




Abstract
Several treatment strategies target the human epidermal growth factor receptor 2 (HER2) in breast carcinomas, including monoclonal antibodies directed against HER2’s extracellular domain (ECD) and small molecule inhibitors of its tyrosine kinase activity. Yet, novel therapies are needed that prevent HER2 dimerization with other HER family members, because current treatments are only partially effective.
To test the hypothesis that HER2 activation requires a protein sequence in the HER2-ECD that mediates HER2 homo- and heterodimerization, we introduced a series of deletion mutations in the third subdomain of HER2-ECD. These deletion mutants were retrovirally expressed in breast cancer (BC) cells that naturally overexpress HER2 and in noncancerous, HER2-negative breast epithelial cells. One-factor analysis of variance or Student’s t test were used to analyze differences. All statistical tests were two-sided.
The smallest deletion in the ECD domain of HER2, which removed only 16 amino acids (HER2-ECDΔ451–466), completely disrupted the oncogenic potential of HER2. In contrast to wild-type HER2, the mutant-inhibited anchorage-independent growth (mean number of colonies: mutant, 70, 95% confidence interval [CI] = 55 to 85; wild-type, 400, 95% CI = 320 to 480, P < .001) increased sensitivity to paclitaxel treatment in both transformed and nontransformed cells. Overexpression of HER2Δ451–466 efficiently inhibited activation of HER1, HER2, and HER3 in all cell lines tested.
These findings reveal that an essential “activating” sequence exists in the extracellular domain of HER2. Disruption of this sequence disables the HER2 dimerization loop, blocks subsequent activation of HER2-driven oncogenic signaling, and generates a dominant-negative form of HER2. Reagents specifically against this molecular activation switch may represent a novel targeted approach for the management of HER2-overexpressing carcinomas.
A growing number of anticancer agents have emerged as a result of understanding the mechanisms underlying malignant transformation and metastatic potential. High levels of human epidermal growth factor receptor 2 (HER2) transform cultured cells and clinical studies have shown poorer long-term survival rates for patients whose tumors overexpress HER2, implying that HER2 is a suitable therapeutic target (1–4). Although various approaches have been developed for the treatment of HER2-overexpressing carcinomas, the most prominent strategy involves antibody targeting of its extracellular domain (ECD) (4–8).
Trastuzumab, a monoclonal antibody that binds to the juxtamembrane region of HER2 and inhibits ligand-independent HER2-HER3 heterodimerization (9), has demonstrated clinical activity in a subset of HER2-overexpressing breast cancer patients (10–12). However, not all HER2-overexpressing breast carcinomas respond to treatment with trastuzumab, and its clinical benefit is limited (13–15). Additionally, HER2 activation via ligand-mediated receptor heterodimerization is not affected by trastuzumab (16–20). The monoclonal antibody pertuzumab, which sterically blocks the association between HER2 and HER3, inhibits ligand-activated HER2 signaling and cell growth, regardless of the expression level of HER2 (21–24). An alternative antireceptor strategy involves the development of small-molecule inhibitors that compete with ATP for the ATP-binding domain in the intracellular portion of HER2, interrupting HER2 and HER1 signaling pathways (25), or antibody-drug conjugates such as TDM-1 that allow for the selective delivery of small molecule inhibitors to HER2-overexpressing cells (26). Although these HER-tyrosine kinase inhibitors are therapeutically promising, their clinical responses in HER2-positive breast cancer patients are generally short-lived (27–31). Therefore, novel therapeutic approaches are needed.
While ligand binding stabilizes heterodimers, leading to active signaling (32–34), structural alignment studies demonstrate that subdomain III of HER1, HER2, and HER3 appears to be essential for dimerization (35). As HER2 is the preferred dimerization partner for HER1 and HER3 (36–38) because of its unique conformation, blocking its dimerization capability might have pleiotropic effects that are detrimental to tumor growth. We hypothesized that HER2 activation occurs through a “functional site,” a protein sequence in the HER2-ECD that is ultimately responsible for HER2 homo- and heterodimerization (21,39–41). Consequently, by blocking this essential region, we predicted that HER2 would fail to act either as a substrate for transphosphorylation or as a catalyst for the transmission of mitogenic and/or prosurvival signaling in the absence or presence of HER ligands.
Methods
Generation of HER2-ECD Deletion Mutants
Deletion mutations within subdomain III of the HER2-ECD were generated by a two-step polymerase chain reaction (PCR) method using a 3938-bp wild-type (wt) HER2 cDNA as the starting template. This 3938-bp insert corresponds to nucleotides 34–3972 of the human HER2 cDNA deposited with GenBank (accession #M11730). Further details are provided in the Supplementary Methods (available online).
Generation of MCF10A Cells Expressing HER2-ECD Deletions
Retroviral stocks were generated by cotransfection of the vector plasmid DNA with a packaging plasmid into a high-efficiency transient amphotropic packaging system (TSA54 cell line) using the FuGENE 6 transfection reagent (Roche, Indianapolis, IN), as per manufacturer’s instructions. Medium containing infectious retroviruses was collected from transfected cells after 48 hours, filtered, and stored at -80 oC until utilization. Further details are provided in the Supplementary Methods (available online).
Immunoblotting Analyses
For assaying levels of protein expression and the phosphorylation status of HER1, HER2, HER3, ERK1/2, and AKT, cells were cultured in 24-well dishes. Cells were then washed with cold PBS, placed on ice, and lysed in nondenaturing 1X lysis buffer (Cell Signaling, Beverly, MA) containing 20mM Tris-HCl, 150mM NaCl, 1mM EDTA, 1mM EGTA, 1% Triton X-100, 2.5mM sodium pyrophosphate, 1mM β-glycerophosphate, 1mM Na3VO4, 1 µg/mL leupeptin, and 1mM PMSF. Cells were scraped, added to Eppendorf tubes, and incubated on ice for 20 minutes before debris was removed by a 15-min spin at 14 000rpm at 4o C. A BCA protein reagent kit (Pierce, Rockford, IL) was used to determine total protein levels. Further details are provided in the Supplementary Methods (available online).
Metabolic Status Assessment
Cell viability was determined using a modified MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reduction assay (Cell Titer 96 Aqueous Non-Radioactive Cell Proliferation Assay, Promega Corp.). Briefly, cells in exponential growth were harvested by trypsinization and seeded at a concentration of 3 x 103 cells/200 µl/well into 96-well plates and allowed an overnight period for attachment. The cells were then washed twice with prewarmed PBS and cultured in serum-free medium overnight. The medium was then removed, and fresh medium in the absence or presence of EGF or graded concentrations of paclitaxel (Sigma Chemicals) was added to the cultures as specified. Further details are provided in the Supplementary Methods (available online).
Other Methods
Descriptions of cell lines and cell culture conditions, soft agar colony formation assays, cell cycle analysis, phosphoproteome profiling, surface biotinylation, immunofluorescence, and image acquisition are described in the Supplementary Methods (available online).
Statistical Analysis
One-factor analysis of variance (ANOVA) (using IBM SPSS version 21.0, IBM Corp. 2012, and R software environment, http://www.r-project.org) was used to analyze differences, with the exception of the soft agar assays comparing the mutant with the current treatments of choice where the data were analyzed using Excel 2007 (Microsoft) and Student’s t test. Data shown are the mean and 95% confidence intervals. All statistical tests were two-sided.
Results
Design and Expression of ECD Deletion Derivatives of HER2
The ECD of HER2 has been organized into a four-domain model, in which subdomain III contributes many of the determinants involved in signal transduction because it strongly interacts with subdomain I, producing a constitutively activated dimerization loop (39–44). Therefore, we introduced a series of deletion mutations into subdomain III in an attempt to disable the dimerization loop and subsequently the activation of the receptor. A series of six overlapping deletion mutants in the ECD of HER2 (Figure 1A) were generated and overexpressed in MCF10A cells. We chose this cell line for the initial signaling analyses to avoid cross-talk with endogenous HER2 and to obtain a clear picture of the dimerization/signaling capacities of the various mutants.
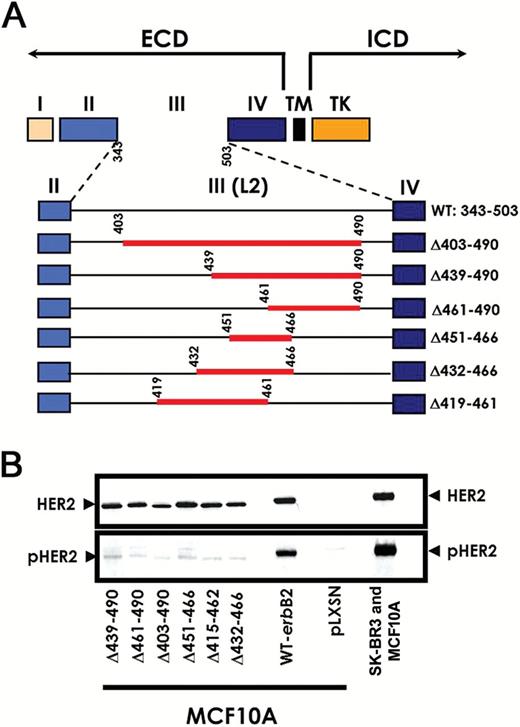
Generation and expression of human epidermal growth factor receptor 2 (HER2)–extracellular domain (ECD) deletion mutants in MCF10A breast epithelial cells. A) Schematic representation of the wild-type HER2 receptor and HER2-ECD deletion mutants. Subdomains II and IV are cysteine cluster domains. Subdomains I and III are putative ligand or dimerization domains. Deletion mutants of the HER2 receptor were generated in the region of subdomain III, a putative functional site. The deleted sequences are indicated as a red line, and the numbers indicate the exact regions that were deleted for each mutant. ECD = extracellular domain; ICD = intracellular domain; TK = tyrosine kinase domain; TM = transmembrane domain. B) Effects of HER2-ECD structural deletions on HER2 autophosphorylation. Twenty μg of total protein from SK-BR3 cells and MCF10A cells (normally HER2-negative) engineered to stably express wt-HER2 (wt-erbB-2), HER2-ECD deletion mutants, or the empty vector pLXSN were subjected to immunoblotting using anti-HER2 and anti-phospho-HER2 (pHER2) antibodies. Shown is an immunoblot representative of three independent experiments.
Effects of Deletions in Subdomain III of the HER2-ECD on Receptor Dimerization and Activation
Expression of wt-HER2, HER2-ECD deletions, or vector control in MCF10A cells was assessed by immunoblotting and demonstrated decreased HER2 phosphorylation/activation of the mutants relative to the constitutively active wt-HER2 or HER2-overexpressing SK-BR3 cells (Figure 1B). As there are no known ligands for HER2, HER2 is phosphorylated either by homodimerization after overexpression or by ligand induced heterodimerization. Therefore, we chose the shortest deletion mutant (HER2-ECDΔ451–466) to analyze the phosphorylation profiles of HER1-3 in MCF10A and SK-BR3 cells. We chose to compare these two cell lines to determine if: 1) HER2 homo- and heterodimerization are affected equally by the HER2-ECD-Δ451–466 mutant, and 2) this particular mutant acts as a dominant-negative. Moreover, MCF10A cells provide a useful in vitro system to analyze the transforming effects of overexpressed HER2 (45,46) as they are dependent on EGF and express only very low levels of HER1, HER2, and HER3. Hence, overexpressing wt-HER2 in MCF10A recapitulates the common clinical HER1 and HER2 phenotype and, upon EGF stimulation, the paracrine/autocrine stimulation of HER receptor–driven signaling.
Indeed, our analysis showed that phosphorylation of the analyzed HER family members was inhibited in both cell lines tested, suggesting impairment of HER2 homo- and heterodimerization upon expression of HER2-ECD-Δ451-466 (Figure 2, A-D). As it is known that HER2 overexpression promotes activation of HER1 in a ligand-independent manner (36,47–50) and HER1 and HER2 are cooverexpressed in breast carcinomas with the worst prognosis (51), our experimental model clearly recapitulates this complex cross-talk between the two receptors because prominent pHER1 was detected in SK-BR3/pBABE and MCF10A/wt-HER2 cells but substantially reduced in the ECD mutant-expressing cells (Figure 2, A and B). Upon ligand binding, HER1 can form heterodimeric complexes with neighboring HER2 receptors and transactivate HER2. Thus, our findings also reveal that HER2-ECD-Δ451–466 impairs the ability of EGF to initiate ligand-activated signaling from HER1/HER2 heterodimers (Figure 2B).
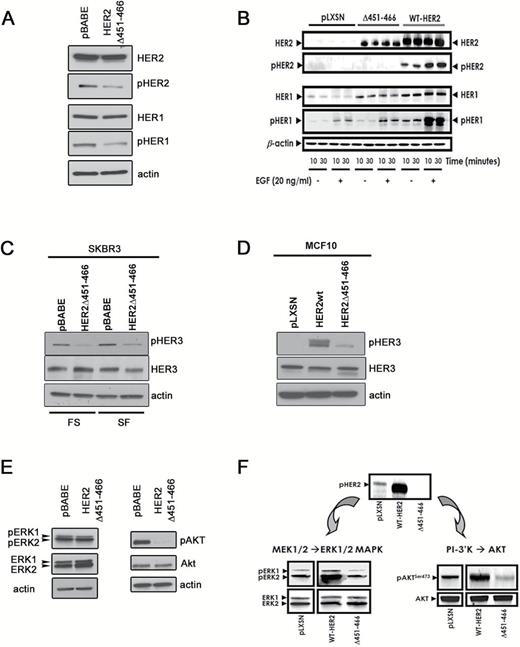
Deletion of the human epidermal growth factor receptor 2 (HER2) dimerization site and HER2-driven signaling. A) Overexpression of HER2-ECD-Δ451–466 interferes with HER1/HER2 crosstalk and acts as a dominant-negative in SK-BR3 cells. Immunoblot analysis of the pTyr profiles of HER1 and HER2 under serum-free (SF) and full serum conditions. β-actin was used as a loading control. Shown is an immunoblot representative of three independent experiments. B) Overexpression of HER2-ECD-Δ451–466 interferes with HER1/HER2 crosstalk in MCF10A cells. Following serum starvation, MCF10A/wt-HER2, MCF10A/HER2-ECD-Δ451–466, and MCF10A/pLXSN control cells were treated with EGF for 10 or 30 minutes. Fifty μg of total protein were subjected to immunoblotting analysis for HER2, pHER2, HER1, and pHER1; β-actin was used as a loading control. Shown is an immunoblot representative of three independent experiments. C and D) Immunoblot analysis of HER3 transactivation in the presence of HER2wt or the HER2-ECD-Δ451–466 mutant in SK-BR3 (C) and MCF10 (D) cells under full serum (SK-BR3 only) and SF conditions (both cell lines), compared with vector controls. Note that for both cells lines, activation of HER3, assessed by its phosphorylation, is dramatically reduced in the cells overexpressing the mutant compared with the HER2wt-expressing cells. C and D) Overexpression of HER2-ECD-Δ451–466 inhibits signaling from downstream effectors in both SK-BR3 (E) and MCF10A (F) cells. Fifty μg of total protein from each cell line stably expressing the empty vector pLXSN, wt-HER2, or HER2-ECD-Δ451–466, as indicated, were subjected to immunoblotting analysis with phospho-specific antibodies for pERK and pAKt, as well as ERK/Akt as loading controls. Shown is an immunoblot representative of three independent experiments for each cell line.
In light of these results, we analyzed the effects of HER2-ECD-Δ451–466 on activation of the HER network in cells expressing physiological levels of HER2. To this end, MCF-7 cells were stably transfected with wt-HER2 (MCF-7/HER2-18 cells) and compared with control-transfected cells (MCF-7/neo cells, which express a neomycin phosphotransferase gene [52]). Phospho-RTK profiling of MCF-7/HER2-18 cells demonstrated that overexpression of wt-HER2 induces HER2 autophosphorylation and remarkably high transactivation of HER1 and HER3. Importantly, MCF-7/HER2-18 cells expressing high levels of HER2-ECD-Δ451–466 displayed a major reduction in tyrosine hyperphosphorylation, not only of HER2 but also of HER1 and HER3 (Supplementary Figure 1, available online).
Taken together, these data clearly demonstrate that manipulation of the HER2 subdomain III prevents HER2 homo- and heterodimerization, inhibits receptor transactivation, and attenuates its oncogenic potential.
HER2-ECD-Δ451–466 as a Dominant-Negative Form of HER2
As our data further suggested that disruption of subdomain III in the HER2-ECD could have a dominant-negative effect, we explored this hypothesis by examining the steady-state levels of the downstream ERK1/2 and PI-3K/AKT growth/survival signaling cascades in SK-BR3 and MCF10A cells (Figure 2, E and F). We predicted that a reduction of HER2 activation in HER2-ECD-∆451-466 expressing cells would also reduce signaling through downstream effectors. Indeed, ERK1/2 activation was reduced in mutant compared with wt-expressing cells. The dominant-negative effect became even more evident upon analysis of the steady-state levels of activated AKT, which were completely repressed in both cell lines harboring the HER2-ECD-∆451-466 mutant (Figure 2, E and F).
While HER1 and HER2 have only a weak ability to bind the p85 adaptor subunit of PI-3K, a critical upstream component of the AKT transduction cascade, the HER2/HER3 dimer provides six docking sites for this subunit, making it one of the most potent activators of the PI-3K/AKT pathway (37,38,53). Therefore, our findings suggest that HER2-ECD-Δ451–466 profoundly affects the ability of HER2 to cross-talk with activated HER1 and, moreover, impairs ligand-induced transactivation of HER2 by HER3. Thus, this 16-amino-acid deletion in subdomain III of the HER2-ECD appears to be sufficient to block HER2 dimerization and subsequently activation of HER2-driven oncogenic signaling cascades.
Based on the above findings, we explored the biological consequences of HER2-ECD-Δ451–466 expression in SK-BR3 breast cancer cells, a widely used in vitro model of HER2 gene amplification, HER2 overexpression, and dependence (54). Stable coexpression of HER2-ECD-Δ451–466 statistically significantly blocked HER2 activation/signaling and inhibited the ability of the cells to form colonies in an anchorage-independent growth assay (mean number of colonies: mutant, 70, 95% confidence interval [CI] = 55 to 85; wild-type, 400, 95% CI = 320 to 480; P < .001) (Figure 3A). Importantly, expression of the HER2-ECD-Δ451-466 mutant also sensitized the cells to chemotherapy as the IC50 value for paclitaxel, a commonly used drug for breast cancer treatment, decreased statistically significantly relative to control cells (mean IC50 in
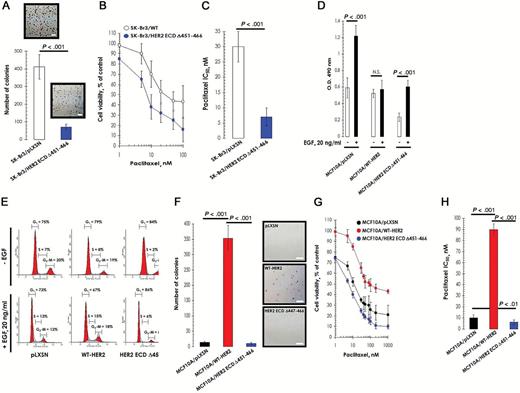
Overexpression of human epidermal growth factor receptor 2 (HER2)–ECD-Δ451–466 and canonical oncogenic properties of HER2. A) Anchorage-independent phenotype of HER2-overexpressing SK-BR3 breast cancer cells. SK-BR3 cells, naturally bearing HER2 gene amplification and exhibiting HER2 overexpression and activation, were engineered to stably express HER2-ECD-Δ451–466 (SK-BR3/HER2-ECD-Δ451–466 cells) or the empty vector pLXSN (SK-BR3/pLXSN control cells). SK-BR3/HER2-ECD-Δ451–466 cells and SK-BR3/pLXSN cells were seeded in soft agar and incubated for 14 days. Colonies were then counted as described in the Methods. Histogram values represent the mean colony number, and error bars are 95% confidence intervals from three separate experiments in triplicate. P < .001 for SK-BR3/pLXSN cells vs SK-BR3/HER2-ECD-Δ451–466 cells. Scale bar = 100 μm. B) Chemotherapy-induced cell damage in SK-BR3 cells. SK-BR3/pLXSN and SK-BR3/HER2-ECD-Δ451–466 cells were cultured in the absence or presence of paclitaxel, and cell viability was determined by MTT assay. Dose-response curves were plotted as percentages of the control cells’ absorbance (=100%), which was obtained from cells treated with appropriate concentrations of paclitaxel vehicle. Data are means, and error bars are 95% confidence intervals. C) Quantitation of three independent experiments as shown in (B). Histogram values represent the mean IC50 value, and error bars are 95% confidence intervals from three separate experiments in triplicate. P < .001 for SK-BR3/pLXSN cells vs SK-BR3/ HER2-ECD-Δ451–466 cells. D) Inhibition of EGF-promoted metabolic activity upon deletion of the HER2 dimerization domain. MCF10A/WT-HER2, MCF10A/HER2-ECD-Δ451–466, and MCF10/pLXSN cells were cultured in the presence or absence of EGF for 5 days, exposed to MTT reagent, and the absorbance at 490nm was measured. Data are presented as means, and error bars are 95% confidence intervals of four independent experiments performed in triplicate. P < .001 compared with control untreated group. E) Overexpression of HER2-Δ451–466 causes G1 arrest in EGF-stimulated MCF10A cells. After serum starvation and refeeding, MCF10A/wt-HER2, MCF10A/HER2-ECD-Δ451–466, and MCF10/pLXSN cells were exposed to EGF (20ng/mL) for 24 hours. Panels show representative flow cytometry profiles of the distribution of cells in different phases of the cell cycle. Standard deviation for percentages in each experimental condition and/or cell cycle phase was less than 2% in three independent experiments. F) Deletion of the HER2 dimerization regions blocks anchorage-independent growth of MCF10A cells. MCF10A/wt-HER2, MCF10A/HER2-ECD-Δ451–466, and MCF10/pLXSN cells were seeded in soft agar, and the number of colonies was assessed after 14 days in medium supplemented with EGF. Histogram values represent the mean colony number, and error bars are 95% confidence intervals from three separate experiments in triplicate. Representative images for each clone are shown on the right. Scale bar = 100 μm. G) Interfering with HER2 dimerization reduces cell viability. Cells were cultured in the absence or presence of paclitaxel, and cell viability was determined by MTT assay as shown in (B). Data are means, and error bars are 95% confidence intervals. H) Quantitation of three separate experiments as described in (G). Histogram values represent the mean IC50 value, and error bars are 95% confidence intervals from three separate experiments in triplicate. P values from one-factor analysis of variance are shown above each column. All statistical tests were two-sided.
HER2wt expressing cells: 30nM, 95% CI = 25 to 35nM; mutant expressing cells: 7nM, 95% CI = 4 to 10nM in, P < .001) (Figure 3, B and C).
Effects of HER2-ECD-Δ451–466 on HER2-Driven Independence of EGF and Cell Cycle Arrest
The biological consequences of the HER2-ECD-Δ451–466 mutant in HER2-driven malignancy were further investigated by evaluating its transforming capability in normal breast epithelial cells (44,45). First, we assessed the effect of the mutant on the ability to subvert EGF dependence in MCF10A cells. As expected, MCF10A/pLSXN control cells were statistically significantly stimulated by EGF, as determined by measurement of metabolic status using the MTT assay, whereas MCF10A cells overexpressing HER2-wt were unaffected (Figure 3D). Interestingly, this HER2-promoted EGF independence was disrupted in the mutant-expressing cells. Remarkably, the absorbance reading in the mutant-expressing cells observed in the absence of EGF was statistically significantly lower than in matched control MCF-10A/pLXSN cells (mean OD490nm in vector control, without EGF: 0.592, 95% CI = 0.472 to 0.712; mutant: 0.236, 95% CI = 0.186 to 0.286; P < .001) (Figure 3D), further supporting the fact that deletion of aa 451–466 in the HER2-ECD promotes a dominant-negative phenotype.
To determine the effects of the mutant on EGF-promoted cell cycle progression, MCF10A/pLXSN, MCF10A/wt-HER2, and MCF10A/HER2-ECD-Δ451–466 cells were incubated in the absence or presence of EGF for 24 hours, and the distribution of cells in different phases of the cell cycle was assessed using flow cytometry (Figure 3E). In the absence of EGF, MCF10A/HER2-ECD-Δ451–466 cells showed a greater-than 70% decrease in the percentage of cells undergoing S phase, which was accompanied by an accumulation of cells in the G1 phase (Figure 3E, top panels). A G0/G1 cell cycle block promoted by HER2-ECD-Δ451–466 was even more evident upon EGF exposure. As shown in Figure 3E (lower panels), EGF reduced the fraction of cells in S phase to approximately 6% in MCF10A/HER2-ECD-Δ451–466 cells, but increased the number of S-phase MCF10A/pLXSN and MCF10A/wt-HER2 cells approximately two-fold. Conversely, 86% of MCF10A/HER2-ECD-Δ451–466 cells accumulated in G1 phase following EGF treatment, while this percentage decreased to 73% and 67% in MCF10A/pLXSN and MCF10A/wt-HER2 cells, respectively (Figure 3E, bottom panels).
Effect of HER2-ECD-Δ451–466 on Transformed and Chemoresistant Phenotypes in Breast Epithelial Cells
Similar to other transforming proteins, HER2 expression induces a more malignant phenotype in MCF10A cells. It is well established that MCF10A cells grow in an anchorage-dependent manner in the absence of oncogenic stimuli, and any colony formation observed in soft agar represents the background level for the colony formation experiment. Therefore, we examined the behavior of MCF10/wt-HER2 and MCF10/HER2-ECD-Δ451–466 in a soft-agar colony formation assay to measure the effects on canonical oncogenic properties. As expected, we observed that wt-HER2 conferred anchorage independence when overexpressed in MCF10A cells (Figure 3F). Strikingly, cells stably expressing the HER2 deletion mutant failed to form colonies in soft agar, similar to MCF10A/pLXSN control cells. Again, these findings suggest that the dimerization of HER2 is indispensable for its oncogenic/transforming capacity.
Previous reports indicated that breast epithelial cells engineered to overexpress HER2 show remarkable differences in their cytotoxic response to chemotherapeutic agents (55–57). Therefore, we also tested the sensitivity of MCF10A/pLXSN, MCF10A/wt-HER2, and MCF10A/HER2-ECD-Δ451–466 cells to paclitaxel. Indeed, HER2 overexpression promoted resistance to paclitaxel, with an approximately nine-fold higher IC50 value than matched control MCF10A/pLXSN cells (Figure 3, G and H). However, this response was lost in HER2-ECD-Δ451–466–expressing cells, with an IC50 value that was even lower than equivalent control cells (Figure 3, G and H). Collectively, these findings strongly suggest that HER2-ECD-Δ451–466 hinders HER2-induced in vitro transformed and chemoresistant phenotypes in MCF10A breast epithelial cells.
In summary, HER2-ECD-Δ451–466 markedly impaired HER2 autophosphorylation while inhibiting the recruitment of activated HER2 into HER2/HER1 and HER2/HER3 heterodimers in breast cancer cells that naturally or ectopically overexpress HER2.
Mechanisms of HER2Δ6-Mediated Inhibition of the HER2 Network
The profound inhibitory effects of the HER2-ECD-Δ451–466 mutant tempted us to speculate that this dominant-negative form may be more efficient in its anti-oncogenic properties than drugs currently in clinical use. We tested this hypothesis by performing anchorage-independent growth assays directly comparing the effects of the mutant to a dose response of trastuzumab (3–300 µg/mL), cetuximab (10–500 µg/mL), and pertuzumab (2.5–250 µg/mL). As shown in Figure 4A, overexpression of the HER2 deletion mutant caused a statistically significant decrease (mean colony number in vector control cells: 1186, 95% CI = 900 to 1472; in mutant expressing cells: 192, 95 % CI = 5 to 380; P < .001) in anchorage independent growth in SK-BR3 cells, which was not achieved by any of the drugs tested.
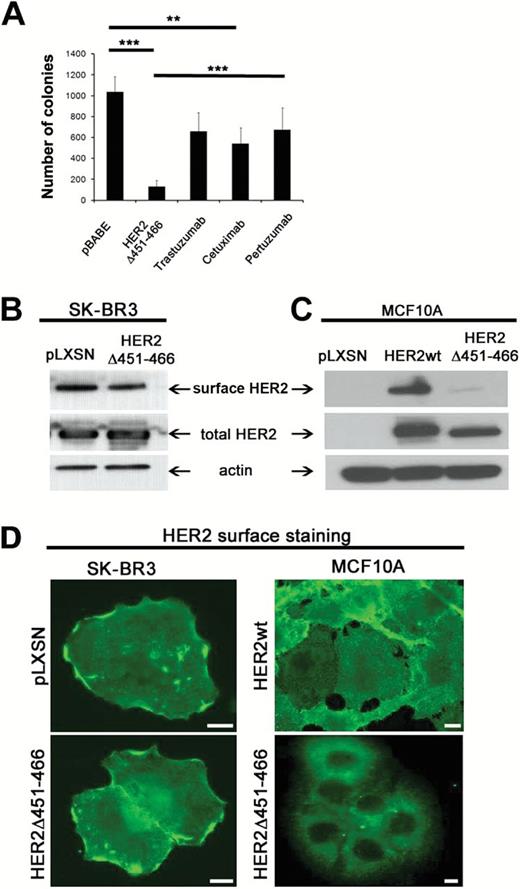
HER-ECD-D451-466 impairs anchorage-independent growth and receptor localization. A) Deletion of the “activating sequence” in HER2 is more effective in interfering with its pro-oncogenic properties than drugs that are currently in clinical use, as assessed by anchorage-independent growth in soft agar. Drug concentrations shown: trastuzumab 30 µg/mL, cetuximab 100 µg/mL, and pertuzumab 25 µg/mL. Data are presented as mean ± SD from three independent experiments, P values (mutant vs drugs or vector control): *** < .002 and ** < .01; Trz treatment was not statistically significant from control. Student’s t test. All statistical tests were two-sided. B and C) Evaluation of HER2 surface levels in SK-BR3 (B) and MCF10A (C) cells assessed by a surface biotinylation assay. Note that HER2 surface association is not affected in SK-BR3 cells, whereas it is greatly diminished in MCF10A cells, suggesting that the mechanisms underlying the biological responses may differ between committed and noncommitted, nontransformed cells. D) Staining of surface HER2 in SK-BR3 (left panels) and MCF10A (right panels) cells ± expression of HER2-ECD-Δ451–466. Surface HER2 was visualized using a specific HER2 antibody directed against the extracellular domain of the receptor without permeabilization of the cells. Images were acquired identically and processed in parallel. Scale bars = 10 μM.
We next sought to explore the potential mechanism(s) contributing to the profound inhibitory effects of this mutant. We hypothesized that the failure to form dimers could be because of structural changes and/or mistargeting of the mutant receptor within the cell, both of which impede receptor-receptor interactions and signaling. Surprisingly, we observed both phenotypes depending on the cell type examined. As shown in Figure 4 (B and C), HER2 mutant–overexpressing SK-BR3 cells exhibited no differences in HER2 surface association relative to control cells (Figure 4B), while the nontransformed MCF10 cells had a clear reduction in surface receptor levels when forced to express the HER2-ECD deletion mutant compared with HER2-wt (Figure 4C). The variation in the plasma membrane–associated HER2 levels was confirmed by immunostaining of surface HER2 levels in both cell types (Figure 4D). The reason for the different trafficking patterns between the two cell lines remains to be determined.
Together, the data presented here provide a strong rationale for clinical evaluation of agents developed against the HER2-ECD sequence between amino acids 451 to 466, particularly in the context of HER2 tumors that have already developed resistance to the current drugs of choice.
Discussion
Here we present evidence that a specific disruption of a small core sequence within subdomain III of the HER2-ECD generates a dominant-negative form of HER2 that is sufficient to disable the dimerization loop that subsequently blocks HER2-driven oncogenic signaling.
Amplification of HER2 occurs in approximately 30% of breast carcinomas and is associated with more aggressive disease progression (1). The association of HER2 levels with enhanced malignant phenotypes, including those with metastatic potential and resistance to endocrine and chemotherapies, explains the poor clinical outcome of breast cancer patients with HER2-overexpressing tumors (2–4). Although the enhanced tyrosine kinase activity of HER2 plays a critical role in the initiation, progression, and outcome of an important subgroup of breast tumors, it should be noted that HER2 is the only HER family receptor without known ligands. In cancer cells, the “ligandless” HER2 can be activated either by overexpression or by ligand-mediated stimulation of another HER receptor, resulting in the formation of receptor homo- or heterodimers for which HER2 is the preferred partner (17–19,36,37,41).
Several HER2-directed agents are in clinical use or at advanced stages of development, but unfortunately, many breast tumors express multiple HER receptors and co-express one or more HER ligands, which negatively impacts the response to current HER2-targeted therapies and highlights an urgent need for novel anti-HER2 molecules to be used alone or in a combination strategy (38).
Here we describe a small deletion of the HER2-ECD that profoundly affects HER2-catalyzed activation of the HER network. Strikingly, expression of HER2-ECD-Δ451–466 drastically reduced the activity of HER2, HER1, and HER3, consequently turning off key signal transduction mediators such as ERK1/2 and PI3K/AKT. Moreover, this deletion clearly affected several canonical oncogenic properties of wild-type HER2 in breast epithelial cells, such as EGF-independent cell growth, colony formation in soft agar, enhanced cell cycle progression, and prevention of cell damage induced by chemotherapeutic agents.
Importantly, elimination of this essential motif within HER2 is more effective in the inhibitory actions than the current clinically used monoclonal antibodies trastuzumab (15,24,58), pertuzumab (21,23,24), and cetuximab. Furthermore, HER2-ECD-Δ451–466 mimics the antityrosine kinase activity of small-molecule inhibitors such as lapatinib (28,29). Notably, the relevance of the ECD domain was highlighted, albeit in HER3, by demonstrating that a SELEX-derived RNA aptamer against the HER3-ECD–inhibited ligand-induced signaling and prevented the higher-order complexes required for proxy phosphorylation of HER2 (2).
How does the specific disruption of only 16 amino acids within the HER2-ECD subdomain III block the HER2 dimerization loop, which in turn precludes the oncogenic activation of intracellular signaling pathways? Part of the answer to this question may be related to the intracellular trafficking behavior of the mutant receptor, at least in nontransformed cells. In these cells, HER2-ECD-Δ451–466 does not associate with the plasma membrane (PM) for reasons that are unknown at present. One can speculate that the mutant receptor is trapped in an intracellular compartment because of a secretion defect (because the receptor is not degraded) or very rapid endocytosis/recycling from the PM (endocytic defect). Differentiating between these possibilities will be the subject of future studies.
With regard to the oncogenic HER2-committed cells, with no differences in receptor surface association, it is plausible that structural differences between the mutant and the wt-HER2 receptor prevent dimerization directly and contribute to the observed dominant-negative and anti-oncogenic effects. Recent works describing the crystal structures of the extracellular region of each HER receptor in various ligand- and therapeutic antibody–bound states (21,23,38,43,44,59–64) have provided new insights into the process of ligand-induced receptor dimerization and biological activity of HER-targeted therapies that may explain, at least in part, our current findings. For example, the crystal structure of ligand-bound HER1 revealed that there is a direct HER1-HER1 interaction promoted by the subdomain II dimerization arm suggesting that, in contrast to other transmembrane receptors, HER dimerization is entirely receptor mediated and ligand independent (43,65–67). Remarkably, the structure of the HER2-ECD appears to be radically different from other HER-ECDs. HER2 has a fixed conformation that resembles the dimerization-competent “open” state in which the dimerization loop in subdomain II is exposed (42), rendering HER2 permanently poised for interaction with other ligand-bound receptors.
Although structural studies are needed to clarify this model, our results imply that, upon deletion of the important 16 aa in the ECD subdomain III of HER2, HER2 can no longer adopt its constitutive dimerization-competent conformation (Figure 5), resulting in failure to converge on the common downstream transduction cascades.
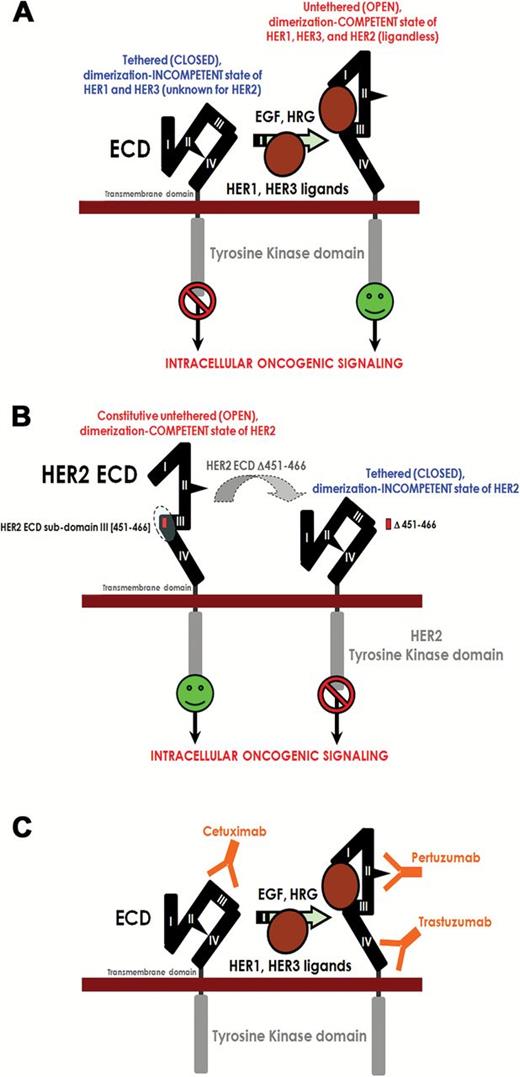
Evidence for a new anti–human epidermal growth factor receptor 2 (HER2) therapeutic strategy based on the disruption of the constitutive dimerization-competent (“open”) conformation of HER2 extracellular domain (ECD) structure: a working model. A) Ligand binding to HER receptors induces the formation of receptor homo- and heterodimers, resulting in phosphorylation/activation on specific tyrosine residues within the cytoplasmic tail. These phosphorylated residues recruit a range of proteins that leads to the activation of intracellular signaling pathways, including ERK1/2- and AKT-driven pathways. The extracellular region of each HER receptor consists of four subdomains (I to IV). It has been proposed that in the absence of ligand, HER1, and HER3 assume a tethered (“closed”) dimerization-incompetent structure. Subdomains I and III are involved in ligand binding, and, following this, the dimerization arms in HER1 and HER3 subdomain II are exposed. None of the ligands bind HER2, but HER2 is the preferred dimerization partner for all other HER receptors because HER2 has a fixed (constitutive) untethered dimerization-competent conformation that resembles the ligand-activated state of HER1 and HER3. B) Our current findings strongly suggest that, upon deletion of a small portion of the HER2-ECD subdomain III, HER2 can no longer adopt its constitutive “open” conformation, making it homo- and heterodimerization-deficient and thus interfering with its signaling capacity. C) The HER2-directed inhibitory antibodies trastuzumab and pertuzumab bind subdomains IV and II, respectively, in the permanently “opened” state of HER2, while the HER1-directed antibody cetuximab binds subdomain III of the autoinhibited tethered state of HER1-ECD. In each case, heterodimerization and downstream signaling are impaired.
We recognize that there are some limitations to the current in vitro study. The demonstration of antitumor activity of HER2Δ451–466 in relevant animal models is an essential prerequisite for progressing to clinical testing. Although new drugs directed against the essential activating sequence in the ECD of HER2 are expected to be highly specific, processes (eg, expression of truncated isoforms of HER2 encompassing most of the HER2-ECD such as p95HER2 [68,69]) or molecules (eg, integrin β1, CD44, MUC4) that mask or prevent high-affinity peptides or antibodies from binding to the HER2-ECD would be predicted to block their activity. In this regard, studies are currently underway to determine whether HER2Δ451–466 could allow developing strategies to prevent or overcome the presence of acquired and de novo resistance in neoadjuvant, adjuvant, and metastatic settings (70,71). Beyond established markers of HER2 overexpression currently employed for selecting patients for adjuvant trastuzumab or neoadjuvant trastuzumab+pertuzumab therapy, the relatedness of predictive biomarkers for HER2Δ451–466–based drugs and those for trastuzumab and pertuzumab, which may differ in early- and late-stage breast cancer, remains to be assessed. Certainly, if trastuzumab-like immune-mediated effector functions play a dominant role in the clinical benefits of HER2Δ451–466–based antibodies, then additional studies focusing on host factors need to be undertaken.
In summary, our study emphasizes the great potential of targeting aa 451–466 in the HER2-ECD, because there is no available HER-targeted therapeutic that specifically affects the strong interaction between subdomains I and III within the HER2-ECD so far. Thus, our findings demonstrating that specific disruption of this essential sequence in the HER2-ECD interferes with the oncogenic potential of the receptor provide a strong rationale for developing this peptide sequence into a valuable anti-HER2 therapeutic drug. Peptides and/or antibodies specific for this functional site should broaden our therapeutic arsenal for the management of HER2-overexpressing carcinomas. We are currently expanding this approach to design soluble, high-affinity peptidomimetic compounds that specifically interact with this essential activating site in the ECD of HER2.
Funding
This work was supported by the US Army Medical Research and Materiel Command under W81XWH-06-1-0686.
The authors thank Drs Silvana De Lorenzo and Zeng Hu for technical support and Ashwani Khurana for the support during editing of the manuscript.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}