
Abstract
The rapid insertion and extraction of Li ions from a cathode material is imperative for the functioning of a Li-ion battery. In many cathode materials such as LiCoO2, lithiation proceeds through solid-solution formation, whereas in other materials such as LiFePO4 lithiation/delithiation is accompanied by a phase transition between Li-rich and Li-poor phases. We demonstrate using scanning transmission X-ray microscopy (STXM) that in individual nanowires of layered V2O5, lithiation gradients observed on Li-ion intercalation arise from electron localization and local structural polarization. Electrons localized on the V2O5 framework couple to local structural distortions, giving rise to small polarons that serves as a bottleneck for further Li-ion insertion. The stabilization of this polaron impedes equilibration of charge density across the nanowire and gives rise to distinctive domains. The enhancement in charge/discharge rates for this material on nanostructuring can be attributed to circumventing challenges with charge transport from polaron formation.
Similar content being viewed by others

Introduction
The inadequacies of Li-ion batteries have emerged as a major constraint in many areas of technological design and can be attributed in large measure to challenges with the identification of optimal cathode chemistries and architectures1,2,3,4. In essence, a cathode material ought to be able to reversibly store a high concentration of inserted ions and, furthermore, the insertion/extraction and intervening diffusion of ions through the host matrix must occur rapidly to facilitate the efficient discharging/charging of the battery. There are numerous other caveats related to charge transfer at interfaces, earth abundance of the constituent elements and safety considerations that are vital for cathode design. Even this simplified description illustrates the critical imperative to carefully match thermodynamic driving forces of charge transfer (the free energy of the ion insertion reaction) with the kinetics of ion diffusion. In the most ubiquitous example of a Li-ion battery, correlated motion of both ions and electrons must often be considered. These correlations can be driven by the chemical composition, crystal structure and/or electrode geometry of the cathode1,5,6. Electron microscopy and microanalysis probes along with local structure characterization methods, such as total scattering, have provided great insight into the transformation of crystal structures on ion insertion and have enabled identification of numerous bottlenecks, for instance, fracture dynamics, formation of deleterious side products7,8,9,10, stabilization of metastable structures or loss of structural homogeneity over repeated charge/discharge cycles. However, the role of electronic structure and its contribution to diffusion barriers for ion migration is less appreciated5,11. Such diffusion barriers are responsible for the limitations of V2O5 as a cathode material at high rates and the remarkable (>100,000-fold) enhancement in the performance of this material on nanostructuring12,13,14. Understanding the origin of these diffusion barriers is imperative for developing fundamental design rules for cathode materials to alleviate charge localization.
V2O5 crystallizes in an orthorhombic layered structure with space group Pmmn with a van der Waals’ separation of 4.368 Å between the layers (Fig. 1a)15,16. Three distinct types of oxygen sites can be identified: vanadyl (V=O) oxygen atoms that point between the layers, and bridging and chaining oxygen atoms that connect the polyhedra. V2O5 was first proposed as a Li-ion intercalation host by Whittingham18, owing to the following: the abundance of interlayer sites that can accommodate Li ions; the readily accessible V5+/V4+ and V4+/V3+ redox couples; and the strong enthalpic driving forces for Li-ion insertion within this structure12,17,18,19. However, despite these promising attributes, the poor high-rate performance of these materials and issues with retention of capacity over prolonged cycling have limited the widespread commercial development of this material. In recent years, this material has enjoyed a resurgence of sorts with the realization that the galleries between V2O5 layers can accommodate not just Li ions but also other main group and transition metal cations of interest to ‘beyond Li ion’ battery chemistries, as well as the understanding that the sluggish kinetics of Li-ion insertion/extraction can be considerably accelerated by scaling to nanometre-sized dimensions19,20,21,22.

As the layered structure of V2O5 is intercalated with Li ions, it undergoes a series of phase transformations, to a puckered ɛ-phase (a); on further lithiation, the ɛ-phase transforms with an in-plane shift to a δ-phase (b). (c) Scanning electron microscopy images depict V2O5 nanowires with lengths spanning hundreds of micrometres (scale bar, 3 μm). (d) High-resolution transmission electron microscopy (TEM) image of an individual V2O5 wire (scale bar, 5 nm), indicating the separation between the (711) lattice planes of orthorhombic V2O5. The top inset shows a low-magnification TEM image of several nanowires (scale bar, 0.2 μm), whereas the bottom inset indicates an indexed selected-area electron diffraction pattern (scale bar, 5 nm−1). (e) XANES measurements of stoichiometrically lithiated V2O5 depict a reduction of the 3dxy resonance at the V L-edge and a diminution of the t2g to eg* ratio at the O K-edge with increasing lithiation.
Electrochemical measurements and spectroscopic probes often reveal the presence of two or more phases when a phase transition accompanies lithiation of a cathode material. In many commercial cathode materials such as LiCoO2 or LiMn2O4, Li insertion occurs with only modest first-order transitions (driven by Li ordering). However, in LiFePO4 a pronounced structural transformation between Li-rich and Li-poor phases is involved. Similarly, a number of intercalated phases can be distinguished for LixV2O5 with varying values of x depending on the concentration of Li ions inserted within the structure. Figure 1a,b show the structural progression of V2O5 with increasing intercalation of Li ions; a slightly distorted α-phase is initially stabilized for x<0.1 and with further lithiation is transformed to the ɛ-phase (Fig. 1a), which is stabilized in the range 0.35<x< 0.8 with initially cubo-octahedral and then tetrahedral coordination of Li ions; with still more lithiation, a puckered δ-phase is stabilized for ca. 0.8<x < 1.0 (Fig. 1b). In this regime, the phase transitions involve increased separation of the V2O5 layers, and their puckering and gliding motions to accommodate the structural distortions induced by an increasing concentration of Li ions without requiring cleavage of V–O bonds1,12,15. Further lithiation (x>1.0) brings about more pronounced structural distortions that involve bond-breaking and inversion of [VO5] polyhedra that are irreversible in the bulk, although there is evidence for recovery of the orthorhombic structure on delithiation for nanostructures21,22 even for x approaching 3 in LixV2O5 (ref. 23). In V2O5, the phase transitions across the structures depicted in Fig. 1a,b are rapidly accelerated, by up to 100,000 times, on scaling to nanometre-sized dimensions, yet there is evidence from combined Raman and powder diffraction studies of substantial variations in the extents of surface and bulk lithiation15,24. Understanding the origin of such phase segregation is imperative for the design of cathode materials and geometries.
On chemical lithiation, the ionized Li ion and the electron must diffuse through the solid matrix with the localization of the latter often bringing about a pronounced structural distortion; the combination of the electron and its structural distortion is termed a small polaron, provided the distortion has a length scale comparable to the primitive unit cell of the host material. The signatures of polaron formation and polaron hopping energies have been predicted theoretically, but direct experimental evidence of polaron formation and the accompanying geometric distortions have hitherto not been examined5,25,26,27,28. Here we present direct evidence of inhomogeneities in charge localization and local structural distortions induced on lithiation using scanning transmission X-ray microscopy (STXM) and corroborate theoretical predictions of a distinctive polaronic state using X-ray absorption near-edge structure and hard-energy X-ray photoemission spectroscopies. The polaron hopping barrier impedes electron diffusion and gives rise to phase inhomogeneity evident as lithiation gradients across an individual particle.
Results
Ensemble X-ray absorption measurements of lithiated V2O5
Chemical lithiation using n-butyllithium is used to model Li-ion insertion within a cathode as per the reaction29.

Figure 1c,d indicate scanning electron microscopy and transmission electron microscopy images of V2O5 nanowires grown by a previously reported hydrothermal method; the nanowires range from 150 to 250 nm in diameter and span several hundred micrometres in length15. The lattice-resolved transmission electron microscopy image in Fig. 1d shows the separation between (711) planes of orthorhombic V2O5 and, along with the accompanying selected area electron diffraction pattern, indicates that the single-crystalline nanowire grows along the crystallographic c-axis direction without any discernible extended defects.
Supplementary Fig. 1 shows an integrated V L- and O K-edge X-ray absorption near edge structure (XANES) spectrum acquired for an individual nanowire of V2O5 along with putative assignments derived from restricted open-shell configuration interaction with singles quantum chemistry calculations reported by Neese and colleagues30 for the V L-edge and our density functional theory (DFT) calculations for the O K-edge, further elaborated below. As an element- and edge-specific probe of unoccupied states, XANES serves as a valuable tool to probe electronic structure and chemical bonding in extended solids and single molecules alike31,32. The V L-edge is characterized by V LIII and V LII spectral features corresponding to transitions from V 2p3/2→V 3d (ca. 518 eV) and V 2p1/2→V 3d (ca. 525 eV) states, respectively30,33,34, which are split by the spin–orbit coupling of the V 2p atomic orbitals of ca. 7 eV. In turn, the O K-edge corresponds to transitions from O 1s states to states with O 2p character. As a result of substantial V 3d–O 2p orbital hybridization, two distinct sets of resonances are observed, reflecting the crystal field splitting of the V 3d orbitals. Assignments of the spectral features illustrated in Supplementary Fig. 1 are derived from DFT modelling and are further corroborated by angle-resolved XANES experiments (wherein the modulation in the intensity of resonances as a function of the polarization reflects the orbital symmetry of the final states)30,33,34.
A Coster–Kronig Auger decay process from a 2p1/2 into a 2p3/2 hole renders the V LII feature less informative due to the associated increase in spectral broadening34; however, the V LIII resonance indicates fine-structure features that strongly depend on the polarization vector; these transitions comprise transitions from the singlet V 2p63d0 into V 2p53d1 states split by crystal field and multiplet effects30. Despite convoluting multiplet effects, restricted open-shell configuration interaction with singles calculations30 indicate that the first two sharp resonances at 515.6 and 516.8 eV, respectively, correspond to final states that have relatively ‘pure’ V 3dxy and 3dxz/yz character and indeed angle-resolved XANES studies bear out these proposed orbital symmetries30,33,34. In contrast, the O K-edge is not convoluted by multiplet effects and can be clearly distinguished as three sets of transitions from O 1s core levels to (a) O 2px and 2py states that engage in π interactions with the t2g (V 3dxz, 3dxy and 3dyz) states of the metal cations (at 529.7 eV); overlapping σ states that represent direct end-on hybridization of (b) O 2px and 2py with V states at 531.6 eV and (c) O 2pz with V states at 533.1 eV. The calculated eXcited-state core hole-X-ray absorption spectroscopy (XCH-XAS) spectrum shown in Supplementary Fig. 1b suggests that the t2g manifold is derived primarily from transitions from O 1s core levels to O 2px/py states of the vanadyl oxygens that are hybridized with V 3dxz and 3dyz states; a lesser contribution to this resonance arises from transitions into O 2py states of bridging oxygen atoms hybridized with V 3dxy states and O 2px states of chaining oxygen atoms hybridized with V 3dxy states. The hybridization of the V=O oxygens with V 3dxz/yz thus dominates the lineshape of the t2g resonance in the O K-edge XANES spectrum with the non-bonding V 3dxy contributing much less, in contrast to the V LIII-edge spectrum, wherein the lowest-lying ‘split-off’ state is primarily V 3dxy in origin30. Thus, these assignments allow for an orbital-specific description of changes in electronic structure as a function of the lithiation of V2O5 and provide unprecedented insight into charge localization phenomena.
Figure 1e shows XANES spectra for a series of intercalated V2O5 samples with increasing values of x in LixV2O5. XANES resonances are collected at magic angle (54.7°) incidence to mitigate specific texturation effects35. Several trends are immediately discernible (as XANES probes empty orbitals, the diminution of a resonance, to first approximation, corresponds to occupation of states that give rise to the resonance): the first resonance in the V LIII spectrum corresponding to transitions to the split-off dxy conduction band of V2O5 is strongly diminished with increasing lithiation consistent with the reduction of V2O5 on lithiation (V5+ to V4+) and indicating the occupation of the lowest-lying conduction band states. Furthermore, at the O K-edge, the relative intensities of the transitions to the t2g and eg* (π* and σ*) states are greatly modified with the former resonances losing spectral weight. The origin of this pronounced modification of O K-edge spectral lineshapes is distinct from filling of the non-bonding V 3dxy states and suggests a pronounced rehybridization of V–O bonding at the vanadyl oxygens (vide infra).
Mapping lithiation inhomogeneities within a V2O5 nanowire
In contrast to ensemble spectra depicted in Fig. 1e, focusing the X-ray beam allows for acquisition of spatially resolved STXM data with ca. 25 nm spatial resolution, thereby allowing us to probe the lithiation of an individual nanowire immersed in a toluene solution of n-butyllithium for 1 min (Fig. 2b). By finely raster scanning the sample, STXM provides a means to construct a spatially resolved map of the local perturbations to the electronic and geometric structure induced by ion intercalation. Indeed, X-ray imaging has contributed greatly to understanding of inhomogeneities in biomaterials and polymeric systems36,37. The absolute energy calibration, detector linearity (Supplementary Fig. 2) and beam point spread function are main sources of error for this technique and have been carefully addressed as described in the Methods section38.

STXM image and integrated XANES spectrum acquired for (a) an individual V2O5 nanowire (scale bar, 500 nm), (b) an individual nanowire after 1 min of chemical lithiation (scale bar, 200 nm) and (c) a lithiated nanowire subjected to delithiation in Br2 solution (scale bar, 500 nm). Pronounced differences are discernible after lithiation including diminution of the V LIII-edge feature attributed to a V 3dxy final state and the reduction of the t2g:eg* ratio. The complete recovery of the electronic structure on delithiation suggests that the spectral changes can be directly attributed to Li-ion intercalation. All spectra have been pre- and post-edge normalized to a unitary absorption cross-section, to depict the relative spectral intensities.
Figure 2a depicts the STXM image and integrated V L- and O K-edge spectra of an individual V2O5 nanowire with a diameter of ca. 200 nm. In contrast to the orientation-averaged ensemble XANES spectra presented in Fig. 1e, well-resolved lineshapes are discernible for an individual single-crystalline V2O5 nanowire and the spectral transitions can be assigned as noted in Supplementary Fig. 1 and discussed above. Figure 2b depicts the STXM image and corresponding integrated spectrum acquired for a V2O5 nanowire after chemical lithiation for 1 min. Pronounced differences are readily discernible in this spectrum; the transition attributed to a V 3dxy final state at 515.6 eV is greatly diminished in intensity. Concomitantly, at the O K-edge, the t2g peaks are broadened and diminished in relative intensity with respect to the eg* peaks. In concordance with the ensemble XANES spectra (Fig. 1e), the integrated element-specific spectrum in Fig. 2b suggests that the electrons donated by the inserted Li ions have been transferred and reside on the V2O5 framework but are localized on the lowest-lying V=O 3dxz/yz–O 2p hybridized states of the conduction band, which have been substantially distorted as a result of lithiation17,39,40,41. Interestingly, on delithiation by immersion in Br2 solution the electronic structure of V2O5 is recovered in its entirety (Fig. 2c), confirming that the electronic structure modulation observed in V2O5 derives directly from lithiation.
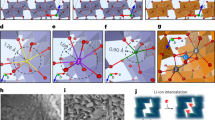
Unlike Fig. 2a–c wherein the spectra show little variation across the span of the nanowires, several distinct spectral contributions are discernible for the lithiated nanowire of Fig. 2b. A region of interest (ROI) analysis allows for identification of three distinct domains that are characterized by spectra individually plotted in Fig. 3a–c; these spectra correspond to different regions of the nanowire shown in Fig. 2b (the spectrum in Fig. 2b captures the integrated spectrum). In going from Fig. 3a–c, the intensity of the t2g resonance is progressively diminished with respect to the eg* resonance, indicating successively greater electronic reduction of the V2O5 framework; the accompanying maps in Fig. 3d–f indicate the spectral intensities of each of these components across the nanowire, suggesting the presence of distinct domains as a result of inhomogeneous lithiation. These maps are derived based on singular value decomposition of the image stack and by using as a reference the ROI spectra identified within different regions of the same image sequence. This operation produces a set of composition maps where intensities represent the signal strength of each of the spectral components (Fig. 3a–c) in that highlighted area. Notably, Supplementary Fig. 3 shows a thickness map of the nanowire (determined after a nonlinearity correction) along with a cross-sectional scanning electron microscopy image of the surface of an individual nanowire, which indicates that the domains visualized in Fig. 3a–c result from inhomogenous lithiation and do not reflect thickness variations. Figure 3d represents the least lithiated domains within this sample and is weighted most strongly at the periphery of the nanowire; based on the ensemble spectra depicted in Fig. 1e and previous angle-resolved spectra acquired for V2O5 nanowires33, an extent of lithiation in the broad range 0.1<x<0.5 can be surmised. It is noteworthy that the intensities at the V LIII edge for the lithiated sample are substantially diminished as compared with unlithiated and delithiated V2O5 as a result of state blocking; occupation of conduction band states diminishes the intensity of the low-energy XANES features. The interiors of the nanowires show two distinct spectral components depicted in Fig. 3e,f with substantially greater extents of lithiation (estimated to be 0.5<x<0.9 and 0.9<x<1.40, respectively). In particular, Fig. 3f defines a highly reduced strip that runs across a large section of the nanowire. The reduction of the 3dxy resonance at the V LIII edge correlates to the occupation of the lowest-lying levels in the conduction band of V2O5 by the electron ionized from the inserted Li atom. The resonance observed after lithiation arises from a superposition of remnant V5+ and reduced vanadium sites. In contrast, the diminished relative intensity of the low-energy t2g peaks at the O K-edge is the result of an induced structural distortion and further polarization of the electron density on V2O5 caused by the heterogeneous insertion of Li ions (vide infra). In other words, the V LIII edge allows for direct evaluation of electron density on the vanadium sites, whereas the O K-edge unveils structural distortion of the vanadyl V=O bonds induced as a result of electron localization.

Three distinct spectral contributions deconvoluted from ROI analysis of Fig. 2b are plotted in a–c in order of increasing lithiation evidenced as a diminution of the V 3dxy resonance at the V LIII-edge and the t2g:eg* ratio at the O K-edge. Intensity maps for each spectral contribution are plotted in d–f (scale bar, 200 nm), respectively, showing inhomogeneous regions of lithiation. A nonlinearity correction has been implemented as described by Collins and Ade and described in the Methods section38. All spectra have been pre- and post-edge normalized to a unitary absorption cross-section to depict the relative spectral intensities. The colour scale bars represent normalized optical density.
The increased lithiation at the core and the reduced lithiation of the surfaces is explicable based on the orientation of the nanowires (Fig. 1d) and the preferred insertion of Li ions between the layers. In essence, the nanowire is being viewed down the crystallographic b-axis and thus is enclosed at the top and bottom by ab planes that are impermeable to lithiation. The pronounced differences in lithiation probably further result from the stage ordering typical of layered materials42,43; the initial stochastic or defect-driven intercalation of Li ions between two specific V2O5 layers results in a local expansion of the interlayer spacing and facilitates insertion of further Li ions within the same layer. Indeed, Supplementary Fig. 4 indicates a sequence of calculated V2O5 structures with insertion of one Li ion (Supplementary Fig. 4a) and then two possibilities for insertion of the second Li ion: within the same layer (Supplementary Fig. 4b) and alternating layers (Supplementary Fig. 4c); insertion of Li ions within the same layer is thermodynamically favoured by −0.235 eV per formula unit.
Structural and electronic distortions induced by lithiation
To better understand the variations in the oxygen XANES spectra of the nanowires, DFT+U calculations with U=3.1 eV44, calculations were employed to examine the evolution of the electronic structure as a function of increasing insertion of Li ions44,45. The on-site Coulomb repulsive energy U is essential to capture the effects of strong electron correlation with vanadium 3d orbitals. Orthorhombic V2O5 (Fig. 1a) is a dielectric material with a bandgap of ca. 2.3 eV16,46,47,48, the conduction band is primarily V 3d in character, whereas the valence band has a significant O 2p contribution16,30. The projected density of states (pDOSs) of pristine V2O5 is shown in Fig. 4a,b, with the valence band maximum aligned at 0. The two spin channels are completely degenerate in this d0 system with pure V5+. In the crystal field of the slightly distorted [VO5] square pyramid, the 3dxy orbitals from the perfectly octahedral t2g group are further split into a high-energy component that overlaps with the degenerate 3dxz and 3dyz orbital, and a lower-energy component that dominates the conduction band edge49,50,51 (at ca. 2 eV above the valence band maximum in Fig. 4a). This lower energy 3dxy orbital is approximately non-bonding and comprises two split-off bands. The prominent feature in the oxygen total pDOS results primarily from the strong hybridization of the V 3dxz and 3dyz orbitals with the vanadyl oxygen atom and indeed these features contribute to the sharp XANES resonance at 529 eV52. The secondary feature from the oxygen pDOS is the typical eg component and gives rise to the absorption peak at 531 eV. Even without core-hole effects, the ground-state pDOS can still reproduce these features in the V2O5 O K-edge spectrum (Supplementary Fig. 1b) and enable understanding of the trends observed on lithiation53. With increasing lithiation, the donated electrons begin to occupy the non-bonding 3dxy component at the band edge. We first consider a simple periodic system with a pure V4.5+ site, where the electron occupies only a quarter of all available 3dxy orbitals (as in α-NaV2O5)51,54. Even at this electron doping level, electron correlation effects are important. The two spin channels are significantly split as a result of the localization of spin states induced by local lattice distortions, which lifts the spin degeneracy as depicted by Fig. 4c. The donated electrons take on 3dxy character and the localized spins51,54,55 are arrayed along the orthorhombic b-axis of V2O5. Contrary to intuition, the observed diminution of t2g intensity is not due to Pauli blocking from electron occupation, as the Fermi level remains far below the main peak position in the quarter-filled case. Instead, the lifted spin degeneracy induced by the correlation effects and lattice distortion plays a much more important role in reducing the t2g peak intensity. In short, the oxygen 2p components that strongly hybridize with the V 3d orbitals are also split into two non-degenerate spin channels, leading to a severe drop in the main peak intensity (Fig. 4d). Further details of the pDOS, illustrating this splitting, are depicted in Supplementary Fig. 5. Both the lifting of spin degeneracy and the lattice distortion caused by lithiation contribute to reduction of the intensity of the t2g peak. The inserted Li ions electrostatically attract the vanadyl oxygens towards them and create a pronounced distortion on the a–c plane (Fig. 1b). Such a distortion further shifts the energetic position of 3dxz orbitals, resulting in a noticeable migration of t2g intensity to higher spectral energies. Based on the abovementioned effects, the pDOS shown in Fig. 4d captures the specific origins of the evolution of the O K-edge spectra.

The GGA+U ground-state pDOS of pristine V2O5 (a,b) and the stoichiometric LixV2O5 (c,d) that adopts the pristine V2O5 vertical stacking order. The upper panels are the pDOS of vanadium in which the grey area indicates the occupied states. In the lower panels, the key components that are mainly responsible for the changes in the main peak intensity at the O K-edge are outlined by solid curves. The total pDOS (black curves) are the summation of px-, py- and pz-components from all three types of oxygen; chaining (Oc), bridging (Ob) and vanadyl (OV), with the corresponding stoichiometric ratio.
The Coulomb interaction between the spin-up and spin-down states, represented by the on-site Coulomb repulsion energy U, favours the removal of spin degeneracy along one spin polarization so as to lower the total energy of the system (Supplementary Fig. 6) and indeed this stabilization counteracts the elastic energy expended to bring about the distortion of the geometric structure depicted in Supplementary Fig. 7 (also see Supplementary Movie 1). In Supplementary Fig. 6, we initiate a supercell with perfect V2O5 lattice symmetry and an added electron, and relax the structure by enforcing spin degeneracy. The electron density also becomes delocalized in this case and this delocalized structure is ca. 0.22 eV higher in energy than the small polaron structure with a localized electron. These results further suggest that the stabilization of the small polaron in V2O5 is energetically favoured, both as a result of lattice distortion as well as the lifting of spin degeneracy56,57. The influence of the Li ion on the small polaron is further discussed below.
To understand the localization of the electron density on Li-ion intercalation, the electronic density difference is calculated from equation (2):

where  is the electron density of the Li-intercalated V2O5,
is the electron density of the Li-intercalated V2O5,  of isolated Li atoms in the same position as in the total system and
of isolated Li atoms in the same position as in the total system and  for V2O5. Supplementary Fig. 4 plots the increase and decrease in electron density of singly lithiated Li0.125V2O5 and doubly lithiated Li0.25V2O5 systems. The increase in electron density traces the contours of a V 3dxy orbital, indicating an electron localized in this lowest-lying state of the conduction band; in contrast, the electron density decrease is localized within bonds along the [VO5] pyramid. To put it differently, the increased electron density localized on the V 3dxy orbitals polarizes the V–O bonds and brings about a pronounced increase of the bond length. The coupled charge localization and distortion of the geometric structure further defines a small polaron as observed in the single-electron reduction case. Supplementary Movie 1 illustrates the localized distortion of the structure wherein the V atom shifts away from the intercalated Li ion; the bridge and chain oxygen atoms distort away from the central vanadium atom reflecting increased bond lengths and the vanadyl oxygen atoms orient towards the intercalated Li ions defining its cubo-octahedral local coordination environment.
for V2O5. Supplementary Fig. 4 plots the increase and decrease in electron density of singly lithiated Li0.125V2O5 and doubly lithiated Li0.25V2O5 systems. The increase in electron density traces the contours of a V 3dxy orbital, indicating an electron localized in this lowest-lying state of the conduction band; in contrast, the electron density decrease is localized within bonds along the [VO5] pyramid. To put it differently, the increased electron density localized on the V 3dxy orbitals polarizes the V–O bonds and brings about a pronounced increase of the bond length. The coupled charge localization and distortion of the geometric structure further defines a small polaron as observed in the single-electron reduction case. Supplementary Movie 1 illustrates the localized distortion of the structure wherein the V atom shifts away from the intercalated Li ion; the bridge and chain oxygen atoms distort away from the central vanadium atom reflecting increased bond lengths and the vanadyl oxygen atoms orient towards the intercalated Li ions defining its cubo-octahedral local coordination environment.
Stabilization of a small polaron
Polaronic confinement in transition-metal oxides has been extensively examined using DFT calculations5,25,26. Ioffe and Patrina27 have previously attempted to correlate the conductivity of V2O5 to small polaron formation using electronic transport measurements. However, direct atomic-scale evidence of polaron formation has thus far been elusive. The clear correlation of transitions related to the final states involving the V 3dxy level on lithiation noted in Fig. 2 and the subsequent effect on the crystal structure of the material shown in the reduction of the t2g/eg* ratio clearly indicates that polaron formation plays a key role in limiting Li diffusion within this material.
The energetic barrier to polaron diffusion within Li0.125V2O5 was calculated. Previous studies have shown strongly disfavoured Li migration along the a and c axes with migration barriers of 1.88 and 1.69 eV, respectively58. Supplementary Fig. 8a,b show a schematic depiction of the path adopted by Li ions between adjacent Li sites along the b axis, with a calculated diffusion barrier of 0.22 eV19. The diffusion of Li ions involves a change in the local coordination environment from 8→3→8 anions. Supplementary Fig. 8c illustrates the constrained trigonal planar transition state; the energetic barrier derives in large part from the substantial change of coordination number and the unfavourable coordination environment in the transition state. To further understand electron–polaron interactions with the intercalated Li ion, the polaron formation energies are calculated for two separate pairs of vanadium positions (Supplementary Fig. 9). In the first case, the electron is localized on the V1–V2 pair (in the proximity of the Li ion), whereas in the second case the electron is localized on the V3–V4 pair (far from the Li ion). The former configuration yields a formation energy of −0.41 eV/V2O5 unit, suggesting that the polaron is stabilized by an attractive interaction with the Li ion. In contrast, the calculated formation energy for the latter configuration with the polaron situated at V3–V4 positions is 0.02 eV/V2O5, clearly a much less stable configuration for the polaron. Supplementary Fig. 9c indicates that the migration barrier for the V1–V2 polaron is 0.34 eV, whereas the comparable value for the V3–V4 pair is 0.03 eV. These calculations thus suggest that small polarons are preferentially stabilized adjacent to the intercalated Li ions but this stabilization also entails a substantial barrier for migration of the polarons along the V2O5 framework56. In other words, the intercalated Li ions play a critical role in stabilizing the polaron and determining its ease of migration.
Now, turning our attention to the electronic consequences of Li-ion intercalation, the calculated orbital pDOSs in Supplementary Fig. 10 suggest that lithiation should be accompanied by the appearance of a ‘mid-gap’ state between the valence and conduction band. To examine the predictions of the appearance of a filled state derived from polaron formation in the upper valence band, hard X-ray photoemission spectroscopy (HAXPES) measurements have been performed for lithiated samples (Fig. 5). The V 2p3/2 spectrum in Fig. 5b clearly indicates the presence of discrete V4+ and V5+ states. Most notably, the inset to Fig. 5a indicates the emergence of a feature not observed for orthorhombic V2O5 at ca. 1.0 eV below the Fermi level in the valence band spectrum that corroborates and serves as a distinctive signature of the mid-gap polaronic state predicted by DFT. The appearance of this state provides definitive experimental evidence for localized electrons corresponding to stabilization of a small polaron.

(a) Valence band spectra for pristine V2O5 (red) and LixV2O5 (black). The right inset depicts a magnification of the region showing the emergence of a feature below the Fermi level. As predicted by theory, HAXPES measurements clearly illustrate the appearance of a polaronic state below the Fermi level. (b) HAXPES performed on these samples demonstrates the existence of V4+ and V5+ sites at the V 2p3/2 peak.
Discussion
The experimental results in concert with the calculations indicate that local structural distortions and the stabilization of small polarons impede electron diffusion within V2O5 and give rise to distinctive lithiation gradients42,59. The STXM images correspond to a map of electron density on the V2O5 framework, which further reflects the lithiation gradient as a result of the close association of localized electrons with Li ions. As noted above, in LixV2O5, the Li-ion stoichiometry x determines the phase of the material and a series of phase transformations are evidenced with increasing lithiation. Barriers to diffusion of Li ions thereby also influence the sequence of structural phase transformations. In other words, STXM provides a view of trapped electron density, which is correlated to lithiation gradients, further reflecting barriers to propagation of phase transformation within an individual LixV2O5 nanowire. The pronounced increase in high-rate performance as observed for nanostructures thus probably results in a much more facile phase nucleation enabled by easier electron and ion diffusion.
In summary, we have mapped the changes in electronic structure and local structural distortions induced by the lithiation of V2O5 using a combination of V L-edge and O K-edge XANES and STXM probes of the conduction band and HAXPES examination of the valence band; the spectra are interpreted with the assistance of DFT+U calculations. Specifically, we note the stabilization of distinctive domains within individual nanowires of lithiated V2O5 corresponding to the emergence of charge density gradients along the nanowires that can be correlated to inhomogeneous lithiation. These measurements provide the first view of highly anisotropic lithiation of layered materials resulting from the peculiarities of their electronic and geometric structure. Spectral assignments verified by DFT calculations suggest that lithiation of V2O5 induces the localized reduction of specific vanadium sites with the electron derived from the ionized Li ion residing in neighbouring V 3dxy orbitals that are the lowest-lying states in the conduction band. In a complementary manner, O K-edge XANES spectra and STXM maps depict the local structural distortions induced by exchange interactions and small polaron formation as a result of strong modification of V–O hybridization along the vanadyl V=O bonds. DFT calculations confirm that electron density localization is sufficient to drive elastic distortion of the local atomic structure. The quasiparticle comprising the trapped electron and the local distortion constitutes a small polaron and polaronic signatures predicted by DFT have been verified by HAXPES studies. Delithiation of V2O5 brings about elimination of the polaron and complete recovery of the electronic structure. The small polaron formation directly evidenced in these studies is thought to be the origin of sluggish diffusion of Li ions through the cathode, with a diffusion barrier of ca. 0.22 eV, limiting high-rate performance. The strongly accelerated kinetics of lithiation observed on scaling to nanometre-sized dimensions can also, in large measure, be attributed to the ability to circumvent the limitations of sluggish small polaron hopping at these sizes. The fundamental limitations to ion diffusion unveiled here suggest that V2O5 cathode materials will benefit from development of quasi-amorphous or highly porous materials where charge is not required to travel large distances or by devising novel lattice frameworks with lower extent of polaronic confinement. Quasi-amorphous or highly porous materials would have a greater contact area with the electrolyte, thereby greatly limiting the range over which small polaron hopping needs to be sustained and mitigating the kinetic impediments imposed by stabilization of a polaron.
Methods
Synthesis and chemical lithiation of V2O5 nanowires
Synthesis and the subsequent lithiation of the V2O5 nanowires were carried out as previously reported15. Briefly, V2O5 nanowires were synthesized via hydrothermal reduction of bulk V2O5 (Sigma-Aldrich, 99.5%) with oxalic acid (J.T. Baker), to prepare V3O7·H2O nanowires, followed by oxidation in air at 300 °C to obtain phase-pure V2O5 nanowires. Lithiation was carried out within a glove bag under Ar ambient via immersion of the powder in molar excess (4:1 Li:V2O5) of 2.5 M n-butyllithium solution in hexanes (Sigma-Aldrich) diluted to 0.025 M in toluene. Delithiation was accomplished by immersion of the lithiated samples in pure liquid Br2 for 2 h, followed by washing with large amounts of hexanes. The samples are sealed within a glovebox for transport to synchrotrons for XANES and STXM measurements.
XANES spectroscopy
XANES measurements were carried out at the National Synchrotron Light Source of Brookhaven National Laboratory at beamline U7A operated by the National Institute of Standards and Technology with a toroidal mirror spherical grating monochromator using a 1,200 lines per mm grating with a nominal energy resolution of 0.25 eV with a slit size of 30 × 30 μm. XANES spectra were collected in partial electron yield mode with a channeltron multiplier near the sample surface; the detector was used with an entrance grid bias of −200 V bias to reject low-energy electrons; a charge compensation gun was used to avert the charging of the samples. The incident beam is linearly polarized 85% in the plane of the synchrotron ring. As XANES uses linearly polarized light and incorporates dipolar transitions, the absorption cross-section transforms as follows:

where  is the Cartesian tensor for the absorption cross-section derived from Fermi’s Golden rule, σa and σb are distribution functions of crystallite orientation and θ is the angle between the polarization vector and sample. If θ=54.7°, the isotropic average is:
is the Cartesian tensor for the absorption cross-section derived from Fermi’s Golden rule, σa and σb are distribution functions of crystallite orientation and θ is the angle between the polarization vector and sample. If θ=54.7°, the isotropic average is:

and thus specific texturation effects are heavily mitigated at this angle.
The partial electron yield signals were normalized using the incident beam intensity, to eliminate the effect of incident beam intensity fluctuations and monochromatic absorption features. The V L- and O K-edge spectra were acquired in a single scan. Data were collected along a metallic vanadium reference mesh for energy calibration. Pre- and post-edge normalization of the spectrum was performed using the Athena suite of programmes.
Scanning transmission X-ray microscopy
STXM measurements were performed at the SM (10-ID1) beamline of the Canadian Light Source, a 2.9-GeV third-generation synchrotron facility. A 25-nm outermost-zone zone plate was used to obtain a diffraction-limited spatial resolution better than 30 nm. A 500-line per mm plane grating monochromator was used to acquire the V L-edge and O K-edge spectral stacks. The incident photon flux (Io) count rate was adjusted to be <20 MHz and optimized to ca. 17 MHz as read by the STXM detector within a hole located close to the sample of interest and measured at 560 eV by adjusting the exit slits to 17/16 μm (dispersive/non-dispersive). The V L- and the O K-edge stacks covered an energy range from 508 to 560 eV with energy steps of 0.2 eV in the ROI and 1 eV in the continuum region beyond the specific elemental edges with dwell time of 1 ms for each section. Right circularly polarized X-rays, generated by an elliptically polarized undulator was used in the experiments. All STXM data were analysed and processed using aXis2000 software (http://unicorn.mcmaster.ca/aXis2000.html). STXM maps are derived based on singular value decomposition of the image stack in aXis2000 and by using as a reference the ROI spectra identified within different regions of the same image sequence. This operation produces a set of composition maps where intensities represent the signal strength of each of the spectral components (Fig. 3a–c) at each specific pixel of that highlighted area. To correct for the nonlinearity of the detector, the flux was measured as a function of dispersive slit width for non-dispersive slit widths of 5, 10, 15 and 25 μm at 560 eV (Supplementary Fig. 2). The resultant curves were fit using the function as per the method described by Collins and Ade38:

where I′ is the measured flux, Is is the detector saturation flux, κ is the rate at which measured flux approaches saturation, x is the dispersive slit width and x0 is a slit width zero offset. The parameters extracted from the fit function allow for the measured flux to be corrected to the actual flux using the following relationship:

From this analysis, the quantum efficiency of the detector can be determined as a function of flux and is plotted in Supplementary Fig. 2 (ref. 38). The experimental spectrum of the lithiated V2O5 nanowires was corrected by first extracting the average measured flux at each pixel from the image stack. The extracted flux was then corrected by calculating the actual flux, as in latter equation, yielding a spectrum representative of the actual flux values at each pixel. Owing to the magnitude of correction being dependent on the measured flux, a correction factor was then calculated for each pixel, which was then multiplied by the stack to yield a corrected image (Fig. 3).
Hard X-ray photoemission spectroscopy
HAXPES measurements were performed at the National Institute of Standards and Technology bending magnet beamline X24 of the National Synchrotron Light Source of Brookhaven National Laboratory. Measurements were performed at a ca. 4 keV photon energy with a pass energy of 500 eV and a Gaussian instrumental broadening of 0.45 eV. The higher excitation of HAXPES circumvents serious charging issues that are common to ultraviolet and soft X-ray photoelectron spectroscopy. No evidence of charging was observed during our measurements. The HAXPES spectra are energy aligned to the Fermi level of a gold foil reference in electrical contact with our samples, unless stated otherwise. To mitigate further energy alignment shifts from beam drift, the Au reference scans were measured before and after each spectrum.
Computational details
The ground-state structural and electronic properties of V2O5 and the lithiated systems are obtained using DFT60,61 with the Vienna ab initio simulation package62. The exchange-correlation energies are calculated within the specific generalized-gradient approximation (GGA) of Perdew–Burke–Ernzerhof 63. The electron–ion interaction is treated with projector-augmented-wave pseudopotentials64,65, using a 400-eV plane-wave kinetic energy cutoff. A rotationally invariant DFT+U approach45 is employed to describe the on-site Coulomb interaction of the spin-up and spin-down electrons, with U=3.1 eV44. To converge the total energy, we sample the first Brillouin zone with a Monkhorst–Pack reciprocal space grid of 6 × 6 × 6 k-points. All the atomic structures being considered have been relaxed until each Cartesian force component is no greater than 0.01 eV Å−1. To guarantee highly resolved pDOS, we calculate Kohn–Sham eigen energies based on the converged electron density on a grid of 24 × 24 × 24 k-points centred at the zone centre (Γ-point). The pDOS is numerically broadened with a Fermi-Dirac smearing of 0.2 eV, approximately mimicking the intrinsic broadening due to the oxygen 1s core-hole lifetime. Higher-resolution pDOS shown in the Supplementary Materials is obtained from the much smaller broadening of 0.03 eV. The Tkatchenko–Scheffler method was used to describe the van der Waals interaction between the layers of V2O5 (ref. 66). Lithium-ion diffusion barriers in α-Li0.125V2O5 are calculated using the nudged-elastic band (NEB) method67 as implemented in the Vienna ab initio simulation package. A total of seven images are calculated between the end points to capture the energy landscape for Li ion diffusion. The end points are optimized to a force tolerance of ±0.001 eV Å−1, whereas the convergence criterion for the forces along the NEB path is 0.1 eV Å−1 (ref. 68).
Data availability
The data that support the findings of this study are available from the corresponding authors (DP and SB) upon request.
Additional information
How to cite this article: De Jesus, L. R. et al. Mapping polaronic states and lithiation gradients in individual V2O5 nanowires. Nat. Commun. 7:12022 doi: 10.1038/ncomms12022 (2016).
References
Whittingham, M. S. Lithium batteries and cathode materials. Chem. Rev. 104, 4271–4301 (2004).
Kang, B. & Ceder, G. Battery materials for ultrafast charging and discharging. Nature 458, 190–193 (2009).
Kim, S. W., Seo, D. H., Ma, X., Ceder, G. & Kang, K. Electrode materials for rechargeable sodium-ion batteries: potential alternatives to current lithium-ion batteries. Adv. Energy Mater. 2, 710–721 (2012).
Tarascon, J.-M. & Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001).
Ping Ong, S., Chevrier, V. L. & Ceder, G. Comparison of small polaron migration and phase separation in olivine LiMnPO4 and LiFePO4 using hybrid density functional theory. Phys. Rev. B 83, 75112/1–75112/7 (2011).
Tarascon, J.-M. Key challenges in future Li-battery research. Philos. Trans. A Math. Phys. Eng. Sci. 368, 3227–3241 (2010).
Ghassemi, H., Au, M., Chen, N., Heiden, P. A. & Yassar, R. S. In situ electrochemical lithiation/delithiation observation of individual amorphous Si nanorods. ACS Nano 5, 7805–7811 (2011).
Gu, M. et al. Demonstration of an electrochemical liquid cell for operando transmission electron microscopy observation of the lithiation/delithiation behavior of Si nanowire battery anodes. Nano Lett. 13, 6106–6112 (2013).
Kirshenbaum, K. et al. In situ visualization of Li/Ag2VP2O8 batteries revealing rate-dependent discharge mechanism. Science 347, 149–154 (2015).
Liu, H. et al. Capturing metastable structures during high-rate cycling of LiFePO4 nanoparticle electrodes. Science 344, 12528171–12528177 (2014).
Castro, L. et al. The spin-polarized electronic structure of LiFePO4 and FePO4 evidenced by in-lab XPS. J. Phys. Chem. C 114, 17995–18000 (2010).
Chernova, N. A., Roppolo, M., Dillon, A. C. & Whittingham, M. S. Layered vanadium and molybdenum oxides: batteries and electrochromics. J. Mater. Chem. 19, 2526–2552 (2009).
Sides, C. R. & Martin, C. R. Nanostructured electrodes and the low-temperature performance of Li-ion batteries. Adv. Mater. 17, 125–128 (2005).
Rui, X. et al. Ultrathin V2O5 nanosheet cathodes: realizing ultrafast reversible lithium storage. Nanoscale 5, 556–560 (2013).
Horrocks, G. A., Likely, M. F., Velazquez, J. M. & Banerjee, S. Finite size effects on the structural progression induced by lithiation of V2O5: a combined diffraction and Raman spectroscopy study. J. Mater. Chem. A 1, 15265–15277 (2013).
Eyert, V. & Höck, K.-H. Electronic structure of V2O5: role of octahedral deformations. Phys. Rev. B 57, 12727–12737 (1998).
Scanlon, D. O., Walsh, A., Morgan, B. J. & Watson, G. W. An ab initio study of reduction of V2O5 through the formation of oxygen vacancies and Li intercalation. J. Phys. Chem. C 112, 9903–9911 (2008).
Whittingham, M. S. The role of ternary phases in cathode reactions. J. Electrochem. Soc. 123, 315–320 (1976).
Marley, P. M., Horrocks, G. A., Pelcher, K. E. & Banerjee, S. Transformers: the changing phases of low-dimensional vanadium oxide bronzes. Chem. Commun. 51, 5181–5198 (2015).
Yoo, H. D. et al. Mg rechargeable batteries: an on-going challenge. Energy Environ. Sci. 6, 2265–2279 (2013).
Wang, Y. & Cao, G. Synthesis and enhanced intercalation properties of nanostructured vanadium oxides. Chem. Mater. 18, 2787–2804 (2006).
Chan, C. K. et al. Fast, completely reversible Li insertion in vanadium pentoxide nanoribbons. Nano Lett. 7, 490–495 (2007).
Galy, J. Vanadium pentoxide and vanadium oxide bronzes—structural chemistry of single (S) and double (D) layer MxV2O5 phases. J. Solid State Chem. 100, 229–245 (1992).
Baddour-Hadjean, R. & Pereira-Ramos, J. -P. Raman microspectrometry applied to the study of electrode materials for lithium batteries. Chem. Rev. 110, 1278–1319 (2010).
Scanlon, D. O., Morgan, B. J. & Watson, G. W. Modeling the polaronic nature of p-type defects in Cu2O: the failure of GGA and GGA + U. J. Chem. Phys. 131, 124703/1–124703/8 (2009).
Franchini, C., Kresse, G. & Podloucky, R. Polaronic hole trapping in doped BaBiO3 . Phys. Rev. Lett. 102, 6–9 (2009).
Ioffe, V. A. & Patrina, I. B. Comparison of the small-polaron theory with the experimental data of current transport in V2O5 . Phys. Stat. Solid. 40, 389–395 (1970).
Reid, O. G. & Rumbles, G. Quantitative transient absorption measurements of polaron yield. J. Phys. Chem. Lett. 4, 2348–2355 (2013).
Whittingham, M. S. & Dines, M. B. n-Butyllithium—an effective, general cathode screening agent. J. Electrochem. Soc. 124, 1387–1388 (1977).
Maganas, D. et al. First principles calculations of the structure and V L-edge X-ray absorption spectra of V2O5 using local pair natural orbital coupled cluster theory and spin-orbit coupled configuration interaction approaches. Phys. Chem. Chem. Phys. 15, 7260–7276 (2013).
Stohr, J. NEXAFS Spectroscopy Springer, (1992).
Schultz, B. J., Dennis, R. V., Lee, V. & Banerjee, S. An electronic structure perspective of graphene interfaces. Nanoscale 6, 3444–3466 (2014).
Velazquez, J. M., Jaye, C., Fischer, D. A. & Banerjee, S. Near edge X-ray absorption fine structure spectroscopy studies of single-crystalline V2O5 nanowire arrays. J. Phys. Chem. C 113, 7639–7645 (2009).
Goering, E., Müller, O., Klemm, M., denBoer, M. L. & Horn, S. Angle dependent soft-X-ray absorption spectroscopy of V2O5 . Philos. Mag. B 75, 229–236 (1997).
Pettifer, R. F. et al. Magic-angle theorem in powder x-ray-absorption spectroscopy. Phys. Rev. B Condens. Matter Mater. Phys. 42, 37–42 (1990).
Ade, H. & Hitchcock, A. P. NEXAFS microscopy and resonant scattering: composition and orientation probed in real and reciprocal space. Polymer 49, 643–675 (2008).
Urquhart, S. G. et al. NEXAFS spectromicroscopy of polymers: overview and quantitative analysis of polyurethane polymers. J. Electron Spectrosc. Relat. Phenom. 100, 119–135 (1999).
Collins, B. A. & Ade, H. Quantitative compositional analysis of organic thin films using transmission NEXAFS spectroscopy in an X-ray microscope. J. Electron Spectrosc. Relat. Phenom. 185, 119–128 (2012).
Marley, P. M. et al. Charge disproportionation and voltage-induced metal-insulator transitions evidenced in β-PbxV2O5 nanowires. Adv. Funct. Mater. 23, 153–160 (2013).
Marley, P. M. et al. Electronic Phase transitions of δ AgxV2O5 nanowires: interplay between geometric and electronic structure. J. Phys. Chem. C 36, 21235–21243 (2014).
Korotin, M. A., Anisimov, V. I., Saha-Dasgupta, T. & Dasgupta, I. Electronic structure and exchange interactions of the ladder vanadates CaV2O5 and MgV2O5 . J. Phys. Condens. Matter 12, 113–124 (2000).
Malik, R., Zhou, F. & Ceder, G. Kinetics of non-equilibrium lithium incorporation in LiFePO4 . Nat. Mater. 10, 587–590 (2011).
Chueh, W. C. et al. Intercalation pathway in many-particle LiFePO4 electrode revealed by nanoscale state-of-charge mapping. Nano Lett. 13, 866–872 (2013).
Wang, L., Maxisch, T. & Ceder, G. Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. B 73, 195107/1–195107/6 (2006).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Zhang, Y. et al. Novel V2O5/S composite cathode material for the advanced secondary lithium batteries. Solid State Ion. 181, 835–838 (2010).
Suli, A., Torok, M. I. & Hevesi, I. On the capacitance of n-V2O5/p-Si heterojunctions. Thin Solid Films 139, 233–246 (1986).
Wang, Y. et al. Synthesis of one-dimensional TiO2/V2O5 branched heterostructures and their visible light photocatalytic activity towards Rhodamine B. Nanotechnology 22, 225702/1–225702/8 (2011).
Chakrabarti, A. et al. Geometric and electronic structure of vanadium pentoxide: a density functional bulk and surface study. Phys. Rev. B 59, 583–590 (1999).
Bhandari, C., Lambrecht, W. R. L. & van Schilfgaarde, M. Quasiparticle self-consistent GW calculations of the electronic band structure of bulk and monolayer V2O5 . Phys. Rev. B 91, 125116/1–125116/12 (2015).
Smolinski, H. et al. NaV2O5 as a quarter-filled ladder compound. Phys. Rev. Lett. 80, 5164–5167 (1998).
Kolczewski, C. & Hermann, K. Ab initio DFT cluster studies of angle-resolved NEXAFS spectra for differently coordinated oxygen at the V2O5(010) surface. Surf. Sci. 552, 98–110 (2004).
De Groot, F. High-resolution X-ray emission and X-ray absorption spectroscopy. Chem. Rev. 101, 1779–1808 (2001).
Konstantinović, M. J. et al. Charge ordering and optical transitions of LiV2O5 and NaV2O5 . Phys. Rev. B 63, 121102/1–121102/4 (2001).
Valentí, R., Saha-Dasgupta, T., Alvarez, J. V., Požgajčić, K. & Gros, C. Modeling the electronic behavior of γ-LiV2O5: a microscopic study. Phys. Rev. Lett. 86, 5381–5384 (2001).
Ding, H. et al. Computational investigation of electron small polarons in α-MoO3 . J. Phys. Chem. C 118, 15565–15572 (2014).
Maxisch, T., Zhou, F. & Ceder, G. Ab initio study of the migration of small polarons in olivine LixFePO and their association with lithium ions and vacancies. Phys. Rev. B 73, 104301/1–104301/6 (2006).
Zhou, B., Shi, H., Cao, R., Zhang, X. & Jiang, Z. Theoretical study on the initial stage of magnesium battery based on V2O5 cathode. Phys. Chem. Chem. Phys. 16, 18578–18585 (2014).
Zhou, F., Marianetti, C. A., Cococcioni, M., Morgan, D. & Ceder, G. Phase separation in LixFePO4 induced by correlation effects. Phys. Rev. B 69, 201101/1–201101/4 (2004).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996)) Errata: 78, 1396 (1997).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Bučko, T., Lebègue, S., Hafner, J. & Ángyán, J. G. Tkatchenko-Scheffler van der Waals correction method with and without self-consistent screening applied to solids. Phys. Rev. B Condens. Matter Mater. Phys. 87, 1–15 (2013).
Sheppard, D., Terrell, R. & Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 128, 1–10 (2008).
Liu, M. et al. Spinel compounds as multivalent battery cathodes: a systematic evaluation based on ab initio calculations. Energy Environ. Sci. 8, 964–974 (2015).
Acknowledgements
This study is based on work supported by the National Science Foundation under DMR 1504702. S.B. further acknowledges support from the Research Corporation for Science Advancement through a Scialog Award. L.D.J. acknowledges support from a National Science Foundation Graduate Research Fellowship under grant number 1252521. Certain commercial names are presented in this Letter for purposes of illustration and do not constitute an endorsement by National Institute of Standards and Technology. We acknowledge Dr Chithra Karunakaran at beam-line 10ID1 of the Canadian Light Source for support and assistance with STXM data collection. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract number DE-AC02-98CH10886. DFT simulations were performed as part of a User Project with Y.L. and D.P. at The Molecular Foundry (TMF), Lawrence Berkeley National Laboratory, and calculations were executed on their Vulcan and Nano compute clusters, administered by the High-Performance Computing Services Group at LBNL. TMF is supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy, under contract number DE-AC02-05CH11231. We thank Dr Joseph Woicik for access and assistance at the X24a end station. Acknowledgement is made to the Donors of the American Chemical Society Petroleum Research Fund (PRF 52827-DNI10) for support of the research at Binghamton University.
Author information
Authors and Affiliations
Contributions
G.A.H. prepared the materials. L.D.J., G.A.H., C.J. and J.W. performed XANES and STXM experiments. L.W. performed HAXPES experiments. Y.L. designed the calculations and performed spectral modelling. L.D.J. and A.P. contributed to modelling. D.F., L.F.J.P., D.P. and S.B. conceptualized the experiments and interpreted the data. All authors contributed to writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-10 and Supplementary References. (PDF 911 kb)
Supplementary Movie 1
Structural Distortion Induced by Lithium Intercalation. Transformation of a single unit cell of V2O5 is characterized by the puckering of the apical oxygen towards the Li-ion, while the vanadium atoms rearrange by moving away. This distortion causes the localization of electron on the vanadium atom, creating a small polaron. (MOV 286 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
De Jesus, L., Horrocks, G., Liang, Y. et al. Mapping polaronic states and lithiation gradients in individual V2O5 nanowires. Nat Commun 7, 12022 (2016). https://doi.org/10.1038/ncomms12022
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms12022
This article is cited by
-
Weakly Polarized Organic Cation-Modified Hydrated Vanadium Oxides for High-Energy Efficiency Aqueous Zinc-Ion Batteries
Nano-Micro Letters (2024)
-
Efficient MoS2/V2O5 Electrocatalyst for Enhanced Oxygen and Hydrogen Evolution Reactions
Electrocatalysis (2023)
-
Selective catalytic oxidation of ammonia to nitric oxide via chemical looping
Nature Communications (2022)
-
A deep learned nanowire segmentation model using synthetic data augmentation
npj Computational Materials (2022)
-
Phase Transformations During Li-Insertion into V2O5 at Elevated Temperature
JOM (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
