




Abstract
Myotonic dystrophy type I (DM1) is an RNA-mediated disease caused by a non-coding CTG repeat expansion. A key feature of the RNA-mediated pathogenesis model for DM is the disrupted splicing of specific pre-mRNA targets. A link has been established between splicing regulation by CUG-BP1, a member of the CELF family of proteins, and DM1 pathogenesis. To determine whether increased CUG-BP1 function was sufficient to model DM, transgenic mice overexpressing CUG-BP1 (MCKCUG-BP1) in heart and skeletal muscle, two tissues affected in DM1, were generated. Histological and electron microscopic analyses of skeletal muscle reveal common pathological features with DM tissues: chains of central nuclei, degenerating fibers and centralized NADH reactivity. MCKCUG-BP1 mice have disrupted splicing of three CELF target pre-mRNAs, cardiac troponin T (Tnnt2), myotubularin-related 1 gene (Mtmr1) and the muscle-specific chloride channel (Clcn1), consistent with that observed in DM heart and skeletal muscle. The results are consistent with a mechanism for DM pathogenesis in which expanded repeats result in increased CUG-BP1 activity and/or other CELF family members and have trans-dominant effects on specific pre-mRNA targets.
INTRODUCTION
Myotonic dystrophy (DM) is a repeat expansion disease in which expanded CTG (type I) or CCTG (type II) repeats cause disease (1,2). The RNA transcribed from the expanded alleles containing CUG or CCUG repeats accumulate in nuclear foci and are proposed to cause trans-dominant effects on splicing by altering the functions of RNA binding proteins (3–5). Consistent with an RNA gain of function model, transgenic mice (HSALR) expressing 250 CUG repeats in the 3′-UTR of human skeletal actin mRNA reproduce the characteristic myotonia, abnormal muscle histology and splicing changes observed in DM1 (5,6). The mechanism by which expanded repeats alter the regulation of alternative splicing is unknown. Two families of RNA binding proteins have been implicated in DM1 pathogenesis based on their abilities to interact with CUG RNA: CUG-BP1 and ETR-3 like factors (CELF) and muscleblind-like (MBNL) proteins (7–10).
MBNL proteins are physically linked with the RNA foci and are functionally linked with the mis-regulation of pre-mRNA targets in DM. MBNL sequestration has been proposed to cause disease because MBNL proteins co-localize with both CUG and CCUG foci and depletion of MBNL proteins by siRNA in tissue culture reproduce the DM pattern of splicing (8,11–13). Mice deficient for Mbnl1 isoforms (MbnlΔ3/Δ3) that specifically bind CUG repeats exhibit myotonia, cataracts and splicing changes similar to individuals with DM (14). The myotonia observed in MbnlΔ3/Δ3 and HSALR mice correlate with splicing mis-regulation of the skeletal muscle chloride ion conductance channel (Clcn1) leading to decreased expression of ClC-1 protein.
CELF proteins associate with CUG repeats in vitro and are functionally linked with the aberrant regulation of alternative splicing in DM (10,15,16). To date, six CELF (also called BRUNOL) genes have been identified in humans (7,17,18). All six CELF proteins have been shown to regulate pre-mRNA alternative splicing and two (CUG-BP1 and ETR-3/CUG-BP2/NAPOR) have been shown to have cytoplasmic RNA processing functions (19,20). CUG-BP1, as well as other CELF family members, regulates alternative splicing of at least three of the pre-mRNAs that are mis-regulated in DM striated muscle, cardiac troponin T (cTNT), insulin receptor (IR) and ClC-1 (21–23). The splicing patterns observed for all three pre-mRNAs are consistent with increased CUG-BP1 activity and the observed increase in CUG-BP1 steady state levels in DM1 striated muscle and skeletal muscle cultures (13,16,21–23).
cTNT minigenes expressed in DM1 muscle cultures or cTNT and IR minigenes co-expressed with CUG repeat RNA in normal cells reproduce the aberrant splicing patterns observed for endogenous mRNAs in DM tissue (21,22). The trans-dominant effect of endogenous or co-expressed CUG repeat RNA on cTNT and IR splicing regulation require the intronic CUG-BP1 binding sites that are also required to regulate splicing of the alternative exons. Thus, binding by CUG-BP1 and/or other CELF family members to intronic regulatory elements is required for induction of aberrant splicing regulation by CUG repeat RNA (21,22).
Transgenic mice that overexpress CUG-BP1 under a modified β-actin promoter display histological abnormalities and differentiation defects in skeletal muscle that are associated with DM1 (24). Several lines were generated expressing different levels of His-CUG-BP1 varying from 2–3-fold to 8–10-fold that of endogenous CUG-BP1. Although the cytoplasmic functions of CUG-BP1 were studied, nuclear functions of CUG-BP1, such as the regulation of alternative splicing, were not evaluated in these transgenic mice.
To determine whether increased CUG-BP1 function was sufficient to reproduce the mis-regulation of splicing observed in DM, we generated transgenic mice (MCKCUG-BP1) that specifically express CUG-BP1 in skeletal muscle and heart, two tissues affected in DM. Similar to the expression of CUG-BP1 under a modified β-actin promoter, our transgenic mice also showed neonatal lethality when CUG-BP1 expression is 4–6-fold above endogenous levels in neonatal heart and skeletal muscle. Histological and electron microscopic (EM) analyses suggest several common histological characteristics with congenital DM skeletal muscle such as central nuclei, chains of nuclei and centralized nicotinamide adenine dinucleotide (NADH) reactivity. Importantly, MCKCUG-BP1 mice reproduce the DM splicing pattern for Tnnt2, Mtmr1 and Clcn1 in striated muscle tissues. Transgenic mice expressing CUG-BP1 have similar splicing patterns for Tnnt2 and Clcn1 to MbnlΔ3/Δ3 mice consistent with a previous demonstration that CELF and MBNL proteins are antagonistic splicing regulators of pre-mRNAs mis-regulated in DM (12). Our results support the possibility that elevated CELF activity, in addition to loss of MBNL activity, contributes to mis-regulation of alternative splicing and DM pathogenesis.
RESULTS
Striated muscle-specific expression of CUG-BP1 results in neonatal lethality
To generate transgenic mice expressing CUG-BP1 only in striated muscle, we subcloned human CUG-BP1 downstream from a mouse genomic fragment containing the creatine kinase (MCK) promoter, enhancer, the untranslated exon 1 and intron 1 (25) (Fig. 1A). The restricted expression of this MCK genomic fragment in heart and skeletal muscle has been well characterized (25,27). The transgene contains an N-terminal FLAG-tagged version of human CUG-BP1 (‘A’ isoform), followed by the bovine growth hormone 3′ flanking genomic segment for proper mRNA 3′ end formation. The FLAG-tagged form of CUG-BP1 is functional in minigene splicing assays (data not shown). Of 33 pups analyzed, seven animals were positive for the transgene. Two successfully established transgenic lines (1032 and 1036) expressing protein and a third founder male (1039), produced 50% transgenic progeny, all of which were dead at birth. Transgenic 1039 pups were normal in size, but were stillborn. It is likely that the 1039 founder male was chimeric.
To determine the relative levels of CUG-BP1 expression, western blot analysis was conducted on serial dilutions of heart and skeletal muscle lysates in the 1039 line (Fig. 1B and C). The 3B1 monoclonal antibody detects both exogenously expressed human CUG-BP1 and endogenously expressed mouse CUG-BP1. Assuming that 3B1 recognizes mouse and human CUG-BP1 with equal affinity, expression of CUG-BP1 is ∼4–6 fold above endogenous levels in both heart and skeletal muscle. The other two lines (1032, 1036) expressed exogenous CUG-BP1 at ∼0.5 and 1.0 times the endogenous level, respectively, in both heart and skeletal muscle (data not shown). Neither of the lower expressing lines had an overt phenotype. Further analyses were conducted on the 1039 line.
Histological and EM analyses of skeletal muscle in transgenic mice
To determine the effects of CUG-BP1 expression on skeletal muscle development, we examined the histology of MCKCUG-BP1 mice by light microscopy. In longitudinal sections, the muscle fascicles of transgenic and normal mice stain red in modified Gomori trichrome preparations and are separated by fibroblasts with delicate processes (Fig. 2A). In the transgenic mice, individual fibers exhibited a central accumulation of red-staining homogenous material, which extended for 20–60 µm and the material is surrounded by muscle nuclei (Fig. 2A). MCKCUG-BP1 muscle fibers showed chains of central nuclei (Fig. 2A, yellow arrowheads) and degeneration (Fig. 2A, black arrowheads). Histochemistry showed a mixture of fiber types in both wild-type and transgenic animals. In the transgenic mice, there was an increased central accumulation of NADH reactivity and acid phosphatase reactivity compared with the wild-type animals (Fig. 2B) (data not shown). This was not apparent with succinate dehydrogenase (SDH) reactivity (data not shown).
EM analysis reveals the presence of irregularly shaped nuclei, centrally located nuclear chains and degeneration of myofibers in MCKCUG-BP1 when compared with wild-type (Fig. 2C and D). Wild-type animals exhibited small fibers with central nuclei consistent with immature skeletal muscle. The transgenic animals exhibited an increased number of internal nuclei in individual fibers.
In congenital DM, affected neonates have CTG repeat lengths greater than 1000, display severe hypotonia and dysphagia, but do not develop myotonia until later in life (28). Skeletal muscle development is impaired in congenital DM and share several common abnormalities such as sarcoplasmic masses, chains of centrally located nuclei, large variations in muscle fiber size and poor fiber type differentiation (29–33). These results suggest that mice expressing CUG-BP1 in skeletal muscle display some pathological features observed in DM tissue. Histological analysis of heart tissue showed no discernable differences from wild-type littermates (data not shown).
Mis-regulated splicing in hearts of MCKCUG-BP1 transgenic mice
To determine whether CUG-BP1 pre-mRNA targets are mis-regulated in neonatal heart, we compared splicing of Tnnt2 in the transgenic 1039 line and non-transgenic littermates by RT–PCR using oligonucleotides that anneal to exons 2 and 6. The primers detect four PCR products due to alternative splicing of both exons 4 and 5 (Fig. 3A). MCKCUG-BP1 transgenic mice displayed increased levels of exon 5 inclusion (73, 76 and 74%) when compared with wild-type (34 and 36%) animals (Fig. 3B). Cardiac tissue from DM patients display an inappropriate retention of the cTNT fetal exon 5, as do DM1 skeletal muscle cultures (21,22). Therefore, the observed pattern of increased Tnnt2 exon 5 inclusion in MCKCUG-BP1 mice is consistent with a DM phenotype.
Alternative splicing of Mtmr1, a member of a phosphatase family, produces three major isoforms: A, B and C (Fig. 4A and B). During mouse heart development, Mtmr1 undergoes a transition from the fetal isoforms, A and B, to the adult isoform, C (26). MTMR1 was recently identified as a target of CELF-mediated regulation when a preferred binding site for ETR-3 was identified in an intron 3′ to exon 2.1 (34). To determine whether Mtmr1 is mis-regulated in MCKCUG-BP1 mice, we compared splicing in transgenic and wild-type littermates in the 1039 line by RT–PCR using oligonucleotides that anneal to exons 2 and 5 (Fig. 4A and B). Analysis of splicing in neonatal heart by RT–PCR indicated that transgenic mice express more of the fetal Mtmr1 isoforms A and B, whereas the predominant isoform in wild-type mice is the C isoform (Fig. 4B). Expression of CUG-BP1 in mouse heart causes splicing mis-regulation of two CELF target pre-mRNAs, Tnnt2 and Mtmr1, and reproduce the aberrant regulation previously observed in DM cells (26).
Mis-regulated splicing in skeletal muscle of MCKCUG-BP1 transgenic mice
Since MTMR1 splicing is also developmentally regulated in skeletal muscle and is mis-regulated in congenital DM1 skeletal muscle cells (26), we examined the splicing of Mtmr1 in skeletal muscle of transgenic mice by RT–PCR. Similar to heart tissue, MCKCUG-BP1 mice display a delay in the expression of the adult isoform (Fig. 5). As a control, we examined adult wild-type mice, which express only the C isoform.
Another pre-mRNA that is mis-regulated in skeletal muscle tissue from individuals with DM1 and in DM mouse models (MbnlΔ3/Δ3, HSALR) is Clcn1 (5,14,23). The myotonia observed in both HSALR and MbnlΔ3/Δ3 mice is associated with inclusion of the fetal exon 7a and decreased ClC-1 protein expression (6,14). To determine whether Clcn1 is mis-regulated in neonatal skeletal muscle from the 1039 line, we analyzed the splicing of exon 7a by RT–PCR (Fig. 6A). MCKCUG-BP1 mice showed increased inclusion of the fetal exon 7a (50, 60 and 64%) when compared with littermates (22 and 26%) (Fig. 6B). Adult wild-type mice express mRNA that predominantly skips exon 7a. ClC-1 immunostaining is not informative at this stage of development because of the normally low expression of ClC-1 in the newborn, and due to the perinatal lethality myotonia could not be assessed. MCKCUG-BP1 mice reproduce the DM splicing pattern for Clcn1 and Mtmr1, consistent with a role for increased CUG-BP1 function in DM pathogenesis (see Discussion).
MTMR1 alternative splicing is mis-regulated in DM heart
Mtmr1 is developmentally regulated during mouse heart development (26) and mis-regulated in MCKCUG-BP1 heart and skeletal muscle (discussed earlier). Human MTMR1 undergoes a similar transition from embryonic to adult isoforms during skeletal muscle differentiation (26). Although the splicing has been examined in fetal human DM skeletal muscle (26), MTMR1 splicing in heart tissue from DM patients has not been examined. We first compared MTMR1 splicing in RNA samples from fetal and adult heart tissue to assess regulation during human heart development. Fetal heart tissues express predominantly the A isoform, whereas adult heart tissues express the adult C isoform suggesting that human MTMR1 undergoes a developmentally regulated transition in heart similar to what is observed in heart and skeletal muscle in mice (Fig. 7). Because MTMR1 splicing is mis-regulated in DM skeletal tissue, we also analyzed the splicing of MTMR1 in adult human DM heart samples. Consistent with the MCK-CUGBP1 mice, MTMR1 splicing is disrupted in DM heart (Fig. 7).
DISCUSSION
Here, we show that transgenic expression of human CUG-BP1 in mice results in neonatal lethality, skeletal muscle histological changes and trans-dominant effects on alternative splicing consistent with features observed in individuals with congenital DM. On the basis of serial dilutions of lysates analyzed by western blots, MCKCUG-BP1 mice express ∼4–6-fold more CUG-BP1 relative to endogenous levels in wild-type mice. The observed neonatal lethality is consistent with the previously reported CUG-BP1 mice in which lines expressing CUG-BP1 8–10 fold above endogenous levels died in utero (24). We examined known CELF pre-mRNA targets and showed that the splicing of Tnnt2, Mtmr1 and Clcn1 is disrupted in striated muscle of MCKCUG-BP1 mice similar to observed patterns in DM tissues. Interestingly, the splicing pattern for Tnnt2 and Clcn1 in transgenic CUG-BP1 mice is similar to MbnlΔ3/Δ3 mice lacking Mbnl1 isoforms, consistent with antagonistic splicing activities of these proteins. Although we have generated only one severe transgenic line, the lethality and histological changes are consistent with published CUG-BP1 transgenic mice and the trans-dominant effects on multiple known targets of CUG-BP1 splicing regulation are very unlikely to be due to transgene integration. The results presented in this manuscript demonstrate that increased CUG-BP1 activity reproduces the DM pattern of splicing and has the potential to play a role in the pathogenesis of DM.
CUG-BP1 regulates alternative splicing in heart and skeletal muscle
Human cTNT and mouse Tnnt2 undergo a developmental transition in which the fetal alternative exon 5 is included in the embryonic heart and skipped in adult (35,36). The isoform transition is influenced by the ratio of activators and repressors of alternative splicing, which change during development (12,35–37). We have previously demonstrated that cTNT is regulated antagonistically by CUG-BP1 and MBNL1 (12). CUG-BP1 activates inclusion and MBNL1 represses inclusion of exon 5 in human and chicken cTNT minigenes. MBNL1 and CUG-BP1 binding sites have been mapped to introns flanking exon 5 and mutation of the protein binding sites correlated with a blunted response to protein overexpression indicating that these proteins regulate splicing by directly binding the pre-mRNA (12,21). siRNA-mediated depletion of MBNL1 or the targeted disruption of Mbnl1 expression in mice results in increased Tnnt2 exon 5 inclusion (12,14). Consistent with increased CELF activity, and activity that is antagonistic to MBNL, MCKCUG-BP1 mice have increased inclusion of Tnnt2 exon 5.
Another CUG-BP1 target pre-mRNA, ClC-1, undergoes developmentally regulated alternative splicing in skeletal muscle (5). ClC-1 is responsible for maintaining the Cl− conductance in skeletal muscle and RT–PCR analysis of neonatal and adult mice indicates that exon 7a is included early in development and skipped in adult (5). We examined Clcn1 splicing in MCKCUG-BP1 mice and find higher levels of exon 7a inclusion compared with age-matched littermates. Clcn1 exon 7a inclusion is associated with increased CELF activity (MCKCUG-BP1 mice) or decreased MBNL activity (MbnlΔ3/Δ3 mice) suggesting that ClC-1 may also be antagonistically regulated by both families of alternative splicing factors.
MTMR1 was identified as a member of a family of phosphatases implicated in X-linked myotubular myopathy (26). Over the course of differentiation in mouse C2C12 myoblasts, there is an increase in expression of the adult C isoform with a parallel decrease in the fetal A and B isoforms. The C isoform contains an additional 17 amino acids at the N-terminal region, but the functional differences between the adult and fetal isoforms are unknown. A similar developmental transition to the C isoform is observed in mouse and human striated muscle (Fig. 7) (26). Systematic evolution of ligands by exponential enrichment (SELEX) analysis of ETR-3, a member of the CELF family, indicated that the preferred binding sites were UG and UGUU motifs (34). These motifs are present in an intron 3′ to exon 2.1 and co-expression of ETR-3 and MTMR1 minigenes suggest that ETR-3 regulates MTMR1 exon 2.1 splicing to generate the B isoform. RT–PCR analysis of mice expressing CUG-BP1 shows that Mtmr1 pre-mRNA in both skeletal and heart tissue shows increased expression of the fetal isoforms A and B and are delayed in switching to the adult C isoform. Our results suggest that increased CELF activity disrupts the Mtmr1 splicing transition in striated muscle. Although MTMR1 is disrupted in human DM cardiac tissue, the variable patterns of splicing between patient samples suggest that factors other than CELF proteins may influence splicing.
We observed alterations in alternative splicing of the CELF targets, Tnnt2, Clcn1 and Mtmr1, in striated muscle that are consistent with increased function of CUG-BP1. For all three pre-mRNAs studied, the transgenic mice expressing CUG-BP1 display the inappropriate embryonic splicing pattern. The predominant CELF proteins expressed in skeletal and cardiac muscle, CUG-BP1 and ETR-3, are significantly downregulated during development despite continued expression of both mRNAs (41). Our results demonstrating that increased expression of CUG-BP1 correlates with retention of embryonic splicing patterns for target pre-mRNAs is consistent with a role for CELF proteins in the regulation of alternative splicing during cardiac and skeletal muscle development.
The role of CUG-BP1 in the pathogenesis of DM
A key feature of the RNA-mediated pathogenesis model for DM is the disrupted splicing of specific pre-mRNA targets. For three targets, ClC-1 intron 2, IR exon 11 and cTNT exon 5, CUG-BP1 binding sites have been mapped to intronic regions and are required for regulation by CUG-BP1 (21–23). The DM splicing patterns observed for all three pre-mRNAs are consistent with increased CUG-BP1 activity and the observed increase in CUG-BP1 steady state levels in DM1 striated muscle cultures and tissue (13,16,21–23). The splicing patterns observed for these three pre-mRNAs are also consistent with decreased MBNL activity. cTNT exon 5 and IR exon 11 are direct targets for MBNL regulation, and Clcn1 intron 2 is retained in MbnlΔ3/Δ3 mice (14) strongly suggesting that, like cTNT and IR, its splicing is antagonistically regulated by CELF and MBNL proteins (12). Similarly, Clcn1 exon 7a is included at significantly higher levels both in MCKCUG-BP1 (Fig. 6) and in MbnlΔ3/Δ3 mice (14) suggesting antagonistic regulation by CELF and MBNL proteins. Therefore, the mis-regulation of these targets is directly consistent with the loss of MBNL and/or gain of CELF activity associated with DM pathogenesis.
Several mouse models of DM have been generated, which reproduce the characteristic myotonia and splicing changes either by an expression of expanded CUG repeat RNA or by a targeted deletion of Mbnl1 isoforms that bind CUG repeats (6,14,38,39). Here, we have generated transgenic mice (MCKCUG-BP1) expressing human CUG-BP1 to determine whether increased CUG-BP1 is sufficient to model DM. MCKCUG-BP1 mice display some of the characteristic histological changes observed in congenital DM skeletal muscle and reproduce the DM splicing patterns for Tnnt2, Clcn1 and Mtmr1 supporting a role for increased CUG-BP1 function in DM.
We propose that expression of CUG repeats alters the functions of alternative splicing regulators, MBNL and CELF proteins, leading to mis-regulation of specific targets. The mechanism for these effects is not yet clear. Co-localization of MBNL proteins with nuclear foci containing expanded CUG and CCUG RNA support a model in which loss of MBNL function results from sequestration (8,14,40). A mechanism for increased CELF activity is not as straightforward. On the basis of siRNA-mediated depletion of MBNL1, the observed CUG-BP1 elevation in DM1 myoblast cultures is independent of MBNL loss of function (13). Similarly, the levels of CUG-BP1 are not affected in MbnlΔ3/Δ3 mice (14). The increase in CUG-BP1 protein steady state levels in response to expanded RNA repeats is likely to involve a signaling event that remains to be determined. One model is that the RNA repeats interfere with a signal, which affects not only CELF protein expression, but also MBNL activities and downstream events. These expanded repeats could be either preventing a normal signaling event for transition to an embryonic pattern or initiating an aberrant event inducing the fetal splicing patterns. These results are consistent with a role for CELF protein expression in developmentally regulated alternative splicing and with an aberrant up-regulation of CELF expression in DM.
MATERIALS AND METHODS
Transgene construction and generation of transgenic mice
Human CUG-BP1, ‘A’ form, containing an N-terminal FLAG epitope tag, was excised with NheI/NotI from the plasmid 3.1FLAGCUGBP and subcloned into SpeI/NotI sites of the plasmid MCKBGH. MCKBGH contains the creatine kinase (MCK) promoter, enhancer, the untranslated exon 1, and intron 1 (25) and bovine growth hormone 3′ flanking genomic segment for proper mRNA 3′ end formation. The transgene (∼8 kb) was released with SacI for microinjection. Transgenic mice were generated by standard techniques and maintained on an FVB background. For genotyping, genomic DNA was extracted using the mouse tail DirectPCR lysis reagent (Viagen Biotech, Los Angeles, CA, USA) and analyzed by multiplex PCR using a β-actin internal control. The following primers were used: (transgene) pcDNA3.1BGH 5′-TAGAAGGCACAGTCGAGG, hCUGBP-E 5′-GCCTACTCGGGTATCCAG and (genomic DNA) actin1 5′-GATGTGCTCCAGGCTAAAGTT, actin2 5′-AGAAACGGAATGTTGTGGAGT.
Histological analysis
Frozen and paraffin sections were prepared from the hind limbs of transgenic and wild-type mice. Paraffin sections were cut and stained with trichrome, hematoxylin/eosin and periodic acid Schiff (PAS). Histochemical reactions for SDH, NADH, ATPase at pH 3.4 and 3.6 acid phosphatase and keratin were also performed.
EM analysis
Muscle for EM was fixed in 3% glutaraldehyde in PIPES buffer overnight at 4°C. After fixation, the muscle was post-fixed with 2% OsO4, stained with uranyl acetate, dehydrated in ethanol and propylene oxide and infused with plastic prior to examination by transmission electron microscopy.
Human heart tissue and pooled RNA
Pooled total RNA from adult (BD Biosciences Clontech, Palo Alto, CA, USA Cat. No. 64025-1 Lot No. 6070658) and fetal (Stratagene, La Jolla, CA, USA Cat. No. 738012, Lot No. 0100704) heart tissue was purchased from commercial sources. The adult pool consisted of tissues from individuals 20–78 years old, and the fetal pool was from tissues of 18- to 21-week-old fetuses. Autopsy samples of DM heart tissue were collected from 26-, 50- and 52-year-old patients. The diagnosis of DM was confirmed by clinical examinations and CTG repeat lengths (>600 CTG).
RNA isolation from tissue
Heart tissue or skeletal muscle from hind limbs was harvested from mice immediately after birth. Total RNA was isolated with Trizol (Invitrogen, Carlsbad, CA, USA) according to manufacturer's instructions. Pieces of human tissue (0.5 cm3) were homogenized with a Tekmar Tissumizer in ice-cold 4 M guanidinium thiocyanate, 20% n-lauryl sarcosine, 25 mM sodium citrate, 100 mM β-mercaptoethanol solution, followed by the addition of 2 M sodium acetate, pH 4.0. Total RNA was isolated with two phenol/chloroform extractions, precipitated with ice-cold isopropanol, pelleted and washed once with ice-cold 70% ethanol. The pellet was dissolved in water and stored at −80°C.
Immunoblot analysis
Tissue was homogenized in protein loading buffer (62.5 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol and 5% 2-β-mercaptoethanol) and the protein concentration was quantitated with the Non-Interfering Protein Assay (Genotech, St Louis, MO, USA). 3B1 CUG-BP1 monoclonal antibody was purchased (Upstate, Waltham, MA, USA) and conjugated to horseradish peroxidase (HRP) for western blot analysis.
RT–PCR analysis of splicing of endogenous genes
The 5′ end-labeled primers were prepared by incubating 2.25 ng forward primer, 10 U T4 Kinase (Invitrogen), 6.5 pmol [γ-(32)]P-ATP (Perkin–Elmer, Wellesley, MA, USA) and forward reaction buffer (Invitrogen) in a total volume of 10 µl at 37°C for 30 min. First strand cDNA was synthesized using 5–10 µg of total RNA, 100 ng oligo(dT) 12–18 primer (GibcoBRL/Invitrogen), 0.5 mM dNTPs and 4 U AMV reverse transcriptase (Life Sciences Inc,, St Petersburg, FL, USA) at 42°C for 1 h. PCR amplification using previously published conditions and primers was carried out using one half of the synthesized cDNA, 200 ng forward primer, 200 ng reverse primer, Taq MgCl2-free buffer (Promega, Madison, WI, USA), 1.75 mM MgCl2, 0.2 mM dNTPs, 2.5 U Taq polymerase (Promega) and 1.5 ng 5′-P(32)-end-labeled forward primer (14,26). RT–PCR products were electrophoretically resolved on 5% non-denaturing polyacrylamide gels, exposed to Biomax MR autoradiography film (Kodak, Rochester, NY, USA) and quantitated on a Phosphorimager (Molecular Dynamics, Piscataway, NJ, USA).
ACKNOWLEDGEMENTS
We thank C.R. Kahn for providing the MCK promoter and Joiner Cartwright for help with electron microscopy. DM tissue was generously provided by T. Ashizawa and C.A. Thornton. This work is supported by the grants from the NIH (R01AR45653 to T.A.C.), the Muscular Dystrophy Association (T.A.C) and a fellowship by the Robert and Janice McNair Foundation (T.H.H.).
Conflict of Interest statement. None declared.
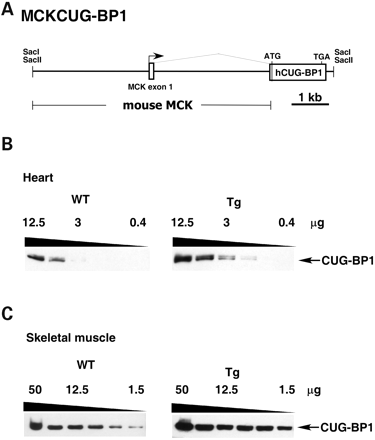
Figure 1. Expression of the MCKCUG-BP1 transgene containing human CUG-BP1. (A) The MCKCUG-BP1 transgene construct contains the mouse muscle creatine kinase promoter and an N-terminal FLAG-tagged version of human CUG-BP1. (B) Western blot analysis of CUG-BP1 expression in neonatal heart tissue from non-transgenic (WT) and transgenic (Tg) mice. The HRP-conjugated 3B1 monoclonal Ab detects both human and mouse CUG-BP1 in (B) and (C). Serial dilutions of protein extract from 12.5 to 0.39 µg were analyzed to compare relative levels of expression. (C) Western blot analysis of CUG-BP1 expression in skeletal muscle of neonatal non-transgenic (WT) and transgenic mice (Tg). Serial 1:2 dilutions of protein extract from 50 to 1.5 µg were analyzed to compare relative levels of expression.
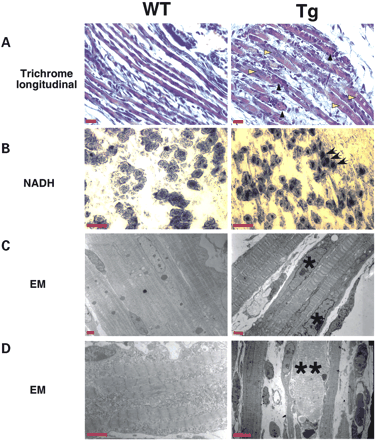
Figure 2. MCKCUG-BP1 mice display abnormal histology, histochemistry and EM, with features resembling the muscle pathology of DM. (A) Gomori trichrome of longitudinal sections of skeletal muscle. The chains of internal nuclei are more prominent in MCKCUG-BP1 muscle (yellow arrowheads). Degenerating fibers are surrounded by nuclei (black arrowheads). Scale bar, 20 µM. (B) NADH histochemical staining of skeletal muscle cross-sections. Transgenic mice showed increased central accumulation of NADH compared with wild-type (black arrows). Scale bar, 30 µM. (C and D) Evaluation of muscle ultrastructure by EM. Transgenic mice displayed irregular nuclei (asterisk) as well increased myofiber degeneration (double asterisk). Scale bar, 2 and 4 µM for (C) and (D), respectively.
![Figure 3. MCKCUG-BP1 mice display the inappropriate splicing pattern for Tnnt2 in heart. (A) Diagram of RT–PCR of cTNT. Forward and reverse PCR primers anneal to exons 2 and 6 (black), respectively. The resulting RT–PCR product that spans exons 2–6 detects the inclusion/exclusion of the alternative fetal exons 4 and 5 (gray). (B) RT–PCR analysis of Tnnt2 in mouse heart. Transgenic mice display increased inclusion of exon 5 when compared with wild-type littermates. Exon inclusion was assayed by RT–PCR. The percent exon inclusion was calculated as [(mRNA+exon 5)/(mRNA−exon 5+mRNA+exon 5)]×100. Each lane represents an individual mouse.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/11/10.1093/hmg/ddi162/2/m_ddi16203.gif?Expires=1716478953&Signature=EKuHiaLgZ8q70Xl0aracUS~NJT~uiq7SLNcAQ-1xPZEwJgNt7CHQ~w5fhy3-g5sovXDADIcaopMvzsp9j5144a2UEyHOFYLA9cpXjsOeQWPoeCW7-G~iLoSwx7Yo5ylLlAbCkmmzfEqxcHxg8VyotH985ZSnIdMIr~4ZxDXaLcjKQLMcoJkM86MolCN7l-ajaMw7TxMvha3hbKwK-vUe2rSqkC58W9nOrCd2GfOwZkkkRQ3nGYtwS1jpqVeGAw13GnNz2lN4Jv8dI9CjU~mNkWdcU~Izs6xDrxGUNHWk35mX0qZ~XuzepPY56MAK2LTtKIw-vjxd~zdtJunGjIjjbg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 3. MCKCUG-BP1 mice display the inappropriate splicing pattern for Tnnt2 in heart. (A) Diagram of RT–PCR of cTNT. Forward and reverse PCR primers anneal to exons 2 and 6 (black), respectively. The resulting RT–PCR product that spans exons 2–6 detects the inclusion/exclusion of the alternative fetal exons 4 and 5 (gray). (B) RT–PCR analysis of Tnnt2 in mouse heart. Transgenic mice display increased inclusion of exon 5 when compared with wild-type littermates. Exon inclusion was assayed by RT–PCR. The percent exon inclusion was calculated as [(mRNA+exon 5)/(mRNA−exon 5+mRNA+exon 5)]×100. Each lane represents an individual mouse.
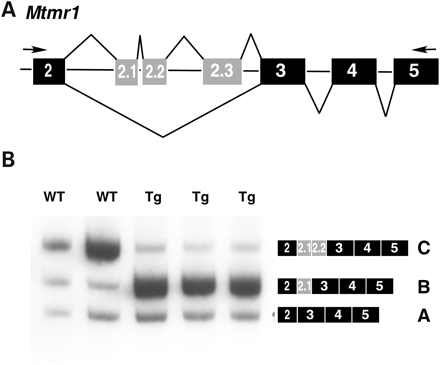
Figure 4. MCKCUG-BP1 mice display an altered Mtmr1 splicing pattern in heart. (A) Diagram of RT–PCR of Mtmr1. Forward and reverse PCR primers anneal to exons 2 and 5 (black), respectively. The resulting RT–PCR product detects the inclusion/exclusion of the alternative exons 2.1, 2.2 and 2.3 (gray). (B) RT–PCR analysis of Mtmr1 in mouse heart. Transgenic mice display increased expression the B isoform when compared with wild-type littermates. Exon inclusion was assayed by RT–PCR. Each lane represents an individual mouse.
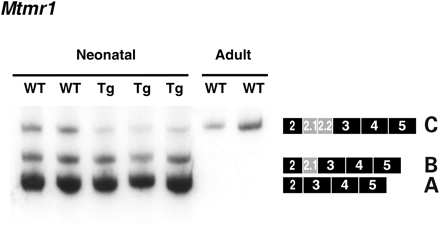
Figure 5. MCKCUG-BP1 mice display an altered Mtmr1 splicing pattern in skeletal muscle. RT–PCR analysis of Mtmr1 splicing in mouse skeletal muscle using primers that anneal to exons 2 and 5. Transgenic mice display increased expression of the fetal isoforms, A and B, when compared with wild-type littermates. As a control, adult wild-type mice were analyzed which express only the adult isoform, C. Each lane represents an individual mouse.
![Figure 6. MCKCUG-BP1 mice display an altered Clcn1 splicing pattern in skeletal muscle. (A) Diagram of RT–PCR of Clcn1. Forward and reverse PCR primers anneal to exons 5 and 8 (black), respectively. The resulting RT–PCR product detects the inclusion/exclusion of the fetal exon 7a (gray). (B) RT–PCR analysis of Clcn1 in mouse skeletal muscle. Transgenic mice display increased inclusion of the fetal exon 7a when compared with wild-type littermates. Adult mice express predominantly Clcn1 mRNA lacking exon 7a (last lane). Exon inclusion was assayed by RT–PCR. The percent exon inclusion was calculated as [(mRNA+exon 7a)/(mRNA−exon 7a+mRNA+exon 7a)]×100. Each lane represents an individual mouse.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/11/10.1093/hmg/ddi162/2/m_ddi16206.gif?Expires=1716478953&Signature=eLe~c5PGs8rKkOYw~BdgBFdO8f7YwCn4VZMBqr2pg4L~c7tGzNsmz60BR8MvRR8wA4xKIM6b6bAspz4r3VzzrSvBpK-cUn6iHLSeYFwZGoHSequawSUBUbrSsEko2WVe478nE1A0TZUMV557a2~5SPSn0qzU4u28MkiEA~TAqAIv0LdoHgJWwiiWcZCPeen~P3-6GMdKls9EZ5f~9tn4Evsl~q50RnLnwTMt2hDBcz1Ocz4IY2dftd2eQduIcdUa1oUg0umlfdFiYP8SMb9F3SW5lbGvYHppvwtXzdrK4o8YXiFvbeh2R3lo2aIaEFXKZzUtorPX2L5vDvwp8z7wvg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 6. MCKCUG-BP1 mice display an altered Clcn1 splicing pattern in skeletal muscle. (A) Diagram of RT–PCR of Clcn1. Forward and reverse PCR primers anneal to exons 5 and 8 (black), respectively. The resulting RT–PCR product detects the inclusion/exclusion of the fetal exon 7a (gray). (B) RT–PCR analysis of Clcn1 in mouse skeletal muscle. Transgenic mice display increased inclusion of the fetal exon 7a when compared with wild-type littermates. Adult mice express predominantly Clcn1 mRNA lacking exon 7a (last lane). Exon inclusion was assayed by RT–PCR. The percent exon inclusion was calculated as [(mRNA+exon 7a)/(mRNA−exon 7a+mRNA+exon 7a)]×100. Each lane represents an individual mouse.
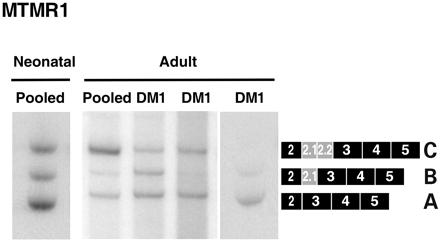
Figure 7. MTMR1 alternative splicing is disrupted in DM1 heart tissue. cDNA was synthesized from pooled commercial RNA or from unrelated individuals with DM. The adult pool consisted of heart tissue from 20–78-year-old individuals and the fetal pool was from heart tissue of 18- to 21-week-old fetuses. Autopsy samples of DM heart tissue were collected from 26-, 50- and 52-year-old patients. RT–PCR analysis using primers the flanking exons 2 and 5 indicates that adult DM tissues show increased retention of the fetal MTMR1 isoforms A and B.
References
Brook, J.D., McCurrach, M.E., Harley, H.G., Buckler, A.J., Church, D., Aburatani, H., Hunter, K., Stanton, V.P., Thirion, J.P., Sohn, R. et al. (
Liquori, C.L., Ricker, K., Moseley, M.L., Jacobsen, J.F., Kress, W., Naylor, S.L., Day, J.W. and Ranum, L.P. (
Ranum, L.P. and Day, J.W. (
Ranum, L.P. and Day, J.W. (
Mankodi, A., Takahashi, M.P., Jiang, H., Beck, C.L., Bowers, W.J., Moxley, R.T., Cannon, S.C. and Thornton, C.A. (
Mankodi, A., Logigian, E., Callahan, L., McClain, C., White, R., Henderson, D., Krym, M. and Thornton, C.A. (
Ladd, A.N., Charlet-B., N. and Cooper, T.A. (
Miller, J.W., Urbinati, C.R., Teng-Umnuay, P., Stenberg, M.G., Byrne, B.J., Thornton, C.A. and Swanson, M.S. (
Fardaei, M., Larkin, K., Brook, J.D. and Hamshere, M.G. (
Timchenko, L.T., Miller, J.W., Timchenko, N.A., Devore, D.R., Datar, K.V., Lin, L.J., Roberts, R., Caskey, C.T. and Swanson, M.S. (
Fardaei, M., Rogers, M.T., Thorpe, H.M., Larkin, K., Hamshere, M.G., Harper, P.S. and Brook, J.D. (
Ho, T.H., Charlet, B.N., Poulos, M.G., Singh, G., Swanson, M.S. and Cooper, T.A. (
Dansithong, W., Paul, S., Comai, L. and Reddy, S. (
Kanadia, R.N., Johnstone, K.A., Mankodi, A., Lungu, C., Thornton, C.A., Esson, D., Timmers, A.M., Hauswirth, W.W. and Swanson, M.S. (
Lu, X., Timchenko, N.A. and Timchenko, L.T. (
Timchenko, N.A., Cai, Z.J., Welm, A.L., Reddy, S., Ashizawa, T. and Timchenko, L.T. (
Good, P.J., Chen, Q., Warner, S.J. and Herring, D.C. (
Ladd, A.N., Nguyen, N.H., Malhotra, K. and Cooper, T.A. (
Timchenko, N.A., Welm, A.L., Lu, X. and Timchenko, L.T. (
Mukhopadhyay, D., Houchen, C.W., Kennedy, S., Dieckgraefe, B.K. and Anant, S. (
Philips, A.V., Timchenko, L.T. and Cooper, T.A. (
Savkur, R.S., Philips, A.V. and Cooper, T.A. (
Charlet-B., N., Savkur, R.S., Singh, G., Philips, A.V., Grice, E.A. and Cooper, T.A. (
Timchenko, N.A., Patel, R., Iakova, P., Cai, Z.J., Quan, L. and Timchenko, L.T. (
Bruning, J.C., Michael, M.D., Winnay, J.N., Hayashi, T., Horsch, D., Accili, D., Goodyear, L.J. and Kahn, C.R. (
Buj-Bello, A., Furling, D., Tronchere, H., Laporte, J., Lerouge, T., Butler-Browne, G.S. and Mandel, J.L. (
Johnson, J.E., Wold, B.J. and Hauschka, S.D. (
Karpati, G., Carpenter, S., Watters, G.V., Eisen, A.A. and Andermann, F. (
Silver, M.M., Vilos, G.A., Silver, M.D., Shaheed, W.S. and Turner, K.L. (
Iannaccone, S.T., Bove, K.E., Vogler, C., Azzarelli, B. and Muller, J. (
Farkas-Bargeton, E., Barbet, J.P., Dancea, S., Wehrle, R., Checouri, A. and Dulac, O. (
Sarnat, H.B. and Silbert, S.W. (
Faustino, N.A. and Cooper, T.A. (
Jin, J.P., Wang, J. and Zhang, J.Y. (
Cooper, T.A. and Ordahl, C.P. (
MacFarland, S.M., Jin, J.P. and Brozovich, F.V. (
Sergeant, N., Sablonniere, B., Schraen-Maschke, S., Ghestem, A., Maurage, C.A., Wattez, A., Vermersch, P. and Delacourte, A. (
Seznec, H., Agbulut, O., Sergeant, N., Savouret, C., Ghestem, A., Tabti, N., Willer, J.C., Ourth, L., Duros, C., Brisson, E. et al. (
Jiang, H., Mankodi, A., Swanson, M.S., Moxley, R.T. and Thornton, C.A. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}