
Abstract
Even though the mammalian sex-determining gene Sry has been intensively studied for the two decades since its discovery, the regions outside the conserved HMG box DNA-binding domain have received less attention due to a lack of sequence conservation and of obvious structural/functional motifs. Here, we summarize the available evidence for function beyond the HMG box, identify the known and postulated biochemical functions of the non-HMG-box domains in sex determination, and present possible explanations for the puzzling diversity of these non-HMG-box domains.
Similar content being viewed by others
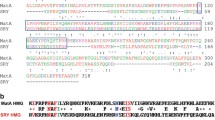

Introduction
Male sex determination in most mammals is initiated by the SRY (sex-determining region on the Y chromosome) gene, expression of which in pre-Sertoli cells drives the differentiation of the bipotential embryonic genital ridges towards testis commitment (Sekido and Lovell-Badge 2009; Kashimada and Koopman 2010). SRY protein binds to the TESCO (testis-specific enhancer of Sox9 core) element of Sox9 and activates the expression of this crucial downstream target (Sekido and Lovell-Badge 2008). In turn, SOX9 protein orchestrates the genetic cascade of testis development (Wilhelm et al. 2007).
In line with a function of SRY proteins as sequence-specific transcription factors (Harley and Goodfellow 1994), all SRY proteins identified to date include an evolutionarily conserved HMG (high-mobility group) DNA-binding domain (Fig. 1), which has taken center stage in research into SRY structure, function and evolution. Nevertheless, all SRY proteins contain additional regions outside the HMG box, albeit sharing much less sequence homology between different species (Fig. 1).

Schematic representation of structures of Sry protein from various mammalian species. The human and mouse SOX3 are included for comparison. Mouse and rat SRY proteins have unique bridge and Q-rich domains at the C terminus. Numbers indicate the identity scores compared to the amino acid sequence of the same domain of human SRY, calculated using Genestream (Pearson et al. 1997)
It has been postulated that the HMG box may be the only functional domain needed for SRY to initiate the male development program (Canning and Lovell-Badge 2002; Lovell-Badge et al. 2002; Sekido and Lovell-Badge 2009). In this review, we challenge this view by summarizing the current knowledge regarding structure and function of SRY’s non-HMG-box domains, and proposing that these domains may be required for the key function of SRY, i.e., directing the testis-determining pathway. We also discuss possible underlying molecular mechanisms involving these domains.
Non-HMG-box domains of SRY are functionally implicated in male sex determination
The HMG box is the signature domain of the SOX (SRY-type HMG box) family of transcription factors (Bowles et al. 2000). It has attracted much of the research attention since Sry was identified as the male sex-determining gene (Koopman et al. 1991), perhaps because it is highly conserved among SRY proteins from different species (Fig. 1). This 79 amino-acid domain contains two nuclear localization signals that allow SRY to access the cell nucleus (Fig. 2; Sim et al. 2008), and provides the structural basis of SRY’s ability to bind (Harley et al. 1992) and bend DNA (Ferrari et al. 1992).

Possible functions of different domains of human and mouse SRY proteins. The nuclear localization signals (NLS), the phosphorylation site (℗), and the interaction sites between human/mouse SRY and SLC9A3R2 or KRAB-O are indicated
In most species, the HMG box is embedded between an N-terminal domain (NTD) typically ∼30–60 amino acids in length, and a C-terminal domain (CTD) of ∼70–100 amino acids. The NTD and CTD are less conserved than the HMG box in different mammalian species (Fig. 1). The exceptions are rat and mouse SRY, which lack all but two amino acids N-terminal to the HMG box and so effectively lack an NTD, and which contain C-terminal to the HMG box a long stretch comprising repeated blocks of glutamine residues separated by short histidine-rich spacers (Fig. 1). This curious domain is referred to as the Q-rich domain. In mouse and rat SRY, the HMG box and Q-rich domains are separated by a 62/63 amino-acid polypeptide known as the bridge domain.
Evidence from a number of transgenic mouse models has been interpreted as suggesting that the HMG box may be the only part required for SRY to initiate testis development (Fig. 3). Chimeric transgenes in which the mouse Sry sequence encoding the HMG box was replaced either with Sox3 or Sox9 HMG box sequence (Bergstrom et al. 2000), or with human SRY sequence encompassing both the NTD and the HMG box, gave XX male sex reversal in mice. Perhaps more surprisingly, transgenic expression of human (Lovell-Badge et al. 2002) or goat SRY (Pannetier et al. 2006) in mice can sex reverse XX embryos, even though neither gene contains non-HMG box sequences even remotely similar to those found in mouse Sry (Fig. 1). Similarly, ectopic expression of Sox3 resulted in XX male sex reversal in mice (Sutton et al. 2011) via a mechanism similar to Sry (i.e., dependent on activation of Sox9), even though similarity between SRY and SOX3 is limited to the HMG box (Fig. 1). Transgenic expression of Sox10 has also been shown to cause XX male sex reversal in mice (Polanco et al. 2010), although it is likely that SOX10 mimics the action of SOX9 rather than SRY in this model. Taken together, these results appear to suggest that expression of any protein bearing an SRY-type HMG box (from any of the SRY proteins found in different mammalian species, or SOX3, -10, or -9) in the embryonic gonads within a critical time window is sufficient to trigger male sex determination. A corollary of that interpretation is that the HMG box is the only essential domain of SRY.

Transgenic constructs and their ability to cause XX sex reversal in mouse embryos. TAD transactivation domain
Balanced against this view, most transcription factors have a bipartite structure, with separable domains responsible for DNA binding and transcriptional modulation, respectively. Further, transcription factors that are members of structurally related families have very similar DNA-binding domains. Although amino acid sequence differences between the conserved domains may contribute to DNA binding site sequence preferences (Mertin et al. 1999), it is reasonable to assume that target specificity is determined, at least in part, by motifs other than that which all the family members have in common (Kamachi et al. 2000; Wilson and Koopman 2002). For SRY, as one of 20 members of the mammalian Sox transcription factor family (Bowles et al. 2000), this assumption points to the involvement of non-HMG-box domains.
Experimental and structure/function evidence supports the involvement of non-box domains in SRY function. Mouse Sry transgenes bearing stop codons introduced into the open reading frame either just 3′ to the HMG box coding sequence (SryStop1; illustrated in Fig. 3) or just 5′ to the Q-rich domain (SryStop2) failed to give XX male sex reversal (Bowles et al. 1999), suggesting that the HMG box alone may not be sufficient and that the Q-rich domain may be required for male sex determination. Nonetheless, the in vivo expression and/or stability of these SRY mutant proteins have not been examined due to lack of suitable antibodies. However, if these truncated proteins are expressed and stable, these results imply that a combination of HMG box and non-box sequences from any species can function together to bring about male sex determination.
Also consistent with the idea that the non-HMG-box domains may be indispensable for SRY to function in male sex determination, mutations within the NTD or CTD of human SRY have been identified in patients with disorders of sex development (DSDs), although these mutations are much rarer than those in the HMG box (Cameron and Sinclair 1997; Shahid et al. 2004).
Mutations in the human SRY NTD can be divided into two subsets. The first subset of mutations (summarized in Assumpção et al. 2002) generate premature stop codons within the NTD that ablate the whole HMG box and CTD, and so are not particularly informative about the functions of NTD. The second subset of NTD mutations (Fig. 4a) are those that result in amino acid substitutions, including S3L (i.e., serine to lysine at amino acid position 3; Gimelli et al. 2006), S18N (Domenice et al. 1998; Canto et al. 2000), R30I (Assumpção et al. 2002), and Q57R (Shahid et al. 2005), all of which were identified in patients with XY gonadal dysgenesis or Turner syndrome with XO/XY mosaicism (Fig. 4b). The first three mutations in the SRY’s NTD are associated with familial gonadal dysgenesis patients with phenotypes of carriers ranging from normal to partial or pure gonadal dysgenesis (Fig. 4b). The variable penetrance of these mutations could not be simply explained by Y mosaicism of fathers. For example, no Y mosaicism in the germ cells of the father was detected in a family bearing the S18N mutation (Domenice et al. 1998). Similarly, a mosaic father is very unlikely in the case of the family bearing the R30I mutation, as three normal brothers also carry the same mutation (Assumpção et al. 2002). Rather, the incomplete penetrance may result from subtle changes in biochemical properties caused by these NTD mutations, which then poise the SRY mutants at the threshold of biological activity and manifest different outcomes in individual carriers, as proposed by a recent study on a familial SRY mutation V60L within the HMG box (Phillips et al. 2011). The fourth NTD mutation, Q57R, located just upstream of the HMG box (Fig. 4a), could potentially affect the function of the HMG box but this has to be tested experimentally. Overall, the identification of these mutations in DSD patients clearly indicates that the NTD plays an essential role in SRY function.

DSD-causing mutations within the human SRY’s non-HMG-box domains, excluding those causing premature stop within the NTD. a Schematic diagram showing the locations of mutated amino acids (marked by asterisks). b Information about the associated DSD patients bearing the mutations. NA not applicable, ND not determined
Mutations in the CTD (Fig. 4a) have also been identified in patients with pure gonadal dysgenesis or Turner syndrome with XO/XY mosaicism (Fig. 4b). Two SRY point mutants with the Serine 143 substituted with either a glycine (S143G, Sánchez-Moreno et al. 2008) or a cysteine (S143C, Shahid et al. 2004) have been reported. Other mutations identified include nonsense or frameshift mutations which generate mutant SRY proteins with either a truncated C terminus (L163X, Tajima et al. 1994) or an altered and shortened C terminus (Q158fsX180 and Q159fsX167, Baldazzi et al. 2003; Shahid et al. 2005). These findings suggest that, within the human SRY CTD, an intact C-terminal tail (amino acid position 163 to 204) and a serine at position 143 are functionally required for male sex determination.
Taken together, the knowledge acquired from both transgenic mouse models and human DSD patients suggests an essential role in male sex determination of the non-HMG-box domains, more specifically, the mouse SRY’s Q-rich domain and the human SRY’s NTD/CTD, respectively.
Functions of the non-HMG-box domains of SRY
What are the likely biochemical functions of these non-box domains? Recent studies on SRY and other SOX proteins have provided important clues as to how the non-HMG-box domains might contribute to SRY function in sex determination, summarized in Fig. 2.
Effects on DNA binding
A number of observations suggest that SRY’s non-HMG-box domains may help to maintain a suitable protein conformation and thus directly contribute to optimal binding to target DNA. Sánchez-Moreno et al. (2008) showed that a CTD-deleted human SRY protein binds less avidly to a SOX binding consensus sequence in in vitro DNA binding assays. Consistent with this idea, the S143 identified as a hotspot of CTD mutations (Fig. 4b) has been proposed to contribute to DNA binding as it is predicted, using a computational protein modeling approach, to be part of a large DNA-interacting cavity and is very close to the bound DNA (Sánchez-Moreno et al. 2009). Furthermore, phosphorylation of the amino acid motif 29RRSSS33 located in the NTD (Fig. 2) has been shown to enhance the DNA binding activity of SRY (Desclozeaux et al. 1998). In accordance with this, the mutation of the R30 within this motif, identified in a familial case of gonadal dysgenesis (Fig. 4b), results in diminished phosphorylation and decreased DNA binding activity of the mutant SRY protein (Assumpção et al. 2002).
Effects on transactivation of Sox9
The main function of SRY is to activate Sox9 transcription in bipotential embryonic genital ridges (Sekido and Lovell-Badge 2009; Kashimada and Koopman 2010). However, it is not known how SRY achieves this. Unlike most other sequence-specific transcription factors including other SOX family members, most SRY proteins do not have an obvious transactivation domain. The exceptions are mouse and rat SRY. Gal4 reporter assays in cultured cells have demonstrated that the Q-rich domain of mouse SRY can function as a transactivation domain in vitro (Dubin and Ostrer 1994). The structurally similar, but much shorter, Q-rich domain of rat SRY might also possess transactivating capacity, but this possibility has yet to be verified experimentally. It is possible that mouse SRY may directly activate Sox9 transcription via its Q-rich domain in vivo. In contrast, human SRY, when fused to a Gal4 DNA-binding domain, did not show any detectable transactivation of a Gal4-responsive reporter in vitro (Dubin and Ostrer 1994). Therefore, human SRY, unlike mouse SRY and other SOX proteins, may have to recruit a partner protein that supplies a transactivation domain to be able to activate the transcription of SOX9 (Dubin and Ostrer 1994). The non-HMG-box domains of SRY are likely to play a role in the recruitment of such a transactivator since the HMG box is common to all SOX proteins and therefore unlikely to mediate an SRY-specific interaction (Wilson and Koopman 2002).
Effects on partner protein binding
Besides the likely contribution to recruiting a transactivator as discussed above, the non-HMG-box domains may also be involved in interactions with other essential SRY partner proteins and thus help to induce and/or stabilize the formation of a ternary complex comprising SRY, its partner proteins, and the target DNA (Kamachi et al. 2000; Wilson and Koopman 2002; Kondoh and Kamachi 2010). Moreover, the contribution of the non-box domains to the interactions with SRY partner proteins may provide the biochemical basis for target selectivity among SOX factors since the HMG box domains from SRY, SOX3, and SOX9 seem to be functionally interchangeable in causing sex reversal in mouse transgenic assays (Fig. 3; Bergstrom et al. 2000).
In support of this, a number of SRY-interacting proteins have been identified (Bernard and Harley 2010), including SLC9A3R2 (also named as SIP-1 or NHERF2) and KRAB-O (protein product of a splicing variant of ZFP748), both of which interact with the human SRY CTD (Poulat et al. 1997; Oh et al. 2005) or with the mouse SRY bridge domain (Oh et al. 2005; Thevenet et al. 2005), respectively (illustrated in Fig. 2). However, the physiological significance of these interactions remains unclear since Slc9a3r2-knockout mice are fertile (Broere et al. 2007) and Zfp748-knockdown mice develop morphologically normal testes (Polanco et al. 2009).
More recently, several groups (Bernard et al. 2008; Tamashiro et al. 2008; Lau and Li 2009) have reported that SRY represses the Rspo1/Wnt/β-catenin signaling pathway that drives the ovarian development (Vainio et al. 1999; Parma et al. 2006; Maatouk et al. 2008). However, the reports do not concur on whether both human and mouse SRY proteins are able to repress, and which domains of the SRY proteins are required for the repression. Bernard et al. (2008) showed that the presence of either the NTD or CTD is required for human SRY to interact with β-catenin. Moreover, the authors showed that the C-terminal truncated, sex-reversing L163X mutant SRY protein (Fig. 4b) failed to inhibit the activity of a Wnt signaling pathway reporter (TOPFLASH), further confirming an essential role of the human SRY CTD in interacting with β-catenin and inhibiting the Wnt pathway. On the other hand, Tamashiro et al. (2008) reported that mouse but not human SRY is capable of repressing TOPFLASH activity and that the repression is dependent on the mouse Q-rich domain. To compound the issue further, Lau and Li (2009) reported that both human and mouse SRY can bind to β-catenin and repress the Wnt pathway, and that mouse SRY can bind to β-catenin via either the HMG box or the Q-rich domain. Nevertheless, these results indicate an important role of the human CTD and mouse Q-rich domain in mediating the SRY/β-catenin interaction and the subsequent repression of the Wnt signaling pathway, at least in vitro. However, the physiological significance of these interactions awaits further examination.
Alternatively, the non-box domains may not harbor the primary protein–protein interaction sites, but instead may help to stabilize the interactions between the HMG box of SRY and its partner proteins. For example, human SRY interacts with WT1 protein via its HMG box (Matsuzawa-Watanabe et al. 2003). However, two SRY mutants lacking the CTD showed significantly decreased binding affinity to WT1 in in vitro pulldown assays, indicating the human SRY CTD contributes to the optimal interaction with WT1. An analogous example comes from studies on the interaction between the vascular endothelial transcriptional regulator SOX18 and its partner protein MEF2C. SOX18 interacts with MEF2C via its HMG box alone. However, the non-HMG-box C terminus of SOX18 was found to influence this interaction, as two SOX18 mutants bearing an altered C terminus failed to interact with MEF2C (Hosking et al. 2001).
Why are non-HMG-box domains of SRY proteins so diverse?
Given the weight of evidence that the mouse Q-rich domain and the human NTD/CTD are both functional and necessary for the SRY protein to carry out its molecular roles in sex determination, it is surprising that these domains are relatively poorly conserved during mammalian evolution. How can this apparent paradox be explained?
It has been suggested that the non-box domains of SRY may have acquired species-specific functions (Sekido and Lovell-Badge 2009). For instance, mouse SRY’s Q-rich domain functions as a transactivation domain, at least in vitro (Dubin and Ostrer 1994). As discussed above, mouse SRY may directly activate the transcription of Sox9, whereas human SRY (and SRY proteins from other species lack of such a potential transactivation domain) may have to recruit a transactivator to fulfill its job (Dubin and Ostrer 1994). Also, it might be possible that the Q-rich domain of mouse SRY may compensate for the lack of NTD, fulfilling some roles that in human SRY are played through its NTD. However, this possibility remains to be tested experimentally.
We propose an additional possibility, namely that the functions of the human and mouse non-HMG-box domains may be conserved even though the sequences encoding these domains are not. In other words, the non-box domains may evolve freely as long as they fulfill essential functions, such as maintaining a suitable protein conformation and/or contributing to interaction with essential partner proteins so as to bind to target DNA sequence and thereby activate SOX9 transcription. In support of this concept, the human SRY’s CTD and mouse SRY’s C-terminal region (bridge + Q-rich domains) appear to be functionally interchangeable: both the intact human SRY or the human/mouse chimera consisting of human NTD + HMG box fused to mouse bridge + Q-rich domains (Fig. 3) can cause XX male sex reversal of transgenic mouse embryos (Lovell-Badge et al. 2002). Also, both the human SRY CTD and the mouse SRY bridge domain interact with SLC9A3R2 and KRAB-O putative partner proteins (Fig. 2), although via different interaction sites (Poulat et al. 1997; Oh et al. 2005; Thevenet et al. 2005), suggesting that these two domains may have similar functions despite having little sequence homology.
We suggest that some parts of the non-HMG-box domains of human and mouse SRY may be functionally equivalent, for example in maximizing target DNA binding affinity, stabilizing the interactions with essential partner proteins and supplementing a transactivation domain (Fig. 2). The lack of obvious sequence homology between human and mouse SRY may result from the high variation and rapid drifting of the Y chromosome genes due to the inability of the Y to recombine with the X chromosome (Graves 2002, 2006). SRY has been suggested to evolve from a hybrid of DGCR8 and SOX3 (Katoh and Miyata 1999; Sato et al. 2010). However, the low sequence homology either between the SRY NTD and the N-terminal region of DGCR8 (Sato et al. 2010), or between the C-terminal regions of SRY and SOX3 (Fig. 1) clearly indicates that rapid sequence drift has indeed affected SRY.
Concluding remarks
Evidence accumulated over the last two decades from studies on transgenic mouse models and molecular analyses of human DSD patients indicates that the non-HMG-box domains, namely the human NTD/CTD and mouse bridge and Q-rich domains, respectively, are required for human/mouse SRY to function properly in male sex determination. In transgenic experiments, although apparently any SRY-type HMG box-containing protein seems to be able to direct male sex determination when expressed in the bipotential genital ridges, the successful transgenic constructs always contain additional regions. We suggest that the exact primary sequence of these regions is immaterial, as long as certain essential functions are provided. These functions likely include contributing directly or indirectly to optimal binding to target DNA and essential partner proteins and to transcriptional activation. It will be useful to determine the structures of full-length human and mouse SRY proteins in order to better understand of the molecular mechanisms underlying SRY’s function and to help evaluate the hypothesis that functional interchangeability of non-box domains is conserved between rodents and other mammals. This information should also shed light on how the SRY gene has evolved.
To explicitly address the essentiality of the SRY’s non-box domains for male sex determination, more definitive studies are required. For example, the mouse C-terminal truncated Sry mutants lacking the Q-rich domain or the bridge + Q-rich domains (Bowles et al. 1999) may be revisited using Sry transgenes tagged with an epitope to facilitate the detection of mutant proteins. This would firmly exclude the possibility that the loss of sex reversing ability of these Sry mutants was due to lack of protein expression or protein instability in vivo. Human SRY can also be functionally dissected using a similar transgenic approach, to confirm the requirements for the NTD and CTD in sex determination and to pinpoint the important sequence motifs.
There are still many outstanding questions about the structure, function, and evolution of SRY’s non-HMG-box domains. For example, do these non-box-domains directly contribute to the optimal binding of SRY to TESCO in vivo? Or do they contribute to the recruitment of the yet-to-be-identified SRY partner proteins to TESCO and in turn activate the Sox9 expression? If so, what are these essential partners and where are the protein–protein binding interfaces located? More specifically, how does the mouse Q-rich domain contribute to the transactivation of Sox9? Does it function as a transactivation domain in vivo? If so, how does human SRY (and other SRY proteins lack of a transactivation domain) activate SOX9 transcription? Does human SRY recruit a partner transactivator to do so? Answers to these questions should provide valuable insights as to how the SRY genes have evolved and how the non-HMG-box mutations affect human SRY function and lead to human DSDs.
Abbreviations
- CTD:
-
C-terminal domain
- DSD(s):
-
Disorder(s) of sex development
- HMG:
-
High-mobility group
- NTD:
-
N-terminal domain
- SOX:
-
SRY-type HMG box
- SRY:
-
Sex-determining region on the Y chromosome
- TESCO:
-
Testis-specific enhancer of Sox9 core element
References
Assumpção JG, Benedetti CE, Maciel-Guerra AT et al (2002) Novel mutations affecting SRY DNA-binding activity: the HMG box N65H associated with 46, XY pure gonadal dysgenesis and the familial non-HMG box R30I associated with variable phenotypes. J Mol Med (Berl) 80:782–790
Baldazzi L, Nicolettic A, Gennari M et al (2003) Two new point mutations of the SRY gene identified in two Italian 46, XY females with gonadal dysgenesis. Clin Genet 64:258–260
Bergstrom DE, Young M, Albrecht KH, Eicher EM (2000) Related function of mouse SOX3, SOX9, and SRY HMG domains assayed by male sex determination. Genesis 28:111–124
Bernard P, Harley VR (2010) Acquisition of SOX transcription factor specificity through protein–protein interaction, modulation of Wnt signalling and post-translational modification. Int J Biochem Cell Biol 42:400–410
Bernard P, Sim H, Knower K, Vilain E, Harley V (2008) Human SRY inhibits β-catenin-mediated transcription. Int J Biochem Cell Biol 40:2889–2900
Bowles J, Cooper L, Berkman J, Koopman P (1999) Sry requires a CAG repeat domain for male sex determination in Mus musculus. Nat Genet 22:405–408
Bowles J, Schepers G, Koopman P (2000) Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev Biol 227:239–255
Broere N, Hillesheim J, Tuo B et al (2007) Cystic fibrosis transmembrane conductance regulator activation is reduced in the small intestine of Na+/H+ exchanger 3 regulatory factor 1 (NHERF-1)- but not NHERF-2-deficient mice. J Biol Chem 282:37575–37584
Cameron FJ, Sinclair AH (1997) Mutations in SRY and SOX9: testis-determining genes. Hum Mutat 9:388–395
Canning CA, Lovell-Badge R (2002) Sry and sex determination: how lazy can it be? Trends Genet 18:111–113
Canto P, de la Chesnaye E, López M et al (2000) A mutation in the 5′ non-high mobility group box region of the SRY gene in patients with Turner syndrome and Y mosaicism. J Clin Endocrinol Metab 85:1908–1911
Desclozeaux M, Poulat F, de Santa BP et al (1998) Phosphorylation of an N-terminal motif enhances DNA-binding activity of the human SRY protein. J Biol Chem 273:7988–7995
Domenice S, Yumie Nishi M, Correia Billerbeck AE et al (1998) A novel missense mutation (S18N) in the 5′ non-HMG box region of the SRY gene in a patient with partial gonadal dysgenesis and his normal male relatives. Hum Genet 102:213–215
Dubin R, Ostrer H (1994) Sry is a transcriptional activator. Mol Endocrinol 8:1182–1192
Ferrari S, Harley VR, Pontiggia A et al (1992) SRY, like HMG1, recognizes sharp angles in DNA. EMBO J 11:4497–4506
Gimelli G, Gimelli S, Dimasi N et al (2006) Identification and molecular modelling of a novel familial mutation in the SRY gene implicated in the pure gonadal dysgenesis. Eur J Hum Genet 15:76–80
Graves JA (2002) Evolution of the testis-determining gene—the rise and fall of SRY. Novartis Found Symp 244:86–97, discussion 97–101, 203–106, 253–107
Graves JA (2006) Sex chromosome specialization and degeneration in mammals. Cell 124:901–914
Harley VR, Goodfellow PN (1994) The biochemical role of SRY in sex determination. Mol Reprod Dev 39:184–193
Harley V, Jackson D, Hextall P et al (1992) DNA binding activity of recombinant SRY from normal males and XY females. Science 255:453–456
Hosking BM, Wang SC, Chen SL et al (2001) SOX18 directly interacts with MEF2C in endothelial cells. Biochem Biophys Res Commun 287:493–500
Kamachi Y, Uchikawa M, Kondoh H (2000) Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet 16:182–187
Kashimada K, Koopman P (2010) Sry: the master switch in mammalian sex determination. Development 137:3921–3930
Katoh K, Miyata T (1999) A heuristic approach of maximum likelihood method for inferring phylogenetic tree and an application to the mammalian SOX-3 origin of the testis-determining gene SRY. FEBS Lett 463:129–132
Kondoh H, Kamachi Y (2010) SOX-partner code for cell specification: regulatory target selection and underlying molecular mechanisms. Int J Biochem Cell Biol 42:391–399
Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R (1991) Male development of chromosomally female mice transgenic for Sry. Nature 351:117–121
Lau Y-FC, Li Y (2009) The human and mouse sex-determining SRY genes repress the Rspol/β-catenin signaling. J Genet Genomics 36:193–202
Lovell-Badge R, Canning C, Sekido R (2002) Sex-determining genes in mice: building pathways. Novartis Found Symp 244:4–18, discussion 18-22, 35-42, 253-257
Maatouk DM, DiNapoli L, Alvers A et al (2008) Stabilization of β-catenin in XY gonads causes male-to-female sex-reversal. Hum Mol Genet 17:2949–2955
Matsuzawa-Watanabe Y, J-i I, Semba K (2003) Transcriptional activity of testis-determining factor SRY is modulated by the Wilms’ tumor 1 gene product, WT1. Oncogene 22:7900–7904
Mertin S, McDowall SG, Harley VR (1999) The DNA-binding specificity of SOX9 and other SOX proteins. Nucleic Acids Res 27:1359–1364
Oh HJ, Li Y, Lau YF (2005) Sry associates with the heterochromatin protein 1 complex by interacting with a KRAB domain protein. Biol Reprod 72:407–415
Pannetier M, Tilly G, Kocer A et al (2006) Goat SRY induces testis development in XX transgenic mice. FEBS Lett 580:3715–3720
Parma P, Radi O, Vidal V et al (2006) R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet 38:1304–1309
Pearson WR, Wood T, Zhang Z, Miller W (1997) Comparison of DNA sequences with protein sequences. Genomics 46:24–36
Phillips NB, Racca J, Chen Y-S et al (2011) SRY and the enigma of inherited human sex reversal. Frustrated induced fit in a bent protein–DNA complex. J Biol Chem. doi:10.1074/jbc.M111.260091
Polanco JC, Wilhelm D, Mizusaki H et al (2009) Functional analysis of the SRY-KRAB interaction in mouse sex determination. Biol Cell 101:55–67
Polanco JC, Wilhelm D, Davidson T-L, Knight D, Koopman P (2010) Sox10 gain-of-function causes XX sex reversal in mice: implications for human 22q-linked disorders of sex development. Hum Mol Genet 19:506–516
Poulat F, de Santa BP, Desclozeaux M et al (1997) The human testis determining factor SRY binds a nuclear factor containing PDZ protein interaction domains. J Biol Chem 272:7167–7172
Sánchez-Moreno I, Coral-Vázquez R, Méndez JP, Canto P (2008) Full-length SRY protein is essential for DNA binding. Mol Hum Reprod 14:325–330
Sánchez-Moreno I, Canto P, Munguía P et al (2009) DNA binding activity studies and computational approach of mutant SRY in patients with 46, XY complete pure gonadal dysgenesis. Mol Cell Endocrinol 299:212–218
Sato Y, Shinka T, Sakamoto K, Ewis A, Nakahori Y (2010) The male-determining gene SRY is a hybrid of DGCR8 and SOX3, and is regulated by the transcription factor CP2. Mol Cell Biochem 337:267–275
Sekido R, Lovell-Badge R (2008) Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 453:930–934
Sekido R, Lovell-Badge R (2009) Sex determination and SRY: down to a wink and a nudge? Trends Genet 25:19–29
Shahid M, Dhillion VS, Jain N et al (2004) Two new novel point mutations localized upstream and downstream of the HMG box region of the SRY gene in three Indian 46, XY females with sex reversal and gonadal tumour formation. Mol Hum Reprod 10:521–526
Shahid M, Dhillon VS, Aslam M, Husain SA (2005) Three new novel point mutations localized within and downstream of high-mobility group-box region in SRY gene in three Indian females with Turner syndrome. J Clin Endocrinol Metab 90:2429–2435
Sim H, Argentaro A, Harley VR (2008) Boys, girls and shuttling of SRY and SOX9. Trends Endocrinol Metab 19:213–222
Sutton E, Hughes J, White S et al (2011) Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest 121:328–341
Tajima T, Nakae J, Shinohara N, Fujieda K (1994) A novel mutation localized in the 3′ non-HMG box region of the SRY gene in 46, XY gonadal dysgenesis. Hum Mol Genet 3:1187–1189
Tamashiro DA, Alarcón VB, Marikawa Y (2008) Ectopic expression of mouse Sry interferes with Wnt/β-catenin signaling in mouse embryonal carcinoma cell lines. Biochim Biophys Acta 1780:1395–1402
Thevenet L, Albrecht KH, Malki S et al (2005) NHERF2/SIP-1 interacts with mouse SRY via a different mechanism than human SRY. J Biol Chem 280:38625–38630
Vainio S, Heikkila M, Kispert A, Chin N, McMahon AP (1999) Female development in mammals is regulated by Wnt-4 signalling. Nature 397:405–409
Wilhelm D, Palmer S, Koopman P (2007) Sex determination and gonadal development in mammals. Physiol Rev 87:1–28
Wilson M, Koopman P (2002) Matching SOX: partner proteins and co-factors of the SOX family of transcriptional regulators. Curr Opin Genet Dev 12:441–446
Acknowledgments
We thank Terje Svingen and Josephine Bowles for critical reading of the manuscript. This work is supported by research grants from the Australian Research Council (ARC) and National Health and Medical Research Council of Australia. P.K. is a Federation Fellow of the ARC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editors: Tariq Ezaz and Jennifer Graves.
Rights and permissions
About this article
Cite this article
Zhao, L., Koopman, P. SRY protein function in sex determination: thinking outside the box. Chromosome Res 20, 153–162 (2012). https://doi.org/10.1007/s10577-011-9256-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-011-9256-x