




Abstract
DNA polymerase gamma (pol γ) is required for replication and repair of mitochondrial DNA. Over 80 mutations in POLG, the gene encoding the catalytic subunit of pol γ, have been linked with disease. The W748S mutation in POLG is the most common mutation in ataxia-neuropathy spectrum disorders and is generally found in cis with the common E1143G polymorphism. It has been unclear whether E1143G participates in the disease process. We investigated the biochemical consequences of pol γ proteins containing W748S or E1143G, or both. W748S pol γ exhibited low DNA polymerase activity, low processivity and a severe DNA-binding defect. However, interactions between the catalytic and accessory subunits were normal. Despite the benefits derived from binding with the accessory subunit, catalytic activities did not reach wild-type (WT) levels. Also, nucleotide selectivity decreased 2.1-fold compared with WT. Surprisingly, pol γ containing only E1143G was 1.4-fold more active than WT, and this increased polymerase activity could be due to higher thermal stability for E1143G pol γ. The E1143G substitution partially rescued the deleterious effects of the W748S mutation, as DNA binding, catalytic activity and fidelity values were intermediate for W748S-E1143G. However, W748S-E1143G had a notably lower change in enthalpy for protein folding than W748S alone. We suggest that when E1143G is in cis with other pathogenic mutations, it can modulate the effects of these mutations. For W748S-E1143G pol γ, the benefits bestowed by E1143G include increased DNA binding and polymerase activity; however, E1143G was somewhat detrimental to protein stability.
INTRODUCTION
DNA polymerase γ (pol γ) is the only known DNA polymerase found in animal cell mitochondria and thus bears sole responsibility for DNA synthesis in replication and repair transactions involving mitochondrial DNA (mtDNA) (1). In humans, pol γ is composed of a heterotrimer consisting of one p140 catalytic subunit encoded by the POLG gene and two p55 accessory subunits encoded by the POLG2 gene (2). The p140 catalytic subunit contains DNA polymerase activity, exonuclease proofreading function and dRP lyase activity, whereas the p55 accessory subunit is required for enhanced DNA binding and processive DNA synthesis (1). Depletion of mtDNA and the accumulation of deletions and point mutations in mtDNA have been observed during aging and in several human disorders (reviewed in 3). Mutations in the POLG gene are linked with a wide variety of mitochondrial diseases, including the devastating childhood onset Alpers syndrome, as well as the later onset diseases such as progressive external ophthalmoplegia (PEO), Parkinsonism and ataxia-neuropathy spectrum (ANS) disorders. Previously, we characterized the most common mutation in POLG, the A467T mutation, which is associated with Alpers syndrome, ANS and PEO. The A467T amino acid substitution occurs in the early linker region of the catalytic subunit, between the polymerase and exonuclease domains. This mutation reduces the affinity of p140 for DNA and diminishes DNA polymerase activity to only 4% compared with wild-type (WT) p140. Furthermore, A467T is currently the only known mutation in the catalytic subunit of human pol γ that disrupts interaction with the accessory subunit. This weak interaction contributes to the decreased catalytic efficiency of this enzyme, giving rise to the mitochondrial diseases observed (4). Recently, our group reported the first disease mutation in the gene for the accessory subunit, POLG2, which also disrupts physical interaction with the catalytic subunit (5).
The W748S-E1143G genotype (Fig. 1A) is associated with a spectrum of disease phenotypes (Alpers syndrome, ANS and PEO), similar to A467T (6–15). The W748S substitution is located in the late linker region and arises because of a G-to-C mutation at nucleotide 2243 in exon 13. The W748S mutation is generally found in cis with the E1143G polymorphism, which is caused by an A-to-G transition at nucleotide 3428 in exon 21 of POLG. Both W748 and E1143 are conserved residues within pol γ (Fig. 1B). E1143G is adjacent to motif C in the polymerase domain of pol γ and has an allele frequency of 3% in the general population (16). Current reports suggest that E1143G is unlikely to cause disease, as individuals homozygous for E1143G do not have mitochondrial disease (7,11,15). W748S-E1143G heterozygotes are also unaffected (10); however, one recent report revealed a heterozygous patient with late onset ataxia and PEO/ptosis with features of Parkinsonism (13).
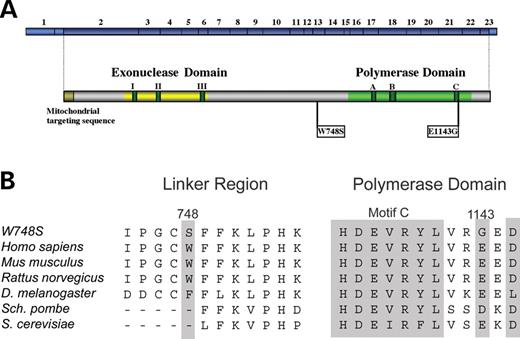
Schematic diagram of human pol γ. (A) Conserved amino acid motifs I, II and III in the exonuclease domain and motifs A, B and C in the polymerase domain flank the linker region of the human pol γ catalytic subunit. The relative positions of the 23 exons in POLG are shown together with the positions of the pathogenic W748S amino acid substitution within the linker region and the E1143G polymorphism within the polymerase domain. (B) Conservation of the W748 and E1143 amino acids across species.
In Alpers syndrome, W748S-E1143G alleles are found paired in trans with R232H, L244P, A467T, G848S, M1163R or Y1210fs1216X mutations (Table 1) (6–8,13,17). For PEO, patients may also have the genotype W748S-E1143G/A467T, or may be homozygous for W748S-E1143G (13). Hakonen et al. (12) recently showed that W748S-E1143G was the major pol γ allele associated with inherited ataxia in Finland, with a carrier frequency for the W748S-E1143G allele of 1:125. The majority of ANS patients have W748S-E1143G in either one, but generally both POLG alleles (12). Other ANS genotypes consist of one W748S-E1143G allele paired in trans with L304R, A467T or Q497H (Table 1) (10,11,13–15). The majority of probands with autosomal recessive ataxia involving the W748S-E1143G alleles are of European origin, having descended from a common founder (12). However, as the E1143G polymorphism has also been identified in patients with other disease mutations, it has been suggested to be a non-neutral polymorphism, as it may aggravate the defect caused by the W748S mutation and other POLG mutations by increasing clinical prevalence and severity (13,15,18). Thus, it is currently unclear whether the E1143G mutation affects patients with W748S or other POLG mutations, and if so, whether this mutation has any pathological or beneficial role in the general population in the absence of other POLG mutations. One computer algorithm that analyzes the impact of single-nucleotide polymorphisms on protein structure and function by sequence analysis predicted the E1143G polymorphism to be damaging to function (http://www.snps3d.org/). In contrast, another computer algorithm predicted this variant to be benign (http://genetics.bwh.harvard.edu/pph/). Until now, however, this substitution has not been biochemically characterized. As more patients are found to have the W748S-E1143G genotype, it is imperative that we understand the mechanism by which these mutations cause mtDNA instability and ultimately mitochondrial disease. In this report, we investigated the biochemical consequences of pol γ proteins containing W748S or E1143G, or both, in order to determine the contributions of each mutation towards pathology.
POLG mutations found in trans with W748S-E1143G and their associated diseases
| POLG mutations | POLG domains | Phenotype | Reference |
|---|---|---|---|
| R232H | Exo | Alpers | (17) |
| L244P | Exo | Alpers | (7) |
| L304R | Exo | ANS | (14) |
| A467Ta | Linker | Alpers/ANS/PEO | (10,12,13,15,17) |
| W748S+E1143G | Linker | ANS / PEO | (10,12,13) |
| G848S | Pol | Alpers | (6,9) |
| M1163R | Pol | Alpers | (17) |
| Y1210fs1216X | Pol | Alpers | (7) |
| POLG mutations | POLG domains | Phenotype | Reference |
|---|---|---|---|
| R232H | Exo | Alpers | (17) |
| L244P | Exo | Alpers | (7) |
| L304R | Exo | ANS | (14) |
| A467Ta | Linker | Alpers/ANS/PEO | (10,12,13,15,17) |
| W748S+E1143G | Linker | ANS / PEO | (10,12,13) |
| G848S | Pol | Alpers | (6,9) |
| M1163R | Pol | Alpers | (17) |
| Y1210fs1216X | Pol | Alpers | (7) |
Data derived from http://dir-apps.niehs.nih.gov/polg/
aA467T/W748S was recently identified in two patients by Horvath et al. (15). The E1143G polymorphism was not reported.
Exo, exonuclease domain; Linker, linker region; Pol, polymerase domain.
POLG mutations found in trans with W748S-E1143G and their associated diseases
| POLG mutations | POLG domains | Phenotype | Reference |
|---|---|---|---|
| R232H | Exo | Alpers | (17) |
| L244P | Exo | Alpers | (7) |
| L304R | Exo | ANS | (14) |
| A467Ta | Linker | Alpers/ANS/PEO | (10,12,13,15,17) |
| W748S+E1143G | Linker | ANS / PEO | (10,12,13) |
| G848S | Pol | Alpers | (6,9) |
| M1163R | Pol | Alpers | (17) |
| Y1210fs1216X | Pol | Alpers | (7) |
| POLG mutations | POLG domains | Phenotype | Reference |
|---|---|---|---|
| R232H | Exo | Alpers | (17) |
| L244P | Exo | Alpers | (7) |
| L304R | Exo | ANS | (14) |
| A467Ta | Linker | Alpers/ANS/PEO | (10,12,13,15,17) |
| W748S+E1143G | Linker | ANS / PEO | (10,12,13) |
| G848S | Pol | Alpers | (6,9) |
| M1163R | Pol | Alpers | (17) |
| Y1210fs1216X | Pol | Alpers | (7) |
Data derived from http://dir-apps.niehs.nih.gov/polg/
aA467T/W748S was recently identified in two patients by Horvath et al. (15). The E1143G polymorphism was not reported.
Exo, exonuclease domain; Linker, linker region; Pol, polymerase domain.
RESULTS
To study the biochemical properties of the W748S-E1143G pol γ variant commonly found in mitochondrial disorders, we utilized site-directed mutagenesis to make pol γ derivatives bearing the W748S substitution, the E1143G substitution, or both. Each protein was constructed from the D198A/E200A exonuclease-deficient (Exo−) pol γ (19), as degradation of DNA substrates by the exonuclease function can complicate in vitro analyses. WT and mutant proteins were overproduced in baculovirus-infected insect cells and purified as described (19,20). Expression and overall yield of recombinant W748S and E1143G p140 were similar to those for WT p140 (19). Immunoblot analysis confirmed the identity of the full-length recombinant proteins and revealed no proteolytic degradation. The effects of the W748S, E1143G and W748S-E1143G point mutations on the secondary structure of p140 were assessed by circular dichroism (CD) spectroscopy (Fig. 2). The similarity of the spectra indicated that the mutations did not cause gross alterations in protein conformation. In addition, pol γ has a high α-helical content and has a similar CD profile to that of DNA polymerase β (21).
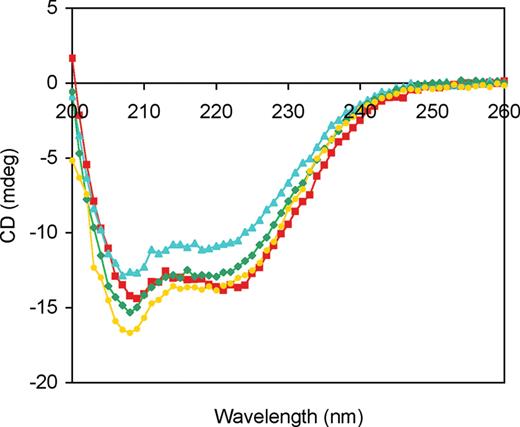
CD spectra of WT and mutated p140 proteins. WT p140 (red squares), W748S p140 (blue triangles), E1143G p140 (green diamonds) and W748S-E1143G p140 (yellow circles) were diluted to a protein concentration of 10 µg/ml, and CD spectra were measured as described in the Materials and Methods section.
Initial screening of p140 mutants by processivity assay
Given its close proximity to the conserved polymerase motif C within POLG (Fig. 1B), the E1143G substitution may alter the polymerase function of pol γ. Similarly, since the A467T linker region mutation in POLG significantly reduces both polymerase function and the ability of the catalytic and accessory subunits to associate (4), the W748S substitution has the potential to cause complex changes in pol γ function. An in vitro assay of polymerase processivity was chosen for the initial screening of the p140 mutants, because this assay is able to score both polymerase function and the ability of the subunits to productively associate (19,22). Each time the catalytic subunit binds productively to a DNA primer-template, it synthesizes 50–100 nt prior to dissociating from the DNA (19). The processivity of the polymerase is increased as much as 50-fold upon association of the accessory subunit, which reduces dissociation from the primer-template by increasing the affinity of the protein complex to DNA (22). In this gel-based, primer extension assay under conditions that permit multiple binding events (Fig. 3), WT p140 extended the primer roughly 100 nt at 0 mm NaCl (lane 1), and this activity was moderately inhibited by 0.1 M NaCl (lane 2). Distinct pausing at ∼90 and ∼125 nt was due to template secondary structure, and addition of the accessory subunit stimulated primer extension at both salt concentrations (lanes 3, 4), as observed previously (19,20). The polymerase activity of W748S p140 was somewhat reduced and did not permit substantial primer extension past the first major pause site (lane 5), and polymerase activity was completely inhibited by physiological salt concentrations (lane 6). Inclusion of p55 stimulated primer extension by W748S p140 at both salt concentrations (lanes 7, 8), although partial sensitivity to 100 mm NaCl was still apparent (lane 8). The overall efficiency of the E1143G p140 reactions (lanes 9–12) was slightly higher than that of WT p140, and the pattern of primer extension products for E1143G p140 resembled those of WT p140. Isolated W748S-E1143G p140 was also sensitive to salt (lane 14), as previously observed for W748S p140 (lane 6). Although this enzyme functionally interacted with p55 (lanes 15, 16), residual salt sensitivity was apparent for the complex (lane 16). The true processivity of each enzyme was revealed in identical reactions that also included a DNA trap, conditions under which the polymerase extends the end-labeled primer, dissociates from the primer-template and migrates to the unlabeled DNA trap (22). Although the overall yield of extended primers was considerably less when each enzyme was limited to a single DNA-binding event, the length distribution of extended primers was unchanged for all four enzymes in the presence of p55, and products were only one-half to one-third as long in the absence of p55 (data not shown). These initial experiments revealed that each mutant form of p140 was catalytically active and able to interact functionally with the p55 accessory subunit.
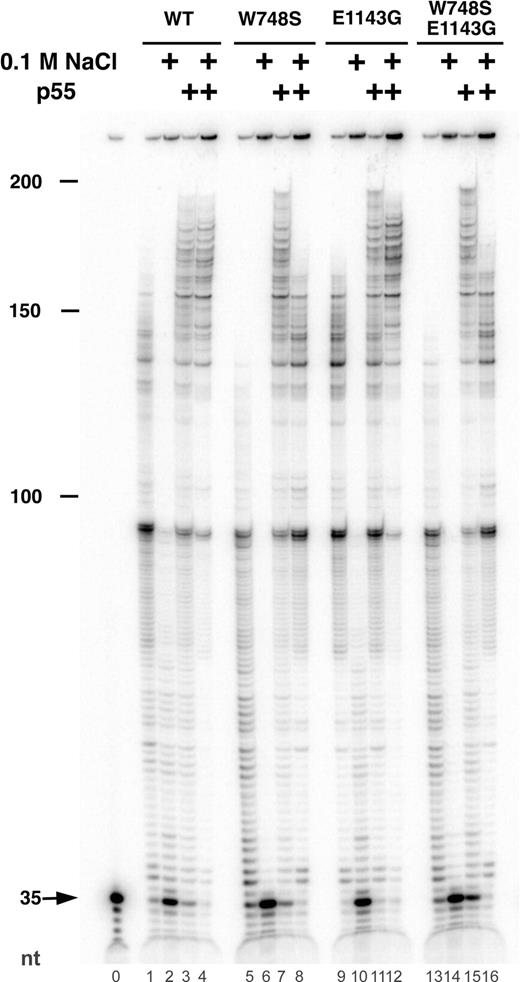
Processivity of WT and mutant p140 proteins on singly primed M13 DNA. Primer extension reactions were performed as described in the Materials and Methods section. Reactions contained 9.6 ng of the p55 accessory subunit (lanes 3, 4, 7, 8, 11, 12, 15, 16) and 12 ng of WT p140 Exo− (lanes 1–4), W748S p140 (lanes 5–8), E1143G p140 (lanes 9–12) or W748S-E1143G p140 (lanes 13–16). Activity was measured at 0 mm NaCl (odd numbered lanes) or 100 mm NaCl (even numbered lanes). Lane 0 had no enzyme. The position of the unextended 35mer primer is indicated by the arrow. Markers indicate the total length of extended primers (nt).
Physical association of mutant p140s withthe accessory subunit
Two additional experiments were undertaken to assess the direct physical interaction of the two subunits. The isolated catalytic subunit of pol γ is extremely sensitive to inhibition by N-ethylmaleimide (NEM) which covalently modifies solvent-accessible cysteine residues (19). We previously established that association with its accessory subunit protects the pol γ catalytic subunit from inactivation by NEM (22,23), and we exploited this protective effect to probe the physical association of the mutant p140 proteins with the accessory subunit. Inhibition of isolated WT p140 by varying concentrations of NEM was reconfirmed in our standard polymerase assay in which 0.2 mm NEM inhibited pol γ activity by ∼90% (Fig. 4). Preassembly of the WT p140 with a 2-fold molar excess of p55 conferred resistance to NEM, most likely through physical protection of critical sulfhydryl groups on p140. As expected, all three isolated mutant forms of pol γ displayed NEM inhibition curves similar to that for the WT enzyme (Fig. 4). Each mutant enzyme was efficiently protected by the addition of the p55 subunit, demonstrating a robust physical interaction between p55 and these mutant p140 proteins.
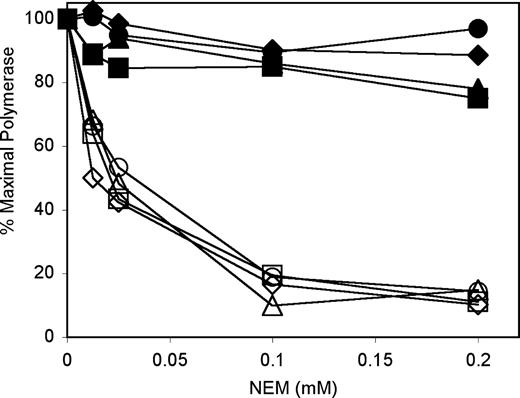
The accessory subunit protects both WT p140 and mutant p140s against inhibition by NEM. Polymerase assays were performed using poly (rA)·oligo(dT)12–18, as described in the Materials and Methods section (2-mercaptoethanol was excluded). Reactions contained 8 ng WT p140 (squares), W748S p140 (triangles), E1143G p140 (diamonds) or W748S-E1143G p140 (circles), 6.3 ng p55 (closed symbols), 75 mm NaCl and the indicated amounts of NEM.
Physical association of the subunits under physiological conditions was also assessed by immunoprecipitation assay, as previously described (4). Protein G-Sepharose beads preloaded with polyclonal rabbit antibodies raised against WT p140 were able to capture the purified WT and mutant derivatives of p140 with equal efficiency. When also incubated with the accessory subunit, immobilized WT and mutant forms of p140 retained similar quantities of p55, despite exhaustive washing of immunoprecipitates (data not shown). On the basis of these assays, we conclude that the W748S, W748S-E1143G and E1143G forms of p140 have no detectable defect in binding their accessory subunit.
W748S substitution significantly reduces DNApolymerase activity
The DNA polymerase activities of the WT and mutant proteins were measured with our standard pol γ assay, utilizing poly(dA)·oligo(dT) as the primer-template (22). Optimization of reaction conditions revealed robust activity for both WT and E1143G p140 at 25 mm NaCl with saturating concentrations of primer-template (66 and 94 pmol dTMP incorporated/h/ng enzyme, respectively). Addition of the p55 accessory subunit mildly stimulated polymerase activity and raised the salt optima to 25–50 mm NaCl. Although polymerase activities of the W748S and W748S-E1143G proteins were greatest at 0–25 mm NaCl, both proteins exhibited markedly reduced specific activities (4.6 and 7.6 pmol dTMP incorporated/h/ng enzyme, respectively), and activity was inhibited by NaCl concentrations >50 mm. Addition of the DNA-binding accessory subunit p55 relieved this inhibition but was unable to restore activity to WT levels. To understand better the nature of the reduced activity of the W748S and W748S-E1143G substituted proteins, as well as the apparent enhanced activity of E1143G p140, the polymerase activity of each enzyme was examined more critically by steady-state kinetic analysis (Table 2). As judged by kcat, the polymerase activity of the E1143G p140 was 1.5-fold higher than that of WT p140. The W748S substitution, however, reduced overall efficiency of DNA synthesis to <3% of WT activity. Compared with WT p140, the W748S substitution increased the Km for the incoming dNTP by a factor of three while reducing the kcat for DNA synthesis by 13-fold. The activity of the W748S-E1143G double mutant was also significantly reduced, although this enzyme retained overall polymerase activity 3-fold higher than the W748S single mutant.
DNA polymerase activities of pol γ variants bearing W748S and E1143G substitutions
| Enzyme | Km(dTTP) (µm) | kcat (s−1) | kcat/Km (s·µm)−1 |
|---|---|---|---|
| WT | 5.5±1.0 | 2.4±0.3 | 0.43 (100%) |
| E1143G | 6.3±0.5 | 3.7±0.4 | 0.59 (137%) |
| W748S | 18±2.0 | 0.18±0.07 | 0.010 (2.3%) |
| W748S-E1143G | 9.4±2.1 | 0.30±0.13 | 0.032 (7.4%) |
| WT+p55 | 3.5±0.4 | 1.7±0.3 | 0.49 (100%) |
| E1143G+p55 | 4.7±0.8 | 3.2±0.7 | 0.68 (139%) |
| W748S+p55 | 17±3.0 | 0.73±0.21 | 0.042 (6.0%) |
| W748S-E1143G+p55 | 5.1±0.9 | 1.0±0.2 | 0.20 (28%) |
| Enzyme | Km(dTTP) (µm) | kcat (s−1) | kcat/Km (s·µm)−1 |
|---|---|---|---|
| WT | 5.5±1.0 | 2.4±0.3 | 0.43 (100%) |
| E1143G | 6.3±0.5 | 3.7±0.4 | 0.59 (137%) |
| W748S | 18±2.0 | 0.18±0.07 | 0.010 (2.3%) |
| W748S-E1143G | 9.4±2.1 | 0.30±0.13 | 0.032 (7.4%) |
| WT+p55 | 3.5±0.4 | 1.7±0.3 | 0.49 (100%) |
| E1143G+p55 | 4.7±0.8 | 3.2±0.7 | 0.68 (139%) |
| W748S+p55 | 17±3.0 | 0.73±0.21 | 0.042 (6.0%) |
| W748S-E1143G+p55 | 5.1±0.9 | 1.0±0.2 | 0.20 (28%) |
Steady-state kinetic values for the incorporation of dTTP into poly (dA)·oligo(dT) were determined for the indicated enzymes, as described in the Materials and Methods section. All values are the averages of at least two independent measurements.
DNA polymerase activities of pol γ variants bearing W748S and E1143G substitutions
| Enzyme | Km(dTTP) (µm) | kcat (s−1) | kcat/Km (s·µm)−1 |
|---|---|---|---|
| WT | 5.5±1.0 | 2.4±0.3 | 0.43 (100%) |
| E1143G | 6.3±0.5 | 3.7±0.4 | 0.59 (137%) |
| W748S | 18±2.0 | 0.18±0.07 | 0.010 (2.3%) |
| W748S-E1143G | 9.4±2.1 | 0.30±0.13 | 0.032 (7.4%) |
| WT+p55 | 3.5±0.4 | 1.7±0.3 | 0.49 (100%) |
| E1143G+p55 | 4.7±0.8 | 3.2±0.7 | 0.68 (139%) |
| W748S+p55 | 17±3.0 | 0.73±0.21 | 0.042 (6.0%) |
| W748S-E1143G+p55 | 5.1±0.9 | 1.0±0.2 | 0.20 (28%) |
| Enzyme | Km(dTTP) (µm) | kcat (s−1) | kcat/Km (s·µm)−1 |
|---|---|---|---|
| WT | 5.5±1.0 | 2.4±0.3 | 0.43 (100%) |
| E1143G | 6.3±0.5 | 3.7±0.4 | 0.59 (137%) |
| W748S | 18±2.0 | 0.18±0.07 | 0.010 (2.3%) |
| W748S-E1143G | 9.4±2.1 | 0.30±0.13 | 0.032 (7.4%) |
| WT+p55 | 3.5±0.4 | 1.7±0.3 | 0.49 (100%) |
| E1143G+p55 | 4.7±0.8 | 3.2±0.7 | 0.68 (139%) |
| W748S+p55 | 17±3.0 | 0.73±0.21 | 0.042 (6.0%) |
| W748S-E1143G+p55 | 5.1±0.9 | 1.0±0.2 | 0.20 (28%) |
Steady-state kinetic values for the incorporation of dTTP into poly (dA)·oligo(dT) were determined for the indicated enzymes, as described in the Materials and Methods section. All values are the averages of at least two independent measurements.
Addition of the p55 accessory subunit in this steady-state kinetic assay produced only a modest stimulation of overall activity for the WT and E1143G forms of pol γ (Table 2). However, supplying the W748S protein with p55 led to a 4-fold increase in kcat, and a similar effect was noted upon addition of p55 to the W748S-E1143G double mutant. Two noteworthy effects are evident from this experiment. First, the W748S substitution appears to be detrimental to the polymerase activity of both the W748S and W748S-E1143G proteins, although the p55 subunit is able to compensate somewhat for this negative effect. Secondly, in the presence or absence of p55, the W748S-E1143G double mutant exhibited a reduced Km and an elevated kcat relative to the W748S values, suggesting the E1143G substitution also partially compensates for the harmful effects of the W748S mutation.
DNA-binding defect of W748S substitution partially rescued by E1143G polymorphism
The ability of the p55 accessory factor to enhance the affinity of the pol γ complex to DNA (22) and to relieve the salt sensitivity of the W748S and W748S-E1143G enzymes suggested their low activity could be caused by a partial defect in DNA binding. Therefore, we measured the affinity of each form of p140 to DNA by electrophoretic mobility shift assay. Various concentrations of enzyme were combined with radiolabeled, double-stranded oligonucleotide substrates, and mixtures were resolved by native PAGE. Reciprocal plots of the fraction of DNA shifted at different enzyme concentrations were utilized to calculate apparent Kd(DNA) values (Fig. 5). Both WT and E1143G pol γ had strong affinities for DNA (Kd(DNA) values of 147 and 159 nM, respectively), similar to the affinities we previously reported for WT p140 and several mutant variants of pol γ associated with dominant forms of PEO (20). However, the W748S-substituted p140 displayed an 8-fold decrease in DNA-binding strength (Kd(DNA)=1130 nM), which is the largest DNA-binding defect due to a pathogenic POLG mutation yet observed. This severe DNA-binding deficiency helps to explain the salt sensitivity and poor catalytic activity of W748S p140, as well as the ability of the p55 subunit to stimulate W748S p140. Interestingly, the W748S-E1143G protein had an intermediate affinity (Kd(DNA)=364 nM) that was only 2.5-fold weaker than WT, suggesting that the E1143G substitution in cis partially rescues the DNA-binding defect induced by the W748S mutation.
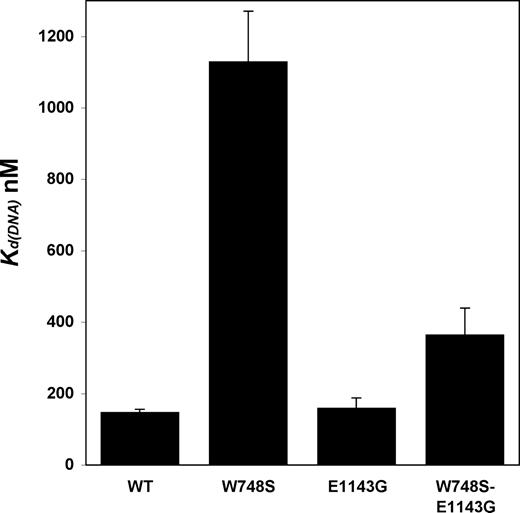
WT and E1143G p140s have similar DNA-binding abilities, whereas W748S and W748S-E1143G p140s have much lower DNA-binding capabilities. Kd(DNA) values were estimated by electrophoretic mobility shift assay, as described in the Materials and Methods section. All values are the average of two independent determinations.
Nucleotide selection by the mutated forms of pol γ
The pathogenicity of some POLG mutations is mediated, in part, through diminished accuracy of DNA replication by pol γ. Whereas mutation of certain residues in the polymerase domain that do not directly interact with the incoming dNTP increases the error frequency of pol γ by only 2- to 4-fold, mutation of residues that stabilize the incoming dNTP increases the error frequency as much as 45-fold (20,24). To understand better their influence on pathogenicity, we wished to measure the impact, if any, of the W748S, E1143G and W748S-E1143G mutations on the fidelity of DNA replication by pol γ. The fidelity of these polymerases was estimated by measuring the relative efficiency of inserting cognate or non-cognate nucleotides onto a 22mer/40mer oligonucleotide primer-template in vitro. Non-linear curve-fitting was used to calculate steady-state kinetic parameters (Km and kcat) for inserting correct (dTTP) and incorrect (dATP) nucleotides, and overall efficiency was represented as kcat/Km (Table 3). Replication error frequencies for a given enzyme are derived from the ratio of incorrect and correct insertional efficiencies [f=(kcat/Km)incorrect/(kcat/Km)correct] in this DNA sequence context. Different polymerases were compared by normalizing misincorporation frequencies to that of WT (fmutant/fWT), such that a value below 1 reflects a more accurate enzyme, whereas a value above 1 reflects a less faithful enzyme. All the mutant enzymes, W748S, E1143G and W748S-E1143G, displayed near-equal replication error frequencies with WT p140 in the absence of p55, thus these mutations in p140 had insignificant effects on fidelity. Upon addition of p55, E1143G p140 had an error frequency similar to that of WT p140. However, W748S p140 and W748S-E1143G p140 displayed 2.1- and 1.4-fold increases, respectively, in nucleotide misinsertion upon addition of p55. Thus, addition of the accessory subunit improved DNA binding of the pol γ complex containing W748S and W748S-E1143G p140s, but this in turn led to greater nucleotide misinsertion. As seen with our other assays, the E1143G polymorphism neutralizes the W748S effect, in this case nucleotide misinsertion effect caused by p55 binding.
Fidelity of DNA synthesis by pol γ variants bearing W748S and E1143G substitutions
| Enzyme | (kcat/Km)dTTP (min·µm)−1 | (kcat/Km)dATP (min·µm)−1 | f=(kcat/Km)dATP/(kcat/Km)dTTP | fmutant/fWT |
|---|---|---|---|---|
| WT | 74 | 0.19 | 0.0026 | 1 |
| E1143G | 95 | 0.27 | 0.0029 | 1.1 |
| W748S | 12 | 0.028 | 0.0024 | 0.90 |
| W748S-E1143G | 6.4 | 0.014 | 0.0022 | 0.83 |
| WT+p55 | 150 | 0.29 | 0.0019 | 1 |
| E1143G+p55 | 160 | 0.30 | 0.0019 | 0.99 |
| W748S+p55 | 67 | 0.26 | 0.0039 | 2.1 |
| W748S-E1143G+p55 | 57 | 0.15 | 0.0027 | 1.4 |
| Enzyme | (kcat/Km)dTTP (min·µm)−1 | (kcat/Km)dATP (min·µm)−1 | f=(kcat/Km)dATP/(kcat/Km)dTTP | fmutant/fWT |
|---|---|---|---|---|
| WT | 74 | 0.19 | 0.0026 | 1 |
| E1143G | 95 | 0.27 | 0.0029 | 1.1 |
| W748S | 12 | 0.028 | 0.0024 | 0.90 |
| W748S-E1143G | 6.4 | 0.014 | 0.0022 | 0.83 |
| WT+p55 | 150 | 0.29 | 0.0019 | 1 |
| E1143G+p55 | 160 | 0.30 | 0.0019 | 0.99 |
| W748S+p55 | 67 | 0.26 | 0.0039 | 2.1 |
| W748S-E1143G+p55 | 57 | 0.15 | 0.0027 | 1.4 |
The steady-state kinetics of incorporating a single correct (dTTP) or incorrect (dATP) nucleotide into a synthetic oligonucleotide primer-template were measured as described in the Materials and Methods section. Error frequencies are derived from the ratio f=(kcat/Km)incorrect/(kcat/Km)correct.
Fidelity of DNA synthesis by pol γ variants bearing W748S and E1143G substitutions
| Enzyme | (kcat/Km)dTTP (min·µm)−1 | (kcat/Km)dATP (min·µm)−1 | f=(kcat/Km)dATP/(kcat/Km)dTTP | fmutant/fWT |
|---|---|---|---|---|
| WT | 74 | 0.19 | 0.0026 | 1 |
| E1143G | 95 | 0.27 | 0.0029 | 1.1 |
| W748S | 12 | 0.028 | 0.0024 | 0.90 |
| W748S-E1143G | 6.4 | 0.014 | 0.0022 | 0.83 |
| WT+p55 | 150 | 0.29 | 0.0019 | 1 |
| E1143G+p55 | 160 | 0.30 | 0.0019 | 0.99 |
| W748S+p55 | 67 | 0.26 | 0.0039 | 2.1 |
| W748S-E1143G+p55 | 57 | 0.15 | 0.0027 | 1.4 |
| Enzyme | (kcat/Km)dTTP (min·µm)−1 | (kcat/Km)dATP (min·µm)−1 | f=(kcat/Km)dATP/(kcat/Km)dTTP | fmutant/fWT |
|---|---|---|---|---|
| WT | 74 | 0.19 | 0.0026 | 1 |
| E1143G | 95 | 0.27 | 0.0029 | 1.1 |
| W748S | 12 | 0.028 | 0.0024 | 0.90 |
| W748S-E1143G | 6.4 | 0.014 | 0.0022 | 0.83 |
| WT+p55 | 150 | 0.29 | 0.0019 | 1 |
| E1143G+p55 | 160 | 0.30 | 0.0019 | 0.99 |
| W748S+p55 | 67 | 0.26 | 0.0039 | 2.1 |
| W748S-E1143G+p55 | 57 | 0.15 | 0.0027 | 1.4 |
The steady-state kinetics of incorporating a single correct (dTTP) or incorrect (dATP) nucleotide into a synthetic oligonucleotide primer-template were measured as described in the Materials and Methods section. Error frequencies are derived from the ratio f=(kcat/Km)incorrect/(kcat/Km)correct.
E1143G modulates the thermostability of WTand W748S p140 proteins
Variability in the DNA polymerase activities of the W748S, E1143G and W748S-E1143G enzymes, together with the apparent ability of the E1143G substitution to partially rescue the DNA-binding deficiency caused by the W748S mutation, suggests these enzymes may have subtle conformational differences. Although these mutations did not cause gross alterations in secondary structure (Fig. 2), small changes in secondary structure may affect the intrinsic stability of each protein. Accordingly, we monitored thermal denaturation of α-helices within each protein by CD spectroscopy (Fig. 6A) and calculated the melting temperature (Tm) and change in enthalpy for protein folding (ΔHm) to estimate the intrinsic stability of each protein, as described (25). The Tms of the four proteins were clustered, with Tm values between 44 and 47°C (Fig. 6B). WT p140 and W748S p140 had similar ΔHm values (38 and 33 kcal/mol, respectively), which were indistinguishable within our experimental error. Surprisingly, the ΔHm value for E1143G p140 was significantly higher (49 kcal/mol), indicating an increased stability compared with the WT protein (Fig. 6B). However, the ΔHm for the W748S-E1143G mutant protein (26 kcal/mol) was significantly lower than WT and W748S p140, which implies that decreased stability contributed to the biochemical defect of this double mutant enzyme.
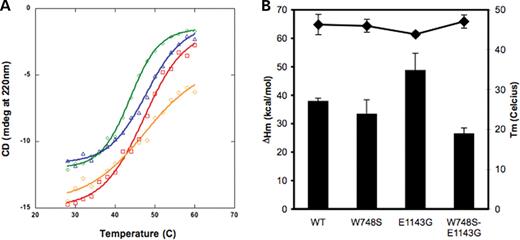
Stability of secondary structure of WT and mutant p140 proteins. CD analysis was performed as described in the Materials and Methods section. (A) Thermal stability of WT and mutant p140 proteins: WT p140 (red squares), W748S p140 (blue triangles), E1143G p140 (green diamonds) and W748S-E1143G (orange circles). The protein concentration of each sample was 10 µg/ml. (B) Melting temperature and enthalpy change of p140 proteins. Diamonds represent the melting temperatures for each p140 protein. Bars represent the change in enthalpy for protein folding at 220 nm. All values are the average of 3 or 4 independent determinations. Standard deviations are represented by error bars.
Molecular modeling of the E1143G substitution
On the basis of homology to the Family A DNA polymerases and the three-dimensional structure of T7 DNA polymerase in complex with an incoming dNTP, a primer-template DNA, and thioredoxin, we built a molecular model of the human pol γ active site (20,26). Encompassing residues 871–1182 of p140, the model differs from the T7 structure by a root mean squared deviation of only 2.44 Å. Although amino acid W748 falls outside this model and is an area with little homology to known protein structures, E1143 is only 7 amino acid residues removed from the two catalytic carboxylic amino acids in polymerase motif C (D1135-E1136) that chelate two catalytic magnesium ions (Fig. 7). The DE amino acids that make up motif C (HDE) are within the most conserved motif in all DNA polymerases. Though E1143 is linearly in close proximity to the active site, it is not within the active site of our molecular model. Instead, E1143 is located at the turn between a β-strand and an α-helix, which appears near the surface of this active site model, distant from the HDE residues. A substitution from glutamic acid to glycine might allow greater flexibility of the downstream α-helix and β-strand (Fig. 7). We do not currently know how the remaining portion of the polymerase molecule interacts with this part of the protein. As the E1143G polymorphism appears to modulate the effects of the W748S mutation, a complete three-dimensional structure of the pol γ catalytic subunit is needed to fully appreciate the consequences of these substitutions.
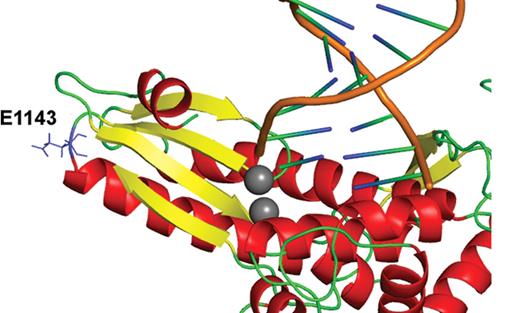
Glu1143 in the pol γ polymerase domain structural homology model. Note the position of E1143—at the turn between a β-strand (yellow) and α-helix (red). The two catalytic Mg2+ ions are colored gray and the DNA primer-template is shown in orange/blue/green. The Glu1143 side chain is exterior to the active site center (labeled).
DISCUSSION
W748S-E1143G is the most common pol γ allele associated with inherited ataxias and is also associated with two other mitochondrial diseases, namely Alpers syndrome and PEO. It has been unclear whether the E1143G polymorphism participates in the disease process. Thus, we characterized the recombinant pol γ proteins containing W748S or E1143G, or both substitutions in vitro, using functional and physical methods to assess whether the E1143G mutation significantly alters the phenotype of W748S.
W748S p140
W748S pol γ had a severe catalytic defect, with poor processive DNA synthesis and primer extension. The severely reduced ability of W748S pol γ to bind DNA appears to contribute greatly to its decreased catalytic efficiency. Accessory subunit binding was normal, and this compensated for some of the DNA-binding defect by anchoring W748S p140 to DNA. Despite some improvement, WT levels of catalysis were not observed. Additionally, a mild mutator effect was observed in the presence of p55. As mutations can have deleterious effects on protein folding, and in turn, enzymatic function, we used CD analysis to assess the secondary structure of pol γ. W748S p140 was similar to WT in terms of Tm and ΔHm (Fig. 6B), and thus the catalytic shortcomings of this enzyme are due to its severe DNA-binding defect, and not to defects of intrinsic protein stability.
E1143G p140
Unexpectedly, E1143G p140 had a consistently higher catalytic activity than WT enzyme, as demonstrated by our polymerase assays. This increase in catalytic activity was largely manifest through an increase in catalytic rate (kcat) for the incoming dNTP. E1143G p140 was similar to WT p140 for processive DNA synthesis and fidelity of nucleotide selection. Accessory subunit binding was also normal. Furthermore, the Tm was not significantly different to WT p140 and W748S p140. Unlike WT p140, however, E1143G p140 had a 1.3-fold higher intrinsic stability. This physical property helps to explain why E1143G pol γ had a higher catalytic activity than WT. It is interesting to note the sharp slope of the denaturation curve for E1143G, which suggests a rapid loss of function once its Tm is reached, compared with the other enzymes. However, the measured Tms are much higher than survivable fever temperatures in humans. Amino acid 1143, although in close linear proximity to the HDE residues, is not predicted to be within the active site of the polymerase domain, as shown by molecular modeling, and this may help to explain why E1143G does not drastically affect polymerase catalytic activity. As there are many conformations in which glycine can rotate within proteins, a substitution to glycine at position 1143 may increase the flexibility of the enzyme at this site. This may also help explain the increased specific activity of E1143G p140 compared with WT p140. In short, p140 protein containing the E1143G polymorphism is a good enzyme, with higher catalytic activity and protein stability compared with WT.
W748S-E1143G p140
As shown earlier, W748S p140 and E1143G p140 have very different characteristics. The W748S-E1143G double mutant had biochemical properties that appear to be a melding of those exhibited by W748S and E1143G p140. The double mutant also had low, but intermediate catalytic activity. Catalytic activity increased upon addition of the accessory subunit, to a much greater extent than for W748S:p55, but not to WT:p55 levels. W748S-E1143G pol γ also had an intermediate Kd(DNA) value, which suggests a partial rescue of the W748S defect by E1143G. Additionally, the double mutant had a slight mutator effect. In contrast to the biochemical assays in which the E1143G polymorphism conferred enhanced DNA binding and activity to the W748S protein, the addition of E1143G proved detrimental to protein stability. W748S-E1143G enzyme was significantly less stable than the rest. The lower ΔHm for W748S-E1143G p140 suggests this protein may unfold more readily at body temperature than the other proteins, which could account for some of its catalytic defect, as our assays are performed at 37°C. The denaturation curve of the double mutant is much broader than the others, which suggests that at 37°C, a greater percentage of this enzyme may be partially unfolded compared with the others.
We previously showed weak binding of A467T pol γ with the accessory subunit (4). As A467T and W748S are both linker region mutations associated with similar disease phenotypes, we first expected the biochemical defects to be similar. The A467T mutation is within the conserved γ1 domain of the linker region, which corresponds with data from Luo and Kaguni (27), who showed decreased processivity and subunit binding when this domain was deleted in Drosophila pol γ. In the same study, Luo and Kaguni also deleted the conserved γ4 region of pol γ linker region in Drosophila homologous to the region in human POLG that contains W748 (corresponding approximately to residues 747–838 in human pol γ, with residue 748 near the beginning of this deletion). In this scenario, disrupted subunit binding was also shown. In contrast, our NEM assay, immunoprecipitation assay, processivity assay and kinetic analysis of the W748S substitution show that binding between the catalytic and accessory subunits was normal. Differences between these two situations are likely due to the limited homology between human and Drosophila pol γ; moreover, a large portion of the Drosophila pol γ linker region was deleted, compared with the single mutation (W748S) in human pol γ. Additionally, W748 in human pol γ corresponds to phenylalanine at this codon in Drosophila pol γ.
W748S and W748S-E1143G enzymes in complex with p55 exhibited reduced discrimination against inserting an incorrect nucleotide. Since residue 748 is not located in the polymerase active site (20), the 1.4- to 2.1-fold effect could be caused by more global structural alterations that affect nucleotide recognition. Alternatively, as the W748S substitution interferes with DNA binding by p140, reduced nucleotide discrimination may be related to altered stability of the ternary complex formed by these enzymes, the incoming nucleotide and DNA. In vivo, this small increase in misinsertion errors by the W748S and the double mutant would likely be masked by the proofreading exonuclease activity. Previous fidelity analysis of human pol γ demonstrates that the contribution of the 3′–5′ exonuclease to overall fidelity is at least 20-fold (28). The Y955C disease variant of pol γ is the most error-prone pol γ documented to date. Analysis of Y955C protein revealed a 45-fold increase in misinsertion errors in the absence of proofreading but only a 2-fold increase in the proofreading-proficient background (24). This suggests that the majority of the errors (95%) produced by Y955C p140 are corrected. The same degree of proofreading would diminish errors in W748S patients. Nonetheless, in addition to the other deleterious effects noted for W748S and W748S-E1143G, this small increase in error frequency may have a slight but significant effect that may aggravate clinical severity.
WT pol γ has been documented in the literature to be rather unstable in vitro, as the isolated catalytic subunit is sensitive to oxidation (29) and has a short half-life at 37°C of only 5 min in the absence of DNA (19). The catalytic subunit can be adequately protected from oxidation and chemical treatment (such as by NEM) by binding with the accessory subunit (22,29). The accessory subunit likely protects the catalytic subunit by physically protecting thermally and chemically sensitive areas on the protein. Ultimately, the catalytic and DNA-binding defects of W748S and W748S-E1143G are mitigated by the accessory subunit. As enhanced DNA binding is the basis of this rescue, this same effect aggravates misinsertion.
Clinical aspects
The nature of the mutation in the second POLG allele in compound heterozygous W748S-E1143G patients may influence severity of disease (Table 1). It appears that mutating amino acids that are highly conserved (such as L244 or G848) and thus likely to be more important for protein structure and function brings about Alpers syndrome, the most devastating mitochondrial disease associated with pol γ mutations, with the earliest age of onset. Transcripts from the allele containing the Y1210fs1216stop mutation are most likely degraded through the nonsense-mediated decay pathway, as we previously documented for another pol γ mutation causing a premature termination codon (E873stop) (30). Mutations of less conserved amino acids, such as L304R, are associated with a later onset disease, ANS.
Similar to A467T p140, W748S-E1143G p140 is associated with a spectrum of disease states. However, unlike homozygous A467T patients, who present with either Alpers syndrome or ANS or PEO, patients homozygous for W748S-E1143G only present with ANS or PEO. Although the overwhelming majority of reports state that W748S-E1143G is a recessive allele, a recent study showed one heterozygous patient (WT/W748S-E1143G) with later onset ANS with PEO/ptosis (13). This patient may not have a monogenic disease, but may have a disease-causing mutation in another allele, such as POLG2, which was not tested in that particular study. Furthermore, we cannot discount the possible effects of environmental factors, drugs (for example, valproic acid has been found to be toxic in Alpers patients, and nucleoside reverse transcriptase inhibitors can also cause mitochondrial dysfunction), as well as the inherited mtDNA mutation load. Additionally, since our biochemical analysis is restricted to the p140 and p55 pol γ subunits, the effect of the W748S and E1143G substitutions on interactions with other proteins involved in mtDNA replication and repair, such as the mitochondrial helicase or the single stranded DNA-binding protein, was not assessed.
The E1143G polymorphism is found in ∼3–4% of the population, but it is not equally dispersed. It is found at this percentage in the European population but was not found at all in the Asian, sub-Saharan African or African American population (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2307441). In one Finnish study, a founder effect was shown for the E1143G substitution in patients harboring the W784S-E1143G allele (12). Although no epidemiology on this mutation has yet been published, it would be interesting to investigate whether the E1143G polymorphism by itself has benefited humans by increasing longevity or improving health.
The prevalence of the E1143G polymorphism in persons with known mitochondrial diseases involving POLG mutations appears to be high (Tables 1 and 4). The addition of E1143G in cis reveals its possible role in disease. For example, the E1143G mutation has been found in cis with A889T in autosomal-dominant PEO (31); however, an earlier study suggested that A889T on its own is a recessive mutation (32). Interactions between the two mutated residues within the same protein molecule may be deleterious, as shown with our CD analysis. Alternatively, partial rescue of function in the W748S p140 by E1143G suggests that the E1143G polymorphism allows a more detrimental mutation to survive selection processes by reducing the burden of the deleterious mutation. The low number of patients observed with only W748S reinforces this theory. Horvath et al. (15) recently reported 38 patients with POLG mutations. Five of these patients (13%) were reported to have the E1143G mutation (Table 4). Although the number of patients in this subcategory is small, the high frequency of E1143G among patients with mitochondrial disease containing other POLG mutations suggests that E1143G may not be neutral, in particular for two patients bearing A467T/E1143G and S433C/E1143G. Because of these results, it has been suggested that the E1143G polymorphism is pathogenic only when it is in compound with other POLG mutations (15,18). We have shown here that E1143G by itself is structurally stable and highly active. However, we also show that E1143G is not a neutral polymorphism in pol γ, as E1143G can both contribute and alleviate the pathogenicity of other mutations in cis (in this case, W748S).
Thus, we propose that, in general, persons with WT/W748S-E1143G retain sufficient polymerase function from the WT allele for survival. Nonetheless, pol γ activity in patients homozygous for W748S-E1143G is inadequate to maintain a sufficient quantity of functional mtDNA as the patient ages. E1143G p140 is a better enzyme than WT p140 in terms of catalytic activity and protein stability. However, when this polymorphism is in cis with other pathogenic mutations in pol γ, it can modulate the effects of these mutations. For W748S-E1143G, the benefits bestowed by E1143G are increased DNA binding and polymerase activity; conversely, E1143G is also detrimental as shown by significantly reduced protein stability.
Genotypes (if known) from patients reported to harbor the E1143G polymorphism
| POLG mutations | Phenotype | Reference |
|---|---|---|
| A889T+E1143G/WT | PEO | (31) |
| E1143Ga | Mitochondrial disease | (36) |
| E1143Ga | mtDNA deletions | (37) |
| E1143Ga | Myasthenia gravis with mtDNA deletions | (38) |
| Q497H+W748S+E1143G/Q497H+W748S+E1143G | ANS | (11,12) |
| S433C/E1143G | PEO | (15,18) |
| A467T/E1143G | PEO | (15) |
| A467T T885S/Q879H+E1143G | Alpers | (15) |
| R627Q+Q1236H/L965X+E1143G | PEO | (15) |
| G517V/E1143G | adANS | (15) |
| POLG mutations | Phenotype | Reference |
|---|---|---|
| A889T+E1143G/WT | PEO | (31) |
| E1143Ga | Mitochondrial disease | (36) |
| E1143Ga | mtDNA deletions | (37) |
| E1143Ga | Myasthenia gravis with mtDNA deletions | (38) |
| Q497H+W748S+E1143G/Q497H+W748S+E1143G | ANS | (11,12) |
| S433C/E1143G | PEO | (15,18) |
| A467T/E1143G | PEO | (15) |
| A467T T885S/Q879H+E1143G | Alpers | (15) |
| R627Q+Q1236H/L965X+E1143G | PEO | (15) |
| G517V/E1143G | adANS | (15) |
These genotypes are in addition to those listed in Table 1.
aThese studies noted E1143G in their patient population; however, E1143G was not ascribed to any patient in particular, thus the full genotypes of these patients are not known.
ad, autosomal dominant.
Genotypes (if known) from patients reported to harbor the E1143G polymorphism
| POLG mutations | Phenotype | Reference |
|---|---|---|
| A889T+E1143G/WT | PEO | (31) |
| E1143Ga | Mitochondrial disease | (36) |
| E1143Ga | mtDNA deletions | (37) |
| E1143Ga | Myasthenia gravis with mtDNA deletions | (38) |
| Q497H+W748S+E1143G/Q497H+W748S+E1143G | ANS | (11,12) |
| S433C/E1143G | PEO | (15,18) |
| A467T/E1143G | PEO | (15) |
| A467T T885S/Q879H+E1143G | Alpers | (15) |
| R627Q+Q1236H/L965X+E1143G | PEO | (15) |
| G517V/E1143G | adANS | (15) |
| POLG mutations | Phenotype | Reference |
|---|---|---|
| A889T+E1143G/WT | PEO | (31) |
| E1143Ga | Mitochondrial disease | (36) |
| E1143Ga | mtDNA deletions | (37) |
| E1143Ga | Myasthenia gravis with mtDNA deletions | (38) |
| Q497H+W748S+E1143G/Q497H+W748S+E1143G | ANS | (11,12) |
| S433C/E1143G | PEO | (15,18) |
| A467T/E1143G | PEO | (15) |
| A467T T885S/Q879H+E1143G | Alpers | (15) |
| R627Q+Q1236H/L965X+E1143G | PEO | (15) |
| G517V/E1143G | adANS | (15) |
These genotypes are in addition to those listed in Table 1.
aThese studies noted E1143G in their patient population; however, E1143G was not ascribed to any patient in particular, thus the full genotypes of these patients are not known.
ad, autosomal dominant.
MATERIALS AND METHODS
Expression and purification
The cDNAs for Exo− WT and mutant forms of the His6 affinity-tagged recombinant catalytic subunit of human pol γ were expressed in baculovirus-infected Sf9 cells as described (19), and the proteins were purified to homogeneity, as described previously (20). The His6 affinity-tagged p55 accessory subunit was expressed in Escherichia coli and purified to homogeneity, as described previously (22). The E1143G mutant derivative of p140 was constructed using the Exo− pQVSL11.4 baculoviral transfer vector encoding p140 Exo− without its mitochondrial targeting sequence (33) as template. Codon 1143 was converted to the E1143G derivative with the QuikChange site-directed mutagenesis kit (Stratagene) and the mutagenic primers 5′-CCT GGT GCG GGG GGA GGA CCG CTA CC′ and 5′-GGT AGC GGT CCT CCC CCC GCA CCA GG-3′. The W748S mutant derivative of p140 was also constructed using the Exo−pQVSL11.4 baculoviral transfer vector (33), and the W748S derivative was made with the QuikChange site-directed mutagenesis kit (Stratagene) and the mutagenic primers 5′-CAT CCC TGG CTG CTC GTT TTT CAA GC-3′ and 5′-GCT TGA AAA ACG AGC AGC CAG GGA TG-3′. The W748S and E1143G mutations were confirmed by DNA sequencing of the baculovirus transfer vector. The E1143G template was then used to make W748S-E1143G derivative. Recombinant baculoviruses expressing each mutant pol γ catalytic subunit were selected and amplified, and Sf9 insect cells from 30 confluent T175 tissue culture flasks were transfected for protein expression and processed as described previously (19,20). MonoQ fractions containing each p140 derivative were frozen in small aliquots in liquid nitrogen and stored at −80°C.
Enzymatic assays
DNA polymerase activity was determined in our standard pol γ assays with either poly(dA)·oligo(dT)12–18 or poly(rA)·oligo(dT)12–18 (Amersham Biosciences) as the primer-template substrate. Steady-state kinetic values were determined with this standard assay, using poly(dA)·oligo(dT)12–18, as previously described (22), and reactions were supplemented with 25 mm NaCl. The two-subunit form of pol γ was reconstituted as described (22). The processivity of WT and mutant forms of pol γ were estimated by primer extension assay on an end-labeled, singly primed M13 DNA substrate, as described (19), without the pre-incubation step. Reaction mixtures (10 µl) contained 25 mm HEPES-KOH, pH 7.6, 5 mm 2-mercaptoethanol, 5 mm MgCl2, 0.05 mg/ml heat-treated BSA, 0 or 100 mm NaCl, 25µm dNTP, 0.02 pmol of the labeled oligonucleotide, 12 ng purified WT or mutant p140 and 9.6 ng of the p55 accessory subunit, as indicated. Following incubation at 37°C for 20 min, reactions were terminated and products were analyzed by denaturing polyacrylamide gel electrophoresis, as described (19). Products were quantified with a Typhoon 9400 PhosphorImager (Molecular Dynamics) and NIH Image software.
Fidelity assay
The fidelity of nucleotide selection by pol γ was determined with a synthetic oligonucleotide substrate in a polyacrylamide gel-based, single nucleotide extension assay, using oligonucleotides to generate the 22mer/40mer primer-template (20,28). Reactions included 1.0 pmol primer-template, with 50–450 fmol (7–64 ng) of the specified Exo− p140 variants, as well as 2 equivalents of the p55 subunit, as indicated. Km and Vmax values were determined by fitting the data to the steady-state Michaelis–Menten model, using KaleidaGraph (version 4.0, Synergy).
Electrophoretic mobility shift assay
The Kd(DNA) value for each form of p140 was determined by electrophoretic mobility shift assay, as described previously (34). Short, double-stranded primer-template molecules were constructed by hybridizing a 5′-end 32P-labeled 38mer (5′-TTA TCG CAC CTA CGT TCA ATA TTA CAG GCG AAC ATA CT-3′) to a 1.2-fold molar excess of an unlabeled, complementary 34mer (5′-GTA TGT TCG CCT GTA ATA TTG AAC GTA GGT GCG A-3′). Binding mixtures (20 µl) were assembled on ice and contained 10 mm HEPES-OH (pH 8.0), 0.2 mg/ml acetylated BSA, 2 mm dithiothreitol, 1 pmol of primer-template and 0–6 pmol of the indicated pol γ protein. Bound and unbound primer-template molecules were resolved at 4°C by polyacrylamide gel electrophoresis through 6% native polyacrylamide (60:1 acrylamide/bis-acrylamide) gels also containing 20 mm HEPES-OH (pH 8.0) and 0.1 mm EDTA. After 3 h at 110 V, gels were dried and exposed to a phosphor storage screen. Radioactivity was imaged on a Typhoon 9400 phosphorimager (GE Healthcare, Piscataway, NJ), and bands were quantified with NIH image software.
CD measurements and analysis
CD studies were performed with a Jasco 810 spectropolarimeter equipped with a Peltier thermal controller (Jasco, Inc., MD), following the method of DeRose et al. (25) with the following modifications. All p140 samples were adjusted to a protein concentration of 10 µg/ml in 10 mm KPO4 buffer (pH 7.5) with 5% glycerol, 0.1 mm EDTA, 0.1 mm 2-mercaptoethanol and 200 mm NaCl and placed in a 1 cm cell. CD spectra were measured at 4°C over a 200–260 nm range. The thermal stabilities of α-helices within the p140 proteins were determined from the ellipticity at 220 nm as the temperature was increased from 28–60°C by 1°C per minute, with stirring. Ellipticity was sampled every 1–2°C, and the melting temperature (Tm) and enthalphy change upon unfolding (ΔHm) for each p140 protein were determined by sigmoidal curve fitting of the thermal denaturation curves (KaleidaGraph). As renaturation was not possible (data not shown), denatured protein samples were discarded after melting curves were obtained.
Molecular modeling
PyMOL (DeLano Scientific, CA) (35) was used to visualize the E1143G substitution in the three-dimensional model of human pol γ catalytic domain (20,26).
ACKNOWLEDGEMENTS
We would like to thank Dr Robert Petrovich of the NIEHS Protein Expression Core Facility, Dr Geoffrey Mueller of the Nuclear Magnetic Resonance group for discussion on the CD studies and Drs Lars Pedersen and Mercedes Arana for critically reading this manuscript. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Conflict of Interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}