0.001; right graph, F = 9.54, P < 0.005, RMANOVA with last 16 trials as the repeated measure). (B) Cumulative probability plots for all trials in each phase show that Calhm1−/− mice have significantly longer latencies in phase 2 (Z = 2.15, P < 0.001, Kolmogorov-Smirnov test). (C) The perseveration ratio is markedly higher in Calhm1−/− mice. (D) Learning scores are unchanged across phases for Calhm1−/− mice, whereas Calhm1+/+ mice show a significantly enhanced score in phase 2. These results show lack of memory flexibility in Calhm1−/− mice. (E) Swimming speeds are comparable between Calhm1−/− and Calhm1+/+ mice across the two phases (left) and probe tests (right). (F) Performance during the first probe test is similar in both genotypes; abbreviations for pool quadrants, L, left, R, right, O, opposite, T, target. (G) Impaired performance of Calhm1−/− mice during the second probe test. Left, the graphs show lower exploration of the target quadrant by Calhm1−/− mice. Middle, representative swim-paths of old mice showing focused search by Calhm1+/+ mouse and broad search by Calhm1−/− mouse. Right, the spatial memory index is markedly lower in Calhm1−/− animals. *P < 0.05; **P < 0.005 (t test).
Disrupted LTP in Calhm1−/− mice
To examine whether synaptic function was altered in young-adult and old Calhm1−/− mice, electrophysiological studies were conducted17,18 in ex vivo slices from the hippocampus, a brain region that is critically involved in memory encoding. Basal synaptic function was examined by recording field excitatory post-synaptic potentials (fEPSP) between CA3–CA1 synapses. Input-output (I-O) functions with stimulation intensity of the CA3 axons as the input and fEPSP slope as the output were similar between genotypes (Fig. 5A; Calhm1+/+, n = 10; Calhm1−/−, n = 12; F = 0.19, P = 0.67, RMANOVA). Analysis of synaptic burst responses produced by high-frequency stimulation (HFS) of CA3 axons showed that the integral of the burst responses was similar between genotypes (Fig. 5B; Calhm1+/+, n = 24; Calhm1−/−, n = 30; t = 0.24, P = 0.8, t test). These results demonstrate that the absence of CALHM1 does not affect the basal function of excitatory CA1 synapses.

(A) Left, traces from old Calhm1+/+ and Calhm1−/− mice show representative fEPSPs at increasing stimulation strengths. Right, plot displays the mean fEPSP slopes vs. stimulation intensities, revealing comparable input-output functions between genotypes. (B) Left, representative traces for a train of high-frequency stimulation (HFS, 100 Hz for 1 sec) from young-adult and old Calhm1+/+ and Calhm1−/− mice, in which the stimulus artifacts have been subtracted. Shaded areas-over-the-curves are used for analysis. Right, total integral of HFS train is comparable across groups. (C) LTP is impaired in young-adult and old Calhm1−/− mice. A comparison at 45 min post-HFS reveals significant differences between genotypes (left, t = 4.23, P < 0.0001; right, t = 3.42, P < 0.005, t test). Inset, traces at 5 min pre- and 45 min post-HFS. (D) LTD is not affected in old Calhm1−/− mice. Inset, traces at 5 min pre- and 60 min post-LFS, low-frequency stimulation (1 Hz for 15 min). (E) BCM curves showing selective deficit of LTP expression (100 Hz train) in Calhm1−/− mice. Each point represents the mean ± SEM at 60 min post-1 Hz, 30 min post-50 Hz, and 45 min post-100 Hz; range = 9–23, young-adult and old experiments combined for each genotype. *P < 0.01, t test. For (C,D), arrows indicate blanked stimulus artifacts; scale, x-axis, 10 msec, y-axis, 1 mV.
Synaptic plasticity was assessed with LTP and long-term depression (LTD) studies in ex vivo hippocampal slices19, prepared from Calhm1−/− mice and Calhm1+/+ controls. Remarkably, Calhm1−/− slices exhibited a significant deficit in LTP when compared to Calhm1+/+ slices, measured 45 min after HFS delivery (Fig. 5C; Calhm1+/+, n = 20, 147.97 ± 8.76%; Calhm1−/−, n = 23, 104.98 ± 3.96%; t = 4.47, P < 0.0001, t test). Moreover, measurement of short-term potentiation (20 min after HFS) indicated that Calhm1−/− slices were also deficient at this earlier point (Calhm1+/+, n = 20, 166.87 ± 5.2%; Calhm1−/−, n = 23, 116.53 ± 5.2%; t = 4.13, P < 0.0001, t test). In contrast, Calhm1−/− slices exhibited normal LTD when compared to Calhm1+/+ slices, measured 60 min after LTD induction (Fig. 5D; Calhm1+/+, n = 10, 63.9 ± 0.4%; Calhm1−/−, n = 9, 64.4 ± 0.7%; t = 0.55, P = 0.59, t test). In addition, we used the induction trains for LTP (100 Hz for 1 sec) and LTD (1 Hz for 15 min), together with an intermediate train (50 Hz for 2 sec) to construct a “BCM curve”19 (Fig. 5E), which clearly showed a shift to the right in the frequency dependence of synaptic plasticity in the Calhm1−/− group and highlighted the selective deficit in LTP expression for Calhm1−/− mice.
Notably, we studied the Calhm1−/− mice in the original hybrid and C57BL/6J backcrossed genetic backgrounds (see Methods), as well as at the young-adult and old stages. Our behavioral and electrophysiological studies revealed that the same deficits due to CALHM1 deficiency were present in the Morris water maze task and LTP measurements (Figs 3, 4, 5). In combination, these data show that CALHM1 controls memory flexibility and plays a selective role in the molecular cascade responsible for the plastic enhancement of CA1 synapses but not in the molecular cascade linked to LTD.
CALHM1 activation controls PKA-mediated NMDAR and AMPAR phosphorylation and glutamate-mediated c-Fos and C/EBPβ expression in neurons
The observed impairment of LTP by CALHM1 deficiency suggests that CALHM1 cross talks with signaling mechanisms relevant to synaptic activity. WB analyses of whole hippocampal homogenates and postsynaptic density (PSD) fractions revealed no changes in the levels of the N-methyl-D-aspartate receptor (NMDAR) subunits, GluN1, GluN2A, and GluN2B, in Calhm1−/− mice (Fig. 6A), showing that CALHM1 deficiency did not affect NMDAR expression or stability. Our previous work has showed that CALHM1 activation in primary neurons (using the CaAB condition) can stimulate intracellular calcium signaling12. One consequence of this stimulation is the increase of the ERK1/2 signaling cascade in neurons [Fig. 6B and ref. 12]. Intracellular calcium elevations can lead to the activation of multiple other signaling kinases20,21, including protein kinase A (PKA), which plays a critical role in activity-dependent gene expression in neurons22. For instance, PKA controls NMDAR trafficking by phosphorylating Ser-897 on GluN123,24. Importantly, we found that CaAB resulted in a significant elevation of phospho-Ser-897 GluN1 levels in Calhm1+/+ primary neurons but not in Calhm1−/− neurons (Fig. 6B,C). Pretreatment with the PKA inhibitor H89 prevented the effect of CaAB on phospho-Ser-897 GluN1 (Fig. 6E). Another well-established target of PKA during synaptic activity is the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor (AMPAR), which is phosphorylated at Ser-845 on the GluA1 subunit25,26. Similarly to NMDAR phosphorylation, AMPAR phosphorylation at Ser-845 on GluA1 was significantly increased by CaAB in Calhm1+/+ neurons. This effect on phospho-Ser-845 GluA1 was absent in Calhm1−/− neurons (Fig. 6B,D) and was fully prevented by H89 (Fig. 6E). Altogether these data show that CALHM1 activation controls NMDAR and AMPAR phosphorylation by PKA.

(A) WB analysis of the levels of the indicated proteins in whole hippocampal homogenates (Total) and PSD fractions obtained from Calhm1+/+ and Calhm1−/− mice. (B) Primary neurons isolated from Calhm1+/+ and Calhm1−/− mice were challenged with the calcium add-back condition (CaAB) or not (Basal). Cell extracts were analyzed by WB for the indicated proteins. Representative results from 4 independent experiments are depicted. (C,D) Densitometric analysis and quantification of the ratio for phospho-Ser-897 GluN1 over total GluN1 (pGluN1/GluN1), (C) and for phospho-Ser-845 GluA1 over actin (pGluA1/actin), (D) from Calhm1+/+ and Calhm1−/− primary neurons treated as in (B). au, arbitrary units (n = 6; *P < 0.05; **P < 0.01; t test). (E) WB analysis of the levels of the indicated proteins in Calhm1+/+ primary neurons pretreated for 30 min with H89 (10 μM) and then challenged with CaAB, as in (B). (F,G) Ratio for phospho-Ser-897 GluN1 over total GluN1 (pGluN1/GluN1), (F) and for phospho-Ser-845 GluA1 over actin (pGluA1/actin), (G) from LTP-stimulated hippocampal slices (n = 10–12; *P < 0.05; t test). (H) WB analysis of the levels of the indicated proteins in Calhm1+/+ and Calhm1−/− primary neurons challenged with CaAB, as in (B), in the absence (Ctrl) or presence of glutamate stimulation (Glu, 20 μM, 1 h incubation for c-Fos, 4 h for c/EBPβ).
To determine whether CALHM1 controls NMDAR and AMPAR phosphorylation in a more physiologically relevant system, we asked whether CALHM1 deficiency affects the levels of phospho-Ser-897 GluN1 and phospho-Ser-845 GluA1 in LTP-stimulated hippocampal slices. In whole hippocampal homogenates at steady states, we could not observe any changes in phospho-Ser-897 GluN1 levels in Calhm1−/− mice (Fig. 6A). However, we found that following LTP, phospho-Ser-897 GluN1 and phospho-Ser-845 GluA1 levels in Calhm1+/+ hippocampal slices were significantly increased (compared to non-tetanized slices), whereas LTP had no effect on GluN1 and GluA1 phosphorylation in slices obtained from Calhm1−/− mice (Fig. 6F,G). These results demonstrate that CALHM1 controls NMDAR and AMPAR phosphorylation in brain slices in response to a physiologically relevant stimulus that activates PKA and synaptic activity.
To go further, we then asked whether CALHM1 is required for glutamate-mediated immediate-early gene (IEG) expression in neurons. We found that CaAB-triggered CALHM1 activation in Calhm1+/+ primary neurons robustly potentiated the effect of glutamate on the expression of c-Fos and also—but to a lesser extent—of C/EBPβ (Fig. 6H), two IEG markers of neuronal activity22. Strikingly, this effect was absent in Calhm1−/− neurons (Fig. 6H), demonstrating that CALHM1 is required for the control of c-Fos and C/EBPβ expression by glutamate exposure in neurons. Thus, CALHM1 activation controls PKA-mediated NMDAR and AMPAR phosphorylation and glutamate-mediated IEG expression in neurons.
Discussion
In this study, we provide the first characterization of CALHM1 function in synaptic activity and cognition in mice. Using behavioral and electrophysiological studies, we reveal that Calhm1−/− mice display significant deficits in LTP and memory flexibility. We further show that under conditions of synaptic activation, CALHM1 controls the phosphorylation by PKA of two key mediators of synaptic transmission in cerebral neurons, NMDAR and AMPAR. Moreover, we demonstrate that CALHM1 potentiates the effect of glutamate on c-Fos and C/EBPβ expression, two markers of neuronal activity. Altogether, these data identify CALHM1 as a novel regulator of neuronal signaling during synaptic activity, which is required for the proper expression of memory flexibility.
The molecular mechanisms of memory flexibility are incompletely understood. Here we show that CALHM1 is required for both memory flexibility and PKA-mediated NMDAR and AMPAR phosphorylation in cerebral neurons. Phosphorylation of NMDAR and AMPAR by PKA is a critical regulatory mechanism of these receptors during activity-dependent synaptic function. NMDAR is essential for the induction of synaptic plasticity and memory formation27, and NMDAR subunit phosphorylation controls the trafficking of this receptor and ultimately its function28. Recently, we have reported that CALHM1 channel activation in neuronal cells primarily signals through the MEK/ERK/MSK/RSK kinase signaling cascade12. A cross talk between ERK1/2 and PKA is also observed during CALHM1 activation12, suggesting that CALHM1 controls neuronal PKA. PKA targets NMDAR by phosphorylating GluN1 at Ser-89728. Recent evidence, obtained in S897A-GluN1 knock-in mice, demonstrates that this phosphorylation is required for both NMDAR- and AMPAR-mediated synaptic transmission and LTP29. These results are in line with the deficits observed in Calhm1−/− mice and suggest that the reduced levels in GluN1 phosphorylation at Ser-897 might be causally linked to the impaired LTP caused by CALHM1 deficiency. Our work therefore sheds light on a new mechanism for controlling memory flexibility.
During synaptic activity, PKA also phosphorylates the GluA1 subunit of the AMPAR at Ser-845 to influence channel function and trafficking30,31,32,33. GluA1 phosphorylation at Ser-845 has been previously linked to LTD and fear memory34,35. In our study, CALHM1 deficiency decreases GluA1 phosphorylation at Ser-845 during synaptic activation; however, Calhm1−/− slices do not show any deficit in LTD. This decoupling suggests that the effect of CALHM1 deficiency on AMPAR phosphorylation is not a central event in its effect on memory. Further studies are required to delineate the exact cross talk between NMDAR and AMPAR phosphorylation during CALHM1-dependent memory formation.
A recent study has already reported results on the effect of CALHM1 deficiency on spatial memory and learning in another Calhm1−/− mouse model11. Cognition in these mice was assessed by using a version of the Morris water maze, which only included the phase 1 of the corresponding task reported in this manuscript (see Fig. 4A). In line with our results, the authors found that CALHM1 deficiency did not affect cognition in phase 1 of the Morris water maze, see Fig. 4A and ref. 11. Memory flexibility was not investigated in this study11.
The specific effect of CALHM1 deficiency on memory malleability is of interest. Indeed, memory flexibility relates to episodic memory and its deterioration is an early and invariable manifestation of AD36,37. The molecular trigger for episodic amnesia at the early stages of AD is unknown. However, several studies have identified defects in brain activity in specific regions, such as the medial temporal and frontal lobes38,39. It will be interesting to determine whether such defects in memory encoding are also apparent in Calhm1−/− mice. This would suggest that CALHM1 loss-of-function in these brain regions might contribute to episodic amnesia at the early stages of AD pathogenesis.
How CALHM1 at the molecular level might influence AD onset is unclear. We have previously reported that CALHM1 expression in cell lines represses the accumulation of Aβ1,5,6. Two independent genetic studies have showed that a CALHM1 variant (P86L) influences Aβ levels in human cerebrospinal fluid8,9. In vitro functional studies further demonstrated that the CALHM1 P86L variant caused a partial loss of CALHM1 function by interfering with CALHM1 ion channel properties and by de-repressing its effect on Aβ accumulation1,12,13,40,41. Collectively, these results support the notion that CALHM1 might control both Aβ metabolism and AD pathogenesis. In the context of the present work, future studies will have to determine whether the observed cognitive deficits in Calhm1−/− mice are mediated, at least in part, by a deregulation in Aβ homeostasis. For example, it will be important to investigate whether CALHM1 deficiency leads to an increase in Aβ levels in brain regions affected by episodic amnesia, such as the medial temporal or frontal lobes. At the molecular level, it will also be interesting to determine whether Aβ cross talks with CALHM1 signaling to control PKA-mediated NMDAR and AMPAR phosphorylation and function during synaptic activity and memory flexibility formation.
Materials and Methods
Calhm1 knockout (KO) mice
All animal experiments were performed according to procedures approved by the Feinstein Institute for Medical Research and Monell Chemical Senses Center Institutional Animal Care and Use Committees. Calhm1+/− breeders were generated at genOway. Deletion was performed by homologous recombination in cells using the PMA1-HR targeting vector (genOway). The targeting vector was electroporated into 129Sv embryonic stem (ES) cells and 307 resistant ES cell clones were isolated, screened by PCR, and confirmed by Southern blot analysis to unambiguously confirm the 5′ and 3′ targeting events. Selected ES cell clones were injected into C57BL/6J blastocytes that were then re-implanted into OF1 pseudo-pregnant females and allowed to develop to term. Successful germline transmission was achieved and agouti pups containing the recombined allele were obtained. Wild type (WT, Calhm1+/+) and Calhm1 KO (Calhm1−/−) littermates of the F2 generation (hybrid 129Sv × C57BL/6J genetic background) were used in this study. Some Calhm1−/− hybrid mice were backcrossed with C57BL/6J mice bearing the EIIa–cre transgene (B6.FVB-Tg (EIIa-cre)C5379Lmgd/J, Jackson Lab) to remove the neomycin resistance cassette by cre-loxP-mediated excision (see schematic representation in Fig. 1A). The resulting Calhm1+/− mice were then backcrossed for 10 generations into the C57BL/6J background, before being made homozygous. The removal of the neomycin cassette was confirmed by PCR. Two age groups of mice carrying the original hybrid genetic background, young-adult (3–6-mo-old) and old (15–17-mo-old), and one group of young-adult backcrossed mice (6-mo-old), were analyzed in this study.
Chemicals and antibodies
H89 and antibodies directed against NeuN and C/EBPβ were purchased from EMD Millipore. Antibodies directed against ERK1/2, phospho-ERK1/2 (pERK1/2, Thr-202/Tyr-204), pGluN1 (Ser-897), GluN2A, GluN2B, pGluA1 (Ser-845), and c-Fos were from Cell Signaling Technology. Anti-actin antibody was from BD Transduction Laboratories. The anti-GluN1 (clone N308/48) and anti-GluA1 (N355/1) monoclonal antibodies were obtained from the UC Davis/NIH NeuroMab Facility.
Brain histochemistry
Sagittal sections (5 μm thick) of formalin-fixed paraffin-embedded brain tissue samples were Nissl-stained with cresyl violet or immunostained with anti-NeuN (1:100 dilution) and anti-GFAP (1:100) antibodies. Immunohistochemistry was performed as described before42, with the following modifications. Sagittal sections of formalin-fixed paraffin-embedded brain tissue were deparaffinized by immersion in xylene and hydration through graded ethanol solutions. Endogenous peroxidase activity was inhibited by incubation in 5% hydrogen peroxide in TBS-T for 30 min at room temperature (RT). After washing twice in TBS-T for 5 min, sections were blocked in 5% fat-free milk in TBS-T for 1 h at RT. Sections were then incubated in the presence of primary antibodies diluted in 5% fat-free milk in TBS-T overnight at 4 °C in a humidified chamber. After washing, the sections were incubated with biotin-coupled anti-mouse IgG1 secondary antibodies (1:1,000 dilution in TBS-T with 20% Superblock, Thermo Fisher Scientific) before incubation with streptavidin-horseradish peroxidase (1:1,000 dilution in 20% Superblock TBS-T, Southern Biotech) and visualization with diaminobenzidine tetrahydrochloride. For in situ hybridization, we used methods described previously16,43. In brief, 10-μm-thick coronal sections of fresh-frozen brains were fixed with 4% PFA, treated with diethylpyrocarbonate, and hybridized with antisense riboprobe at 58 °C. After hybridization, the sections were washed in 0.2 × SSC at 58 °C and incubated with alkaline phosphatase-conjugated anti-digoxigenin antibody (1:500, Roche Diagnostics). Signals were visualized with 4-nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl-phosphate for 16 h at RT. RNA probes generated were to nucleotides 96–1734 of Snap25 (GenBank accession number BC018249) and 1–3789 of Syt1 (GenBank accession number BC042519).
PSD isolation and Western blot (WB) analyses
Forebrains of Calhm1+/+ or Calhm1−/− mice were homogenized in homogenization buffer (10 mM HEPES, pH 7.4, 320 mM sucrose) containing proteases and phosphatases inhibitors (Roche Applied Science). Cells debris and nuclei were removed by 1,000 × g centrifugation. The supernatant was spun for 20 min at 12,000 × g, resulting in supernatant and P2 pellet. The latter was resuspended in a buffer containing 4 mM HEPES, 1 mM EDTA, proteases and phosphatases inhibitors, and spun for 20 min at 12,000 × g. The resulting pellet was resuspended again in HEPES buffer and spun for 20 min at 12,000 × g. The resulting pellet was resuspended in a buffer containing 20 mM HEPES, pH 7.2, 100 mM NaCl, 0.5% Triton X-100, proteases and phosphatases inhibitors, and rotated slowly for 15 min before being spun for 20 min at 12,000 × g. The supernatant was used as the non PSD fraction and the pellet was resuspended in a buffer containing 20 mM HEPES, pH 7.5, 0.15 mM NaCl, 1% Triton X-100, 1% deoxycholic acid, 1% SDS, 1 mM DTT, proteases and phosphatases inhibitors, allowed to rotate gently for 1 h and spun for 15 min at 10,000 × g. The resulting supernatant was used as the PSD fraction. For WB analyses, whole brain and PSD extracts were separated by SDS-PAGE and transferred to nitrocellulose membranes, as described before12.
Ex vivo electrophysiology
We have published these methods elsewhere17,18. Briefly, mice were anesthetized with isoflurane in a mobile anesthesia chamber, then immediately decapitated. The brain was quickly extracted and placed in ice-cold (<2 °C) artificial cerebral spinal fluid (ACSF) containing the following (mM): 126 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 2.4 CaCl2, 1.3 MgCl2, 1.2 NaH2PO4, and 1 kynurenic acid, constantly gassed with 95% O2, 5% CO2 (“carbogen”). The brain was bisected then mounted on a block with ethyl cyanoacrylate glue. Transverse hippocampal slices (400 μm) were cut on a Leica VT1200 brain slicer while bathed in ice-cold ACSF. Slices were incubated (35 °C for 35 min), then allowed to equilibrate back to 25 °C for at least 2 h. For ex vivo field recordings, slices were transferred into a recording chamber continuously perfused with 30 °C ACSF. Picrotoxin (100 μM) was added to block GABAA-mediated activity. Field excitatory postsynaptic potentials (fEPSPs) were recorded with borosilicate glass electrodes (2–3 MΩ tip resistance) placed in hippocampal area CA1’s stratum radiatum. Two bipolar Pt-Ir stimulating electrodes (Frederick Haer & Co., Bowdoinham, ME) were placed over the Schaeffer collateral/commissural axons, so that we could activate (Grass SD9 square-voltage-pulse stimulator) two independent pathways, test and control, in the same slice. To obtain baseline responses, each pathway was stimulated every 10 sec (0.1 Hz). The fEPSPs were amplified (AM Systems 1800), digitized, and stored on a PC running acquisition software (custom programs based on AxoBasic, or WinLTP v2, Bristol, UK). For the input-output functions, stimulation intensity was reduced to a value at which no fEPSP was evoked, and the stimulation was then increased incrementally to elicit fEPSPs until a population spike was detected, which defined the final point of the function. For burst analysis, a stable baseline was recorded for ~10 min and then a single HFS train (100 Hz for 1 sec) was delivered and analyzed offline (Origin v9, OriginLab) by subtracting the stimulus artifacts and integrating the total area-over-the-curve of the response. For 50 Hz trains, following a baseline period (~10 min) a single train (50 Hz for 2 sec) was delivered and responses were further recorded for at least 30 min. For LTP experiments, a stable baseline was recorded (~15 min), followed by one HFS train to induce LTP and at least 45 min of post-HFS responses. For LTD experiments, the baseline recording (~15 min) was followed by LTD induction (LFS protocol of 1 Hz for 15 min, 900 pulses total) and at least 70 min of post-LFS responses.
Morris water maze task
We have published these methods previously17. Importantly, animals were maintained in a reverse lighting schedule (lights on at 9:00AM, lights off at 9:00PM) and were assessed in all the behavioral procedures during the dark cycle. We used a water maze (diameter, 160 cm), which was filled with water (18–20 °C) and surrounded by focally illuminated distal cues that were mounted on the room walls. We used behavioral software (Ethovision v8.5, Noldus) to track and record the animal movement. Each mouse was trained to find a hidden escape platform (diameter, 10 cm) that was submerged 0.5 cm below the surface of the water. A trial was terminated when the animal located the platform, or 60 sec elapsed, in which case the mouse was guided to the platform and allowed to sit on it for ~10 sec. For phase 1, mice received 4 trials per day for 4 days with the platform located at the center of the North quadrant. For phase 2, there were 4 trials per day for 4 days with the platform at the center of the South quadrant.
For analysis, the latencies to find the platform were grouped in blocks of 4 trials each. The perseveration ratio was calculated as the latency of the last 4 trials in phase 1 (L1) subtracted from the latency of the initial 4 trials in phase 2 (L2), divided by their sum [(L2 − L1)/(L1 + L2)]. The learning score was computed as the average of the inverse-of-latency during non-novel blocks (i.e., for phase 1, blocks 2 to 4, for phase 2, blocks 6 to 8). After each phase, mice received a probe trial (duration, 60 sec) in which the platform was removed entirely. To analyze the probe trials, the pool was divided into four imaginary quadrants and the time spent in each sector was measured. The spatial memory index was defined as the mean fraction of total time in the Target quadrant during the probe trial.
Behavioral assessments
Besides the Morris water maze task, all mice were subjected to an observational screen, adapted from ref. 44 and the first stage of the SHIRPA procedure45, the rotarod task, the open field test, and fear conditioning. The tests were separated by at least 1 day and conducted during the dark cycle.
The observational screen started with anatomical parameters (coat length, hair length and hair morphology), followed by observation in a cylindrical glass flask (height 15 cm, diameter 11 cm), which measured body position, spontaneous activity, respiratory rate, tremor occurrence, defecation and urination. Transfer to an arena (55 cm × 33 cm) allowed for measuring of transfer arousal, latency to move in the arena and locomotion in the arena. This was continued with manipulations for measuring piloerection, palpebral closure, startle response, gait, pelvic elevation, tail elevation, touch escape, positional passivity, trunk curl, limb grasping, visual placing, grip strength, body tone, pinna reflex, corneal reflex, toe pinch, body length, tail length, lacrimation, whisker morphology, provoked biting, salivation, heart rate, abdominal tone, skin color and limb tone. Measuring several reflexes (wire maneuver, righting reflex, contact righting, negative geotaxis) completed the screen. Throughout the screen, incidences of fear to the experimenter, irritability, aggressivity to the experimenter, vocalizations and abnormal behavior were recorded. Finally, body weight was measured. We found that CALHM1 deficiency did not affect body weight in the young-adult mouse groups, but slightly reduced it in the old group (Fig. 3A), as recently reported46. The observed parameters were grouped according to five functional categories45, which were: muscle and spinal function; spinocerebellar function; sensory function; neuropsychiatric function; and autonomic function. The summed scores for each function were averaged across mice belonging to the same group (Calhm1+/+ or Calhm1−/−) and these were then subjected to statistical analysis.
For the rotarod test, mice were placed individually on a rotating drum (ENV-576M, Med Associates Inc, St. George, VT, USA), which accelerated from 4 to 40 rpm over a course of 5 min. The time at which the mouse fell off the drum was recorded. The test was repeated 4 times for each mouse with an interval of at least 1 h between trials. The room was illuminated with low-level white lights.
The open field test47 consisted of a single trial (20 min) in which each mouse was placed in a square chamber (40 cm on the side, 30-cm high walls painted gray) with bedding on the floor. The mouse was transported into the darkened experimental room and immediately placed in the experimental chamber. We used behavioral software (Ethovision v8.5) to track the position and movement of the animal. For analysis, a center zone (10-cm square at the center of the chamber) and a periphery zone (7-cm wide corridor adjacent to the walls) were defined (Fig. 3D); the occupancy in these zones was computed and expressed as the percent of total time. The animal’s movement was calculated at 60 sec intervals.
The fear conditioning task48 was implemented in a conditioning chamber (clear Plexiglas, dim light, metal grid floor) and a testing chamber (dark plastic, brightly lit, black floor). Video cameras were mounted on top of the chambers for videotaping and control of stimuli by software (FreezeFrame). The procedure was as follows: on the day before conditioning (day 1), mice were habituated to both chambers for 10 min in a counterbalanced manner to control for order effects. On the day of conditioning (day 2), each mouse was acclimated to the conditioning chamber (3 min) and then given five pairings of a conditional stimulus (tone, 20-sec long, 5 kHz, 80 dB) that co-terminated with an unconditional stimulus (foot shock, 1 sec, 1 mA). The inter-trial interval was 90–120 sec. On the day of testing (day 3), the freezing responses to the conditional stimulus were measured in the testing chamber with five test tones (20 sec, 5 kHz, 80 dB, 100-sec interval), starting from the first tone and lasting until 100 sec after the fifth tone. The freezing response was expressed as the percent of the total time that the animal remained frozen. After 1 h, the mice were placed in the conditioning chamber and were allowed to explore for 5 min (to give them time to recognize the context), after which the duration of freezing was scored for an additional 5 min.
Primary neuronal culture preparation and treatments
Primary neurons were prepared as described previously49. 7–9 DIV neurons were used for the different treatments. CaAB was performed as described previously1,12. Briefly, cells were incubated for 10 min in Ca2+/Mg2+-free Hank’s balanced salt solution (HBSS), supplemented with 20 mM HEPES buffer, 0.5 mM MgCl2, and 0.4 mM MgSO4. Calcium was then added back for 10 min to a final concentration of 1.8 mM. Cells were washed after the different treatments, homogenized, and analyzed by WB.
Semi-quantitative PCR
Calhm1 expression levels in brain tissue were determined by PCR following procedures described previously12.
Statistical Analysis
Datasets are presented as mean ± SEM. We used factorial ANOVA, repeated measures ANOVA, Kolmogorov-Smirnov test, and the Student t test to examine statistical significance, which was defined as P < 0.05.
Additional Information
How to cite this article: Vingtdeux, V. et al. CALHM1 deficiency impairs cerebral neuron activity and memory flexibility in mice. Sci. Rep. 6, 24250; doi: 10.1038/srep24250 (2016).
References
Dreses-Werringloer, U. et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 133, 1149–1161, doi: 10.1016/j.cell.2008.05.048 (2008).
Skrabanek, L. & Campagne, F. TissueInfo: high-throughput identification of tissue expression profiles and specificity. Nucleic Acids Res 29, E102–102 (2001).
Desgranges, B. et al. The neural substrates of episodic memory impairment in Alzheimer’s disease as revealed by FDG-PET: relationship to degree of deterioration. Brain 125, 1116–1124 (2002).
Lambert, J. C. et al. The CALHM1 P86L polymorphism is a genetic modifier of age at onset in Alzheimer’s disease: a meta-analysis study. J Alzheimers Dis 22, 247–255, doi: 10.3233/JAD-2010-100933 (2010).
Vingtdeux, V. et al. CALHM1 ion channel elicits amyloid-β clearance by insulin-degrading enzyme in cell lines and in vivo in the mouse brain. J Cell Sci 128, 2330–2338, doi: 10.1242/jcs.167270 (2015).
Vingtdeux, V. et al. Effect of the CALHM1 G330D and R154H Human Variants on the Control of Cytosolic Ca2+ and Aβ Levels. PLos One 9, e112484, doi: 10.1371/journal.pone.0112484 (2014).
Selkoe, D. J. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81, 741–766 (2001).
Kauwe, J. S. et al. Validating predicted biological effects of Alzheimer’s disease associated SNPs using CSF biomarker levels. J Alzheimers Dis 21, 833–842, doi: 10.3233/JAD-2010-091711 (2010).
Koppel, J. et al. CALHM1 P86L polymorphism modulates CSF Aβ levels in cognitively healthy individuals at risk for Alzheimer’s disease. Mol Med 17, 974–979, doi: 10.2119/molmed.2011.00154 (2011).
Giedraitis, V. et al. CALHM1 P86L polymorphism does not alter amyloid-beta or tau in cerebrospinal fluid. Neurosci Lett 469, 265–267, doi: 10.1016/j.neulet.2009.12.011 (2010).
Wu, J. et al. Generation of Calhm1 knockout mouse and characterization of calhm1 gene expression. Protein Cell 3, 470–480, doi: 10.1007/s13238-012-2932-6 (2012).
Dreses-Werringloer, U. et al. CALHM1 controls the Ca2+-dependent MEK, ERK, RSK and MSK signaling cascade in neurons. J Cell Sci 126, 1199–1206, doi: 10.1242/jcs.117135 (2013).
Ma, Z. et al. Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc Natl Acad Sci USA 109, E1963–1971, doi: 10.1073/pnas.1204023109 (2012).
Tanis, J. E. et al. CLHM-1 is a functionally conserved and conditionally toxic Ca2+-permeable ion channel in Caenorhabditis elegans. J Neurosci 33, 12275–12286, doi: 10.1523/JNEUROSCI.5919-12.2013 (2013).
Rusakov, D. A. & Fine, A. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron 37, 287–297 (2003).
Taruno, A. et al. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature 495, 223–226, doi: 10.1038/nature11906 (2013).
Chang, E. H., Rigotti, A. & Huerta, P. T. Age-related influence of the HDL receptor SR-BI on synaptic plasticity and cognition. Neurobiol Aging 30, 407–419, doi: 10.1016/j.neurobiolaging.2007.07.006 (2009).
Chang, E. H. et al. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci USA 103, 3410–3415, doi: 10.1073/pnas.0507313103 (2006).
Bliss, T. V., Collingridge, G. L. & Morris, R. G. The Hippocampus Book (Chapter 10). 343–474 (Oxford Univ. Press, New York, 2007).
Greer, P. L. & Greenberg, M. E. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860, doi: 10.1016/j.neuron.2008.09.002 (2008).
Wiegert, J. S. & Bading, H. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 49, 296–305, doi: 10.1016/j.ceca.2010.11.009 (2011).
Alberini, C. M. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev 89, 121–145, doi: 10.1152/physrev.00017.2008 (2009).
Scott, D. B., Blanpied, T. A. & Ehlers, M. D. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 45, 755–767 (2003).
Tingley, W. G. et al. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem 272, 5157–5166 (1997).
Roche, K. W., O’Brien, R. J., Mammen, A. L., Bernhardt, J. & Huganir, R. L. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16, 1179–1188 (1996).
Middei, S., Ammassari-Teule, M. & Marie, H. Synaptic plasticity under learning challenge. Neurobiol Learn Mem, doi: 10.1016/j.nlm.2014.08.001 (2014).
Malinow, R. & Malenka, R. C. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25, 103–126, doi: 10.1146/annurev.neuro.25.112701.142758 (2002).
Chen, B. S. & Roche, K. W. Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53, 362–368, doi: 10.1016/j.neuropharm.2007.05.018 (2007).
Li, B. et al. NMDA receptor phosphorylation at a site affected in schizophrenia controls synaptic and behavioral plasticity. J Neurosci 29, 11965–11972, doi: 10.1523/JNEUROSCI.2109-09.2009 (2009).
Goel, A. et al. Phosphorylation of AMPA receptors is required for sensory deprivation-induced homeostatic synaptic plasticity. PLos One 6, e18264, doi: 10.1371/journal.pone.0018264 (2011).
Esteban, J. A. et al. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6, 136–143, doi: 10.1038/nn997 (2003).
He, K. et al. Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1-S845 phosphorylation. Proc Natl Acad Sci USA 106, 20033–20038, doi: 10.1073/pnas.0910338106 (2009).
Oh, M. C., Derkach, V. A., Guire, E. S. & Soderling, T. R. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem 281, 752–758, doi: 10.1074/jbc.M509677200 (2006).
Clem, R. L. & Huganir, R. L. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 330, 1108–1112, doi: 10.1126/science.1195298 (2010).
Lee, H. K., Takamiya, K., He, K., Song, L. & Huganir, R. L. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol 103, 479–489, doi: 10.1152/jn.00835.2009 (2010).
Albert, M. S. et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 270–279, doi: 10.1016/j.jalz.2011.03.008 (2011).
McKhann, G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 263–269, doi: 10.1016/j.jalz.2011.03.005 (2011).
Kopelman, M. D. Disorders of memory. Brain 125, 2152–2190 (2002).
Dubois, B. et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 9, 1118–1127, doi: 10.1016/S1474-4422(10)70223-4 (2010).
Rubio-Moscardo, F. et al. Rare variants in calcium homeostasis modulator 1 (CALHM1) found in early onset Alzheimer’s disease patients alter calcium homeostasis. PLos One 8, e74203, doi: 10.1371/journal.pone.0074203 (2013).
Moreno-Ortega, A. J., Ruiz-Nuño, A., García, A. G. & Cano-Abad, M. F. Mitochondria sense with different kinetics the calcium entering into HeLa cells through calcium channels CALHM1 and mutated P86L-CALHM1. Biochem Biophys Res Commun 391, 722–726, doi: 10.1016/j.bbrc.2009.11.127 (2010).
Vingtdeux, V., Davies, P., Dickson, D. W. & Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol 121, 337–349, doi: 10.1007/s00401-010-0759-x (2011).
Ohmoto, M., Matsumoto, I., Misaka, T. & Abe, K. Taste receptor cells express voltage-dependent potassium channels in a cell age-specific manner. Chem Senses 31, 739–746, doi: 10.1093/chemse/bjl016 (2006).
Irwin, S. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia 13, 222–257 (1968).
Rogers, D. C. et al. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome 8, 711–713 (1997).
Hellekant, G., Schmolling, J., Marambaud, P. & Rose-Hellekant, T. A. CALHM1 Deletion in Mice Affects Glossopharyngeal Taste Responses, Food Intake, Body Weight, and Life Span. Chem Senses 40, 373–379, doi: 10.1093/chemse/bjv009 (2015).
Chang, E. H. & Huerta, P. T. Neurophysiological correlates of object recognition in the dorsal subiculum. Front Behav Neurosci 6, 46, doi: 10.3389/fnbeh.2012.00046 (2012).
Huerta, P. T., Kowal, C., DeGiorgio, L. A., Volpe, B. T. & Diamond, B. Immunity and behavior: antibodies alter emotion. Proc Natl Acad Sci USA 103, 678–683, doi: 10.1073/pnas.0510055103 (2006).
Vingtdeux, V. et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem 285, 9100–9113, doi: 10.1074/jbc.M109.060061 (2010).
Acknowledgements
We thank Tomás S. Huerta, Toby Klein, Jonas Oppenheimer, and Sergio Robbiati for experimental help. This work was supported by the National Institute of Health grants R01AG042508 (to PM) and P01AI073693-06 (to PTH), and Monell Chemical Senses Center Institutional Fund (to IM). Parts of the in situ hybridization analyses were performed at the Monell Histology and Cellular Localization Core, which is supported, in part, by funding from NIH Core Grant P30DC011735 (to Robert F. Margolskee, Monell Chemical Senses Center). MO was a Japan Society for the Promotion of Science Postdoctoral Fellow for Research Abroad.
Author information
Authors and Affiliations
Contributions
V.V., E.H.C., S.A.F., M.O., I.M., P.T.H. and P.M. designed and analyzed experiments. V.V., E.H.C., S.A.F., M.O., I.M., H.Z., P.C., L.A., J.J.S. and E.L.G. performed experiments. V.V., E.H.C., I.M., P.T.H. and P.M. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Vingtdeux, V., Chang, E., Frattini, S. et al. CALHM1 deficiency impairs cerebral neuron activity and memory flexibility in mice. Sci Rep 6, 24250 (2016). https://doi.org/10.1038/srep24250
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep24250
This article is cited by
-
Human CALHM5: Insight in large pore lipid gating ATP channel and associated neurological pathologies
Molecular and Cellular Biochemistry (2021)
-
Proteomic and Transcriptomic Analyses Reveal Pathological Changes in the Entorhinal Cortex Region that Correlate Well with Dysregulation of Ion Transport in Patients with Alzheimer’s Disease
Molecular Neurobiology (2021)
-
Structure and assembly of calcium homeostasis modulator proteins
Nature Structural & Molecular Biology (2020)
-
CALHM1/CALHM3 channel is intrinsically sorted to the basolateral membrane of epithelial cells including taste cells
Scientific Reports (2019)
-
Calhm2 governs astrocytic ATP releasing in the development of depression-like behaviors
Molecular Psychiatry (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
