





Abstract
A current algebraic analysis on genome-wide heterozygosity estimates suggests that correlations between molecular markers and genome-wide heterozygosity, ρ, depend on the ratio between the number of markers used, r, and the number of genome loci, n; that is: 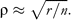 Hence, it is unfeasible to obtain reliable estimates of genome-wide heterozygosity in species of large genome using a few markers. We cast some doubts about this analysis as it assumed that the probability that an individual was heterozygous at a locus is equal to the average heterozygosity of this locus in the population. However, we believe that individual heterozygosity at a given locus depends on individual pedigree. Because the pedigree is common for all loci of an individual, their probabilities of heterozygosity are not independent within the genome. We first performed simulations generating random genomes for 100 individuals. Among these individuals, markers and genome-wide heterozygosities correlated as expected from the above equation. However, when we simulated random mating among these individuals and in successive generations including their descendents, as occur in real populations, the correlations between markers and genome-wide heterozygosity were much higher than those predicted from algebraic analyses, and estimates of genome-wide heterozygosity improved slightly with the increment of the number of loci in the genome.
Hence, it is unfeasible to obtain reliable estimates of genome-wide heterozygosity in species of large genome using a few markers. We cast some doubts about this analysis as it assumed that the probability that an individual was heterozygous at a locus is equal to the average heterozygosity of this locus in the population. However, we believe that individual heterozygosity at a given locus depends on individual pedigree. Because the pedigree is common for all loci of an individual, their probabilities of heterozygosity are not independent within the genome. We first performed simulations generating random genomes for 100 individuals. Among these individuals, markers and genome-wide heterozygosities correlated as expected from the above equation. However, when we simulated random mating among these individuals and in successive generations including their descendents, as occur in real populations, the correlations between markers and genome-wide heterozygosity were much higher than those predicted from algebraic analyses, and estimates of genome-wide heterozygosity improved slightly with the increment of the number of loci in the genome.
It is thought that heterozygous individuals possess fitness advantages with respect to homozygous (e.g., Charlesworth D and Charlesworth B 1987). This idea has attracted the interest of numerous scientists to estimate heterozygosity of the individuals. Since the advent and application of DNA polymorphic markers, researches have measured heterozygosity at a handful of neutral markers as an estimate of genome-wide heterozygosity. However, several theoretical studies have recently questioned this approach because they consider that the expected correlations between markers heterozygosity and genome-wide heterozygosity are presumably very poor. Most of these criticisms come from studies performing simulations that analyzed the relationship between inbreeding and heterozygosity measured at a few neutral markers (Baloux et al. 2004; Slate et al. 2004). These studies concluded that correlates between inbreeding coefficients and markers heterozygosity are so weak that any correlation between heterozygosity measured in different parts of genome should be unexpected. More recently, DeWoody YD and DeWoody JA (2005) carried out an algebraic analysis on this subject, concluding that genome-wide heterozygosity is poorly estimated by microsatellite loci. Therefore, this analytic approach seems to reinforce the thesis of noncorrelation between markers and genome-wide heterozygosity. However, we cast some doubts about the validity of this analysis.
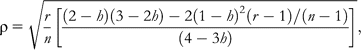 This means that if we use a certain number of microsatellites for estimating markers heterozygosity, we could expect smaller correlations with genome-wide heterozygosity if the genome consists of 10 000 loci than if it consists of only 1000. The result is very disappointing because the number of markers normally used is extremely low in relation to the number of loci included in the genome. For example, in species with a genome consisting of 30 000 loci, the expected correlation coefficients between genome-wide and markers heterozygosity would be around 0.02 if we use a set of 10 or 15 markers, which is a usual number in many current studies.
This means that if we use a certain number of microsatellites for estimating markers heterozygosity, we could expect smaller correlations with genome-wide heterozygosity if the genome consists of 10 000 loci than if it consists of only 1000. The result is very disappointing because the number of markers normally used is extremely low in relation to the number of loci included in the genome. For example, in species with a genome consisting of 30 000 loci, the expected correlation coefficients between genome-wide and markers heterozygosity would be around 0.02 if we use a set of 10 or 15 markers, which is a usual number in many current studies.The reasoning conducted by DeWoody YD and DeWoody JA (2005) from this equation appears to be correct. However, the equation provided is based on a questionable assumption done in the original paper by Chakraborty (1981) inspired on Mitton and Pierce's (1980) simulations. He assumed that the probability that an individual was heterozygous at a locus, i, is equal to the heterozygosity of this locus in the population, hi (Chakraborty 1981, p. 461). Thus, all individuals in the population would have the same probability of being heterozygous in a particular locus, and the probabilities of 2 loci of being heterozygous for a particular individual would be totally independent. However, we believe that in real populations, these assumptions are not attainable because the probability of heterozygosity of an individual for a particular locus is not determined by the population, but by the parental genotypes. To illustrate the error of these assumptions, imagine a cross of 2 siblings in a population. The probability that their offspring was homozygous at any locus is higher than the probability of homozygosity of those loci in other individuals resulting from outbred crosses. Thus, within an individual, the probability that any locus was heterozygous depends on the pedigree of that individual. Because the pedigree is common for all loci of an individual, their probabilities of heterozygosity are not independent.
To show our arguments, we have performed 100 simulations that generate random genomes consisting of 200, 300, …, 1000 coding loci for 100 individuals that represent the founder population. Each ith locus has a random number of alleles (between 2 and 10) in the population, with equal frequency. The probability of bearing any allele at a locus is equal for all individuals. Thus, the probability of being heterozygous, hi, depends on the number of alleles and is equal for all members in the population. Note that this “zero generation” is, therefore, exactly the same as the population imagined by Chakraborty (1981). From the pool of coding loci, we extracted 50 loci that represent the markers used to estimate genome-wide heterozygosity. For each simulation, the average individual heterozygosity obtained from markers was correlated with individual genome-wide heterozygosity. We performed 100 simulations, and found that the average correlation coefficients with genomes ranging from 200 to 1000 loci were closely similar to those predicted by DeWoody YD and DeWoody JA (2005) (Figure 1A).
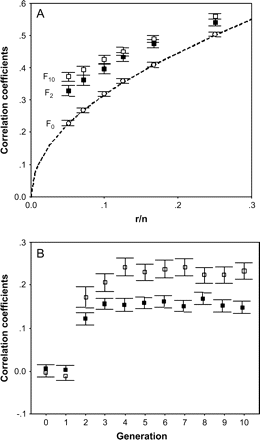
(A) Correlation coefficients between markers and genome-wide heterozygosity in relation to the proportion of number of loci assayed, r, relative to number of loci in the genome, n. Broken line represents the expected correlation coefficients from algebraic analysis by DeWoody YD and DeWoody JA (2005). Circles, filled and open squares and their respective bars represent mean correlation coefficients and standard errors (SEs) obtained at different generations (F0, F2, and F10, respectively) in 100 populations whose founders were endowed with random genome. (B) Mean correlation coefficients between heterozygosity values (±SE) of 1000 coding loci and 10 (open squares) and 50 (full squares) unlinked neutral markers obtained across 10 generations in 100 simulated populations. Note that in Figure 1A, markers were considered as part of the coding genome, whereas in Figure 1B markers were not.
Now, let us consider a second stage of simulations in which successive generations are included. Founder individuals mated randomly and every pair bred once and produced 2 offspring. Obviously, each offspring received an allele from the father and another from the mother for each particular locus whose alleles were also randomly selected among those of the parents. The descendents mated at random to breed again in similar conditions than their parents did (i.e., 1 reproductive event and 2 offspring), and these simulation conditions continued for 10 generations. In this case, we found that correlations between markers and genome-wide heterozygosity were much higher than those predicted by DeWoody YD and DeWoody JA (2005) at equal r/n ratio, even after 2 generations only (Figure 1A). Therefore, simulations considering a few generations with Mendelian allele inheritance, do not support the predictions derived from the analytical development by Chakraborty (1981) and DeWoody YD and DeWoody JA (2005).
In the previous section, we have assumed that markers used to estimate genome-wide heterozygosity are a part of the coding genome following Chakraborty (1981) and DeWoody YD and DeWoody JA (2005). However, the practical interest discussed by DeWoody YD and DeWoody JA (2005) is about estimating heterozygosity from the whole coding genome using neutral markers, such as microsatellites, rather than directly using coding loci. Because we normally use neutral marker loci, our sample is not included within coding genome for which we are estimating heterozygosity. Therefore, the expected correlation predicted by the algebraic analysis should be zero. However, under identity disequilibrium (e.g., Bierne et al. 2000; Hansson and Westerberg 2002) originated by the effects of mating after a few generations, we could expect correlations between genome-wide and markers heterozygosities although both parts of the genome are not physically linked. To illustrate our arguments, we also included 50 additional loci in our simulations that represent neutral markers and, therefore, they are not part of the coding genome. Then, we examined correlations between heterozygosity at these markers and heterozygosity at the 1000 simulated coding loci. The correlation was zero in the F0 generation, as expected, as genomes were randomly generated. Nevertheless, only 3 generations were enough to produce significant correlations between markers and coding loci heterozygosity (Figure 1B). The observed correlations occurred because of identity disequilibrium generated by inbreeding variance (e.g., Balloux et al. 2004; Slate et al. 2004), but not by sampling markers as a part of the coding genome.
Another aspect of the algebraic analysis is that the reliability of genome-wide heterozygosity estimates would decrease with the increase of the number of loci included in the genome. However, this conclusion is only true under the conditions assumed above, when those molecular markers are part of the pool of the genome (Chakraborty 1981; DeWoody YD and DeWoody JA 2005). In this case, the greater proportion of genome sampled the higher correlation. We wondered if those correlations would be poorer when genome consists of a great number of coding loci and markers are not part of the coding genome. To explore this, we performed 35 simulations with a variable number of coding loci (up to 20 000), and also with different set of neutral markers (10, 20, 30, and 50). Correlations between genome and markers heterozygosity increased with both number of markers (F1,33 = 176, P < 0.0001) and number of coding loci in the genome (F1,33 = 77, P < 0.0001) for any set of markers (Figure 2). To explain this positive correlation, we should take into account that we are using neutral marker loci, and our sample is not included within the coding genome for which we are estimating heterozygosity. Thus, correlations between coding genome and neutral markers heterozygosity are only due to identity disequilibrium originated by inbreeding variance that affects heterozygosity of both, markers and coding loci. The association between inbreeding and heterozygosity in both, coding and marker loci, is disturbed by random effects due to Mendelian segregation and these random effects are relatively less important when the number of loci is greater than when smaller. Therefore, the higher the number of loci in neutral markers or genome, the better is the correlation between their heterozygosities.
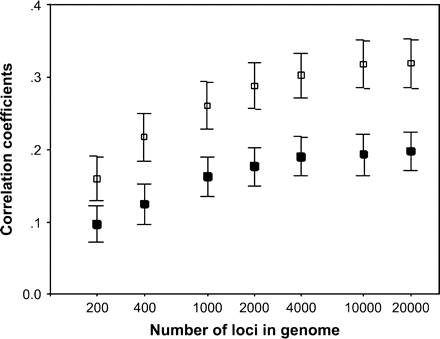
Mean correlation coefficients (±SE) between neutral markers (filled squares: 10 markers; open squares: 50 markers) and coding loci heterozygosity obtained in the 10th generation of simulated populations with a variable number of loci in the genome.
In conclusion, it seems obvious that genome-wide heterozygosity is not a random product, but the result of individual pedigree in real populations. The simplification that the probability of heterozygosity was the same for all individuals in a population makes easier the algebraic analysis but it is an unreal assumption that leads to false conclusions as shown. Assuming that individuals inherit one allele per locus from its father and another from its mother, and considering only a few generations, we have demonstrated that correlations between markers and genome-wide heterozygosity are expected to be higher than those predicted from algebraic analyses. Furthermore, neutral markers allow predicting more reliable heterozygosity of larger than of smaller genomes.
We received financial support from the projects: CGL2005-05611-C02-02/BOS (Ministerio de Educación Ciencia) and PAI05-053 (Junta de Comunidades de Castilla-La Mancha). During this work, J.O. was supported by a predoctoral fellowship from the Junta de Comunidades de Castilla-La Mancha and the European Social Fund.
References
Author notes
Corresponding Editor: Stephen J. O'Brien

{kind=link}
{kind=link}