
Abstract
Aims/hypothesis
Inflammatory alterations in white adipose tissue appear to underlie complications of obesity including diabetes mellitus. Polyunsaturated fatty acids (PUFA), particularly those of the n−3 series, modulate immune responses and may ameliorate insulin sensitivity. In this study, we investigated how PUFA affect white adipose tissue inflammation and gene expression in obese diabetic animals.
Materials and methods
We treated db/db mice as well as lean non-diabetic mice (db/+) with either low-fat standard diet (LF) or high-fat diets rich in (1) saturated/monounsaturated fatty acids (HF/S), (2) n−6 PUFA (HF/6) and (3) the latter including purified marine n−3 PUFA (HF/3).
Results
Many genes involved in inflammatory alterations were upregulated in db/db mice on HF/S compared with LF in parallel with phosphorylation of c-Jun N-terminal kinase (JNK). In parallel, adipose tissue infiltration with macrophages was markedly enhanced by HF/S. When compared with HF/S, HF/6 showed only marginal effects on adipose tissue inflammation. However, inclusion of n−3 PUFA in the diet (HF/3) completely prevented macrophage infiltration induced by high-fat diet and changes in inflammatory gene expression, also tending to reduce JNK phosphorylation (p<0.1) in diabetic mice despite unreduced body weight. Moreover, high-fat diets (HF/S, HF/6) downregulated expression and reduced serum concentrations of adiponectin, but this was not the case with n−3 PUFA.
Conclusions/interpretation
n−3 PUFA prevent adipose tissue inflammation induced by high-fat diet in obese diabetic mice, thereby dissecting obesity from adipose tissue inflammation. These data suggest that beneficial effects of n−3 PUFA on diabetes development could be mediated by their effect on adipose tissue inflammation.
Similar content being viewed by others


Introduction
Obesity is associated with chronic low-grade inflammation that is characterised by increased serum concentrations of acute phase reactants and combines with metabolic alterations such as type 2 diabetes mellitus [1]. The inflammatory response in the presence of obesity appears to be triggered by and to reside predominantly in adipose tissue [2]. Notably, low-grade inflammation is involved in the pathogenesis of atherosclerosis that might explain the association of obesity with cardiovascular complications [3–5]. Obesity and insulin resistance are correlated with activation of peripheral blood monocytes, showing increased expression of a variety of cytokines including migration inhibitory factor, IL-6 and TNF-α [6]. In addition, blood-glucose-lowering drugs metformin and rosiglitazone reduce inflammatory alterations, indicating that part of their beneficial effect, including that of metformin on cardiovascular complications, could be due to modulation of the inflammatory response [7–9].
Obesity-associated inflammatory changes in white adipose tissue are predominantly due to macrophage infiltration in human obesity as well as in genetic- and high-fat-diet-induced mouse models [10, 11]. These studies used high-fat diets rich in monounsaturated fatty acids. However, dietary polyunsaturated fatty acids (PUFA) seem to postpone diabetes development [12] and have considerable impact on gene expression in a variety of tissues, including adipose tissue, where they regulate genes involved in adipocyte differentiation and lipid metabolism [13, 14]. In addition, PUFA, particularly those of the n−3 fatty acids that are found in marine fish oils, exert immunomodulatory effects, e.g. in inflammatory joint and bowel diseases [15]. Immunomodulation by PUFA has been studied mainly in T lymphocytes and T cell-mediated diseases, but n−3 PUFA have been found to affect monocyte and macrophage functions as well [16]. Therefore, we studied the effect of marine n−3 PUFA on white adipose tissue inflammation and gene expression in db/db mice on a high-fat diet. Compared with animals on a diet rich in saturated and monounsaturated fatty acids, n−3 PUFA completely prevented high-fat diet-induced inflammatory alterations, whereas n−6 PUFA had only marginal effects. Notably, interference with white adipose tissue inflammation occurred in the absence of body weight reduction, indicating that n−3 PUFA dissect obesity from inflammatory changes.
Materials and methods
Animals and diets
Male C57BL/KsJ-leprdb/leprdb diabetic (db/db) mice and their non-diabetic littermates (db/+) were purchased from Charles River Laboratories (Sulzfeld, Germany) at 7 weeks of age and were routinely fed a low-fat standard rodent diet (LF; 3 kcal% fat; N1324; Altromin, Lage, Germany) with water freely available. For the experiment, db/db and db/+ mice (n=5–8/group) were fed for 6 weeks with either the LF or high-fat diets that included 30% of calories from lard oil (rich in saturated and monounsaturated fatty acids; HF/S), safflower oil (rich in n−6 PUFA; HF/6) or the latter with 40% of oil volume being replaced by a concentrate of highly purified n−3 PUFA eicosapentaenoic acid (20:5n−3) and docosahexaenoic acid (22:6n−3) re-esterified to triglycerides (EPAX6000TG; HF/3) generously provided by Pronova Biocare, Lysaker, Norway. High-fat diets were isocaloric and all fat supplements included 4 mg/g α-tocopherol (Sigma, St Louis, MO, USA). Animals on high-fat diets were pair-fed to ensure equivalent food intake. Epididymal and subcutaneous white adipose tissue was immediately snap-frozen in liquid nitrogen. Blood glucose, plasma cholesterol and triglycerides were measured on an automated analyser, NEFA with a commercial kit (Wako Chemicals, Richmond, VA, USA). Adiponectin, macrophage chemotactic protein (MCP)-1, IL-6 (both R&D Systems, Wiesbaden-Nordenstadt, Germany) and C-reactive protein (CRP) (Immunology Consultants Lab, Newberg, OR, USA) were determined in blood plasma using commercially available ELISA kits. The study protocol was approved by the local ethics committee for animal experiments, and followed the guidelines on accommodation and care of animals formulated by the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes.
Microarray hybridisation and data analysis
Total RNA was prepared using TRIzol (Invitrogen, Carlsbad, CA, USA) and repurified with an RNeasy MinElute kit (Qiagen, Valencia, CA, USA). Following in vitro transcription, labelled cRNA was hybridised to U74Av2 GeneChips (Affymetrix, Santa Clara, CA, USA) representing more than 12,000 transcripts. Scanned images were analysed with Microarray Suite version 5.0 software (Affymetrix). Normalisation was performed by global scaling to an average intensity of 100 arbitrary units. Gene abundances were estimated by robust multi-array analysis [17] using the probe-level modelling (affyPLM) package from Bioconductor (http://www.bioconductor.org). We used a strategy similar to ANOVA and computed a consensus estimate for the variability among all groups as a basis for t-test statistics. Genes with absolute fold change of at least 1.5 (increase or decrease) were selected at p<0.01.
Quantitative real-time PCR
One microgram of total RNA was treated with DNase I and reverse transcribed into cDNA by Superscript II using random hexamer priming (all Invitrogen). Real-time RT-PCR was performed on an ABI PRISM 7000 Cycler using gene-specific FAM-TAMRA-labelled commercial Assays-on-Demand (Applied Biosystems, Foster City, CA, USA) normalised to 18S VIC-labelled endogenous control (Applied Biosystems).
Immunofluorescence
Five-micron paraffin-embedded tissue samples were deparaffinised, treated with Antigen Unmasking Solution (Vector Labs, Burlingame, CA, USA) and stained for macrophages using MAC-2 antibody (Cerderlane Laboratories, ON, Canada) followed by Alexa Fluor 594 goat anti-rat IgG (Molecular Probes, Eugene, OR, USA) and nuclei counterstaining with DAPI. F4/80-positive macrophages were detected on frozen tissue sections stained with Alexa488-labelled F4/80 antibody (clone CI:A3-1; Serotec, Düsseldorf, Germany) and nuclei counterstained with propidium iodide.
Fatty acid analyses
Fatty acids from diets were analysed as described previously [18] with some modifications. Briefly, methanol (Merck, Darmstadt, Germany)/benzene (Riedel-de Haën, Seelze, Germany) 4:1 (v/v) extracts including heptadecanoic acid as internal standard were methanolysed. Fatty acid methyl esters were separated by gas chromatography on a DB-23 column (J & W Scientific, Folsom, CA, USA) and detected by electron-impact ionisation mass spectroscopy using fatty acid methyl esters of the highest available quality (Alltech, Deerfield, IL, USA) as standards.
Immunoblotting
Adipose tissue was homogenised and lysed on ice for 30 min in TBS, pH 7.4, containing 1% Triton X-100 (Pierce, Rockford, IL, USA), phosphatase (2 mmol/l sodium orthovanadate, 20 mmol/l NaF, 10 mmol/l sodium pyrophosphate, 50 mmol/l β-glycerophosphate, and 10 mmol/l EDTA) and protease inhibitors. The tissue extract was cleared from fat, nuclei and debris by centrifugation at 13,000×g for 10 min. Protein content was determined by BCA assay (Pierce) and identical amounts of proteins (30 μg/lane) were separated by SDS-PAGE and blotted onto nitrocellulose membranes (Hybond ECL, Amersham, UK). Equal blotting was verified by Ponceau red staining (Pierce) (data not shown). Phosphorylation of c-Jun N-terminal kinase (JNK) (Thr-183, Tyr-185) was assessed using a mouse monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and JNK was detected using a rabbit polyclonal antibody (Cell Signaling, Danvers, MA, USA), both followed by respective horseradish peroxidase-labelled secondary antibodies (Accurate, Westbury, NY, USA). Chemiluminescence was generated by BM chemiluminescence substrate (Roche, Mannheim, Germany) and quantified on a Lumi-Imager (Roche). Densitometric data from both JNK isoforms (p46 and p54) were combined for numerical analyses.
Statistical analyses
Data are given as means±SEM unless indicated otherwise. Expression data from quantitative RT-PCR and microarrays as well as other data exhibiting unequality of variances between groups according to Levene’s test were log-transformed before ANOVA. Except for evaluation of expression profiles (see above), study groups within a genotype were compared with mice fed HF/S by univariate ANOVA using Dunnett’s t-test for post hoc analysis. The use of HF/S-fed animals as a control allowed simultaneous statistical evaluation of the effect of high-fat diet per se (i.e. compared with LF) and modifications of this effect by inclusion of PUFA (i.e. compared with HF/6 or HF/3). As an exception, the effect of obesity and diabetes was evaluated by comparing db/+ vs db/db mice, both on LF, resulting in its reciprocal (diabetes−1) in order to facilitate direct comparisons with a normalising effect of PUFA.
Results
Body weight and metabolic changes by diets with different fat composition
To elucidate the impact of high-fat diets with different fatty acid composition on diabetes-induced alterations in white adipose tissue we treated obese diabetic db/db mice and lean non-diabetic littermates (db/+) with either LF or high-fat diets enriched in saturated and monounsaturated fatty acids (HF/S), n−6 PUFA (HF/6), or a mixture of n−3 and n−6 PUFA (HF/3) that included 25.1 mg/g long-chain marine n−3 PUFA, mainly eicosapentaenoic (20:5n−3) and docosahexaenoic acid (22:6n−3; data not shown). All groups within a genotype were compared with mice fed HF/S in order to allow simultaneous statistical evaluation of the effect of high-fat diet per se and modifications of this effect by inclusion of different PUFA.
Body weight at the end of the study was somewhat higher in db/db mice fed HF/3 compared with HF/S (Table 1). Plasma triglyceride concentrations were significantly lowered by HF/3 compared with HF/S in diabetic and non-diabetic animals. Plasma NEFA concentrations tended to be lowered by n−3 PUFA in db/db animals (p=0.13), whereas blood glucose concentrations did not change. Markers of systemic inflammation such as plasma concentrations of MCP-1 (CCL2), IL-6 and CRP showed minor alterations in obese compared with lean mice and different high-fat diets, respectively, indicating that systemic inflammation was not enhanced in obese animals under the conditions studied (Table 1). Only opposing alterations of CRP by n−6 and n−3 PUFA in obese mice and reduction of MCP-1 by n−3 compared with n−6 PUFA in lean mice turned out to be statistically significant.
Gene expression profiling
Gene expression profiles from db/db mice fed LF and different PUFA-enriched diets were compared with those fed HF/S (Table 2). HF/S treatment predominantly upregulated gene expression in db/db animals (244 up, 21 down) whereas effects of n−3 PUFA in combination with n−6 PUFA (n−3 & 6 PUFA effect) or on their own (n−3 PUFA effect) predominantly decreased gene expression. Compared with HF/S, n−6 PUFA did not significantly change gene expression profiles and was omitted from further analyses. A complete list of genes regulated by any diet effect is given in the electronic supplementary material (ESM). When analysing overlaps of expression profiles, we found that many HF/S-induced changes were discordant with the diabetes−1 effect, revealing aggravation of diabetes-associated changes, whereas n−3 PUFA were associated with normalisation of gene expression, i.e. alterations in the direction to non-diabetic mice. Moreover, high-fat diet effects were strictly discordant with those of n−3 & 6 PUFA (r 2=0.8325, p<0.0005; Fig. 1) and n−3 PUFA (r 2=0.6814, p<0.0005; data not shown), indicating that n−3 PUFA prevent gene expression alterations induced by HF/S. Thus, consumption of a high-fat diet aggravated diabetes-induced alterations in white adipose tissue gene expression, but inclusion of n−3 PUFA in the high-fat diet abrogated this effect.

Discordant changes of adipose tissue gene expression in response to high-fat diets that lack or include n−3 polyunsaturated fatty acids (PUFA). Obese diabetic db/db mice were fed either low-fat (LF) or high-fat (HF) diets rich in saturated and monounsaturated fatty acids (HF/S), n−6 polyunsaturated fatty acids (HF/6) or the latter including n−3 PUFA (HF/3) for 6 weeks. Gene expression was evaluated in gonadal white adipose tissue by oligonucleotide microarrays (n=3/group). Alterations in gene expression given in log2 by high-fat diet (expression in HF/S vs LF) are plotted against those by n−3 & 6 PUFA (HF/3 vs HF/S) for all 671 genes achieving a significant regulation at p<0.01 for at least one effect. Pearson’s r 2=0.8325, p<0.0005. A similar correlation was obtained when effects of n−3 PUFA (HF/3 vs HF/6) were plotted at the ordinate (r 2=0.6814, p<0.0005; data not shown)
Genes involved in the inflammatory response
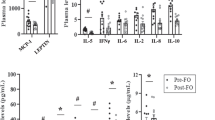
Prototype genes associated with macrophage infiltration include those for the macrophage surface marker CD68 (CD68); macrophage antigen-1 (CD11b; Itgam) that is also expressed on monocytes, neutrophils and NK cells; MCP-1 (Ccl2) that is involved in macrophage infiltration; macrophage scavenger receptor-1 (Msr-1); macrophage-expressed gene 1 (Mpeg1); and the lipopolysaccharide receptor CD14 (CD14) that is expressed on monocytes and macrophages. HF/S considerably increased expression of these (Fig. 2) and a large number of other inflammatory and macrophage genes in gonadal adipose tissue of db/db mice, including those encoding integrins (e.g. CD18), leucocyte surface antigens (e.g. CD14, CD48), chemokines and chemokine receptors (e.g. CCR1, CCR5), Fc receptors and macrophage-specific genes (ESM Table). Macrophage marker genes Ccl2, CD68 and Itgam were significantly upregulated by high-fat diet in db/db animals, whereas TNF-α (Tnf) just failed to reach statistical significance (Fig. 2a). In addition, IL-1 receptor antagonist (Il1rn), an anti-inflammatory cytokine whose expression is increased also in human obesity [19] was upregulated by HF/S (Fig. 2b). The particularly enhanced expression of IL-1 receptor antagonist (Fig. 2) and of arginase (5.6-fold; not shown) in HF/S vs LF point to an alternatively activated phenotype of adipose-tissue-infiltrating macrophages [20]. Notably, inclusion of n−3 PUFA downregulated inflammatory gene expression in db/db mice to levels equal to or even lower than those found in animals on LF (Fig. 2a,b). Changes in subcutaneous adipose tissue (Fig. 3) mirrored those from gonadal fat in db/db mice, revealing that the preventive effect of n−3 PUFA on white adipose tissue inflammation is common to different fat depots. Expression of many genes involved in the inflammatory response, including macrophage marker genes, was already upregulated in db/db compared with db/+ animals, both on LF (Figs. 2 and 3; note different scaling of the ordinate in panels 2c and 3b). In contrast to db/db animals, only minor changes in the expression of inflammatory genes induced by high-fat diets were observed in db/+ animals, including upregulation of Ccl2 selectively in gonadal adipose tissue (Fig. 2c) whose expression was again normalised by dietary n−3 PUFA. Notably, expression of macrophage marker genes in spleen and lung, typical macrophage-rich tissues, was not significantly altered by dietary interventions (Fig. 4), indicating that effects of high-fat diet and n−3 PUFA in obese animals are restricted to adipose tissue. Thus, inflammatory changes in gene expression that were induced by feeding a high-fat diet selectively in white adipose tissue of obese diabetic mice were efficiently abrogated by inclusion of n−3 PUFA in the diet.

n−3 PUFA prevent high-fat diet-induced expression of inflammatory genes in gonadal adipose tissue of diabetic mice. Obese diabetic db/db (a, b) and lean non-diabetic db/+ mice (c) were fed either low-fat (LF) or high-fat diets rich in saturated and monounsaturated fatty acids (HF/S), n−6 PUFA (HF/6) or the latter including n−3 PUFA (HF/3). Relative gene expression compared with animals on LF (set to 100% separately for each genotype) was evaluated by quantitative real-time RT-PCR (a, c, n=4–5/group; normalised to 18S) or oligonucleotide microarrays (b, n=3/group) in gonadal adipose tissue. Genes analysed: a, c black bars, Ccl2, MCP-1 gene; hatched bars, CD68, macrosialin; grey bars, Itgam, integrin α M, CD11b; open bars, Tnf, TNF-α gene; b black bars, Msr1, macrophage scavenger receptor 1; hatched bars, Il1rn, IL-1 receptor antagonist; grey bars, CD14, lipopolysaccharide receptor; open bars, Mpeg1, macrophage-expressed gene 1. Significance of differences from the HF/S group of the same genotype are indicated as follows: *p<0.05; †p<0.01; ‡p<0.001. Compared with db/db animals on LF (set to 100%; a), db/+ animals on the same diet (c) had the following mean relative levels of gene expression: Ccl2, 5.1%; CD68, 6.1%; Itgam, 19.0%; Tnf, 38.9%. All data from db/+ vs db/db animals, both on LF, were significant at p<0.001 (not indicated in the figure). Means±SEM

n−3 PUFA prevent high-fat diet-induced expression of inflammatory genes also in subcutaneous adipose tissue of diabetic mice. Obese diabetic db/db (a) and lean non-diabetic db/+ mice (b) were fed different diets and relative gene expression was analysed as detailed in the legend to Fig. 2. Gene expression of mice fed LF was set to 100% separately for each genotype. Black bars, Ccl2, MCP-1 gene; hatched bars, CD68, macrosialin; open bars, Itgam, integrin α M or CD11b. Significance of differences from the HF/S group of the same genotype are indicated as follows: *p<0.05; †p<0.01; ‡p<0.001. Compared with db/db animals on LF (set to 100%; a), db/+ animals on the same diet (b) had the following mean relative levels of gene expression: Ccl2, 16.2%; CD68, 34.2%; Itgam, 41.6%. All data from db/+ vs db/db animals, both on LF, were significant at p<0.01 (not indicated in the figure). Means±SEM

Expression of inflammatory genes in lung and spleen. Gene expression was analysed in lung (a) and spleen (b) from obese diabetic db/db mice fed different diets as detailed in the legend to Fig. 2, in parallel with tissues from db/+ mice fed LF. Gene expression was evaluated by quantitative RT-PCR and db/db mice fed LF were set to 100% (n=4–5/ group). Black bars, Ccl2, MCP-1 gene; hatched bars, CD68, macrosialin; open bars, Itgam integrin α M, CD11b gene. No significant differences between diets and/or genotypes were detected. Means±SEM
Histological analyses and JNK activation
Histological analyses revealed considerable mononuclear infiltrates in white adipose tissue from db/db mice fed HF/S compared with those on LF (Fig. 5). Most infiltrating cells were macrophages, as they expressed the markers F4/80 and/or MAC-2, although the specificity of the latter has been recently questioned [21, 22]. HF/S increased macrophage numbers in gonadal adipose tissue sections by about three-fold (Fig. 5k). However, inclusion of n−3 PUFA in the high-fat diet completely prevented macrophage infiltration by high-fat diets. These observations indicate that n−3 PUFA downregulate expression of macrophage genes in white adipose tissue predominantly by interfering with macrophage infiltration.

n−3 PUFA prevent high-fat diet-induced aggravation of adipose tissue infiltration with macrophages. Obese diabetic db/db mice were fed either low-fat diet (a, b c; LF) or high-fat diets rich in saturated and monounsaturated fatty acids (d,e,f; HF/S), or including n−3 PUFA (g,h i; HF/3) for 6 weeks. Representative histological sections stained with hematoxylin-eosin (a d,g) and immunofluorescence detection of macrophages performed with antibodies to F4/80 (b,e,h; green; counterstaining of nuclei with propidium iodide, orange) and MAC-2+ (c,f,i; red; counterstaining of nuclei with DAPI, blue) of gonadal white adipose tissue are depicted. Bar=50 μm. k Number of macrophages infiltrating gonadal adipose tissue counted as F4/80+ (black columns) or MAC-2+ cells (open columns), respectively, as percent of the total number of nuclei. *p<0.05; ‡p<0.001 vs HF/S. Means±SEM
To investigate whether the inflammatory changes in adipose tissue are paralleled by alterations in the functional status of JNK, which is activated by phosphorylation and critically involved in the deterioration of insulin sensitivity, we analysed JNK phosphorylation in adipose tissue protein extracts. As shown in Fig. 6, JNK activation, as determined by the phosphorylated JNK:JNK protein ratio, was strongly enhanced in obese compared with lean animals, both on LF (p<0.01). Moreover, feeding HF/S resulted in a further nearly two-fold borderline significant (p<0.1) increase in JNK phosphorylation that was almost completely prevented by the n−3 PUFA-containing diet. Thus, the increased inflammatory gene expression in adipose tissue of db/db animals fed high-fat diets was accompanied by JNK activation that tended not to occur in the presence of n−3 PUFA (p<0.1).

Effect of obesity and different high-fat diets on JNK phosphorylation. Obese diabetic db/db mice were fed low-fat (LF) or different high-fat diets (HF/S, HF/3) as detailed in the legend to Fig. 2 and were analysed for JNK phosphorylation in parallel with lean non-diabetic db/+ mice fed LF. a Immunoblots (IBs) of phosphorylated JNK (p-JNK) and JNK protein from gonadal white adipose tissue extracts. A representative blot is given showing one extract from each group of mice with comparable amounts of blotted JNK protein. b Summary of the densitometric quantification. The ratio of chemiluminescence intensities from pJNK and JNK protein (combining p46 and p54) is depicted for lean (db/+) and obese (db/db) mice on different diets (n=4/group). Data are presented related to those from db/+ LF-diet mice. Significance of differences from the db/db LF diet group are indicated as follows: °p<0.1; †p<0.01. Means±SEM
Adiponectin
Adiponectin is known to improve insulin sensitivity [23, 24]. Adiponectin gene (Acrp30) expression was significantly reduced in db/db mice fed HF/S compared with those on LF (Fig. 7). Notably, high-fat diet-induced downregulation of adiponectin gene expression in db/db animals was paralleled by reduced adiponectin serum concentrations (Table 1). Inclusion of n−3 PUFA efficiently reversed the high-fat diet-induced downregulation of adiponectin in db/db mice and restored adiponectin serum concentrations. Thus, inflammatory changes in white adipose tissue of db/db mice that were induced by high-fat diet and prevented by n−3 PUFA were associated with inverse changes in the expression of the insulin-sensitising adipokine adiponectin.

Adiponectin gene (Acrp 30) expression in gonadal adipose tissue of db/db mice. Mice were fed low-fat (LF) or different high-fat diets (HF/S, HF/6, HF/3) and gene expression was analysed by quantitative RT-PCR as detailed in the legend to Fig. 2 (n=4–5/group). Significance of differences from the HF/S group are indicated as follows: †p<0.01; ‡p<0.001. Means±SEM
Discussion
Obesity is associated with chronic subclinical inflammation and macrophage infiltration of white adipose tissue [2, 6, 10, 11]. This inflammatory response appears to underlie metabolic complications of obesity and hence is of primary importance in the treatment of this disease to prevent development of insulin resistance and type 2 diabetes [25]. Here we show that the proinflammatory effects in white adipose tissue of a high-fat diet with a fatty acid composition similar to that of a common western diet (HF/S) is prevented by inclusion of marine n−3 PUFA. In addition, the downregulation of adiponectin induced by high-fat diet was restored by n−3 PUFA. Hence, interference with adipose tissue inflammation represents a novel concept by which n−3 PUFA could exert beneficial metabolic effects in obesity.
The alterations induced by high-fat diet in inflammatory gene expression along with macrophage infiltration found in white adipose tissue of db/db mice were strikingly similar to those found in recent studies on this issue separately in genetic and diet-induced obesity using a dietary fatty acid composition very similar to HF/S [10, 11]. We used a model of genetic obesity (db/db mice) to compare white adipose tissue alterations in animals closely resembling the metabolic syndrome, although excluding leptin effects. Typical inflammatory cytokines such as TNF-α are known to interfere with insulin sensitivity [26–29] and antagonise the release of the adipocyte-derived hormone adiponectin that improves insulin sensitivity. Only few inflammatory changes were observed in db/+ animals under HF/S, presumably due to the short duration of treatment and the lower dietary fat content that provoked only minimal body weight gain [10, 11]. In particular, these changes included induction of MCP-1 (Ccl2) expression that was shown to precede insulin resistance [10]. Notably, induction of MCP-1 was prevented by inclusion of n−3 PUFA also in db/+ animals.
The mechanism underlying macrophage infiltration of adipose tissue in obesity is still unclear, as are the mechanisms underlying the preventive effect of n−3 PUFA. n−3 PUFA interfere with monocyte and macrophage functions [16] and inhibit inflammatory responses in a variety of disorders including rheumatoid arthritis and inflammatory bowel disease, albeit with limited potency [15, 30, 31]. Adipocytes promote diapedesis of monocytes through microvascular endothelial cells, thereby facilitating macrophage accumulation [32], and n−3 PUFA interfere with monocyte chemotaxis, at least in non-diabetic patients [33]. A variety of molecular mechanisms underlying the anti-inflammatory action of n−3 PUFA have been described, mostly in vitro, including altered synthesis of eicosanoids (prostaglandins, leukotrienes), activation or inhibition of nuclear receptors (e.g. peroxisome proliferator-activated receptor γ [PPARγ], liver X receptors) and alterations of membrane lipid rafts [15, 18, 34]. Despite anti-inflammatory effects of PPARγ agonists in obese animals and humans [8, 10] there is no evidence for a selectivity of PPARγ for n−3 PUFA [35]. Hence, the molecular mechanisms by which PUFA inhibit adipose tissue inflammation warrant further investigation.
n−3 PUFA could inhibit adipose tissue inflammation by directly inhibiting macrophage immigration and activation. However, published data on n−3 PUFA effects on macrophage function revealed moderate effects [16] compared with the complete abolishment of high-fat diet-induced macrophage infiltration of adipose tissue shown here. On the other hand, n−3 PUFA could prevent adipose tissue inflammation by predominantly interfering with adipocyte function. It could be speculated that n−3 PUFA ameliorate adipocyte stress and normalise adipocyte metabolism, thereby hindering adipocyte expression of pro-inflammatory cytokines [25], by still unresolved mechanisms. We did not detect alterations in the expression of genes related to inflammatory pathways interfering with insulin sensitivity, such as inducible nitric oxide synthase, JNK (JNK1) and its interacting protein (JIP1; all data not shown). However, JNK activation as reflected by its phosphorylation paralleled inflammatory alterations in adipose tissue in lean mice on LF and obese mice fed different diets. Activation of this central player that links inflammatory pathways with deterioration of insulin sensitivity indicates functional alterations in adipose tissue that could lead to insulin resistance. Accordingly, adiponectin expression was restored by n−3 PUFA in our experiments and this finding could be taken as a marker of normalised adipocyte function. Moreover, expression of some adipocyte metabolic genes, e.g. fatty acid synthase and hormone-sensitive lipase, were downregulated in db/db mice when fed HF/S but not in the presence of n−3 PUFA (ESM Table and data not shown). Even though n−3 PUFA treatment of db/db mice resulted in only limited alterations of systemic metabolism in our study, local improvements in adipocyte function and amelioration of adipocyte stress response could prevent adipose tissue inflammation.
Treatment with n−3 PUFA separated obesity from the development of adipose tissue inflammation in obese mice. This result emphasises that obesity and obesity-associated complications are based on different mechanisms. Thus, reduction of body weight is not the only means to reduce adipose tissue inflammation [36], and hence complications in obese patients. Indeed, our results with n−3 PUFA and data with rosiglitazone [10] suggest that agents affecting macrophage infiltration of adipose tissue could be an effective way to prevent metabolic consequences. This finding is of particular importance in view of the well-known low compliance of patients to dietary calorie restriction.
Dietary PUFA appear to exert preventive effects on the development of insulin resistance and diabetes [12, 37] particularly when compared with saturated fatty acids that promote diabetes development [38, 39]. However, most human intervention studies are not adequately powered (reviewed in [12]) or have limited follow-up periods, since the beneficial effects may take many months to occur [38]. In addition, human studies could be complicated by genetic variations. For example, the Pro12Ala polymorphism of the PPARγ2 gene, the isoform expressed predominantly in adipose tissue, significantly affects the lipid metabolic response by n−3 PUFA, and similar appears to be true for insulin sensitivity [40–42]. Moreover, particularly obese patients with metabolic syndrome and high systemic inflammatory signs at baseline would be predicted to respond to n−3 PUFA treatment according to our study, whereas others may not, as suggested by recent human data [43]. Beneficial effects of n−3 PUFA on insulin resistance are particularly significant in rodents [44, 45] and this could in part be due to the higher dose of experimental n−3 PUFA intake compared with human studies. Blood glucose concentrations were not significantly changed by HF/S in our study, making it difficult to detect significant improvements by n−3 PUFA that appear to only prevent HF/S-induced changes. However, adiponectin, which is positively correlated with insulin sensitivity and negatively associated with obesity [24, 29, 46, 47], was expressed to a significantly lesser extent in adipose tissue of db/db mice fed HF/S but restored by n−3 PUFA (Fig. 7). We have primarily investigated effects of n−3 PUFA on adipose tissue inflammation in mice that were already obese and diabetic at the beginning of the treatment, and saw rapid changes in adipose tissue inflammation. Possible beneficial effects of n−3 PUFA on the onset of insulin resistance in obesity remain to be investigated, e.g. using rodent models of diet-induced obesity. Published human intervention studies with n−3 PUFA were mostly performed with fish oil preparations or meals with oily fish [48]. However, the recent availability of highly enriched re-esterified n−3 PUFA concentrates, such as the one used in our experiments, also warrant adequately powered clinical trials on diabetes prevention with an effective daily dose of n−3 PUFA.
In conclusion, the inflammatory response in white adipose tissue induced by a high-fat diet in obese diabetic animals is prevented by inclusion of n−3 PUFA. Moreover, detrimental effects of a high-fat diet on adiponectin expression are restored in parallel. Thus, specific anti-inflammatory properties of n−3 PUFA could play a role in restoring adipocyte function in obese subjects, and this could extend to improved insulin sensitivity when n−3 PUFA are given in an appropriate dose over a prolonged period of time.
Abbreviations
- CRP:
-
C-reactive protein
- ESM:
-
Electronic supplementary material
- HF/3:
-
HF/6 including n−3 PUFA
- HF/6:
-
high-fat diet rich in n−6 PUFA
- HF/S:
-
high-fat diet rich in saturated/monounsaturated fatty acids
- JNK:
-
c-junN-terminal kinase
- LF:
-
low-fat diet
- MCP:
-
macrophage chemotactic protein
- PPARγ:
-
peroxisome proliferator-activated receptor γ
- PUFA:
-
polyunsaturated fatty acids
References
Dandona P, Aljada A, Bandyopadhyay A (2004) Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 25:4–7
Wellen KE, Hotamisligil GS (2003) Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 112:1785–1788
Pickup JC, Mattock MB, Chusney GD, Burt D (1997) NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 40:1286–1292
Pradhan AD, Ridker PM (2002) Do atherosclerosis and type 2 diabetes share a common inflammatory basis? Eur Heart J 23:831–834
Pai JK, Pischon T, Ma J et al (2004) Inflammatory markers and the risk of coronary heart disease in men and women. N Engl J Med 351:2599–2610
Ghanim H, Aljada A, Hofmeyer D, Syed T, Mohanty P, Dandona P (2004) Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation 110:1564–1571
Ghanim H, Garg R, Aljada A et al (2001) Suppression of nuclear factor-kappaB and stimulation of inhibitor kappaB by troglitazone: evidence for an anti-inflammatory effect and a potential antiatherosclerotic effect in the obese. J Clin Endocrinol Metab 86:1306–1312
Mohanty P, Aljada A, Ghanim H et al (2004) Evidence for a potent antiinflammatory effect of rosiglitazone. J Clin Endocrinol Metab 89:2728–2735
Dandona P, Aljada A, Ghanim H et al (2004) Increased plasma concentration of macrophage migration inhibitory factor (MIF) and MIF mRNA in mononuclear cells in the obese and the suppressive action of metformin. J Clin Endocrinol Metab 89:5043–5047
Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808
Rivellese AA, Lilli S (2003) Quality of dietary fatty acids, insulin sensitivity and type 2 diabetes. Biomed Pharmacother 57:84–87
Sessler AM, Ntambi JM (1998) Polyunsaturated fatty acid regulation of gene expression. J Nutr 128:923–926
Jump DB (2002) The biochemistry of N3-polyunsaturated fatty acids. J Biol Chem 277:8755–8758
Stulnig TM (2003) Immunomodulation by polyunsaturated fatty acids: mechanisms and effects. Int Arch Allergy Immunol 132:310–321
Hughes DA (1998) In vitro and in vivo effects of n−3 polyunsaturated fatty acids on human monocyte function. Proc Nutr Soc 57:521–525
Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP (2003) Summaries of Affymetrix GeneChip probe level data. Nucl Acids Res 31:e15
Stulnig TM, Huber J, Leitinger N et al (2001) Polyunsaturated eicosapentaenoic acid displaces proteins from membrane rafts by altering raft lipid composition. J Biol Chem 276:37335–37340
Juge-Aubry CE, Somm E, Giusti V et al (2003) Adipose tissue is a major source of interleukin-1 receptor antagonist: upregulation in obesity and inflammation. Diabetes 52:1104–1110
Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3:23–35
Inoue T, Plieth D, Venkov CD, Xu C, Neilson EG (2005) Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int 67:2488–2493
Le Hir M, Kaissling B (2005) Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int 68:2400
Berg AH, Combs TP, Du X, Brownlee M, Scherer PE (2001) The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 7:947–953
Maeda N, Shimomura I, Kishida K et al (2002) Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med 8:731–737
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119
Torti FM, Dieckmann B, Beutler B, Cerami A, Ringold GM (1985) A macrophage factor inhibits adipocyte gene expression: an in vitro model of cachexia. Science 229:867–869
Beutler B, Greenwald D, Hulmes JD et al (1985) Identity of tumour necrosis factor and the macrophage-secreted factor cachectin. Nature 316:552–554
Roden M, Price TB, Perseghin G et al (1996) Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 97:2859–2865
Ruan H, Miles PD, Ladd CM et al (2002) Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: implications for insulin resistance. Diabetes 51:3176–3188
Calder PC (2001) Polyunsaturated fatty acids, inflammation, and immunity. Lipids 36:1007–1024
Calder PC, Zurier RB (2001) Polyunsaturated fatty acids and rheumatoid arthritis. Curr Opin Clin Nutr Metab Care 4:115–121
Curat CA, Miranville A, Sengenes C et al (2004) From blood monocytes to adipose tissue-resident macrophages: induction of diapedesis by human mature adipocytes. Diabetes 53:1285–1292
Schmidt EB, Varming K, Pedersen JO et al (1992) Long-term supplementation with n−3 fatty acids. II. Effect on neutrophil and monocyte chemotaxis. Scand J Clin Lab Invest 52:229–236
Stulnig TM, Berger M, Sigmund T, Raederstorff D, Stockinger H, Waldhäusl W (1998) Polyunsaturated fatty acids inhibit T cell signal transduction by modification of detergent-insoluble membrane domains. J Cell Biol 143:637–644
Kliewer SA, Sundseth SS, Jones SA et al (1997) Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci U S A 94:4318–4323
Clément K, Viguerie N, Poitou C et al (2004) Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J 18:1657–1669
Vessby B, Aro A, Skarfors E, Berglund L, Salminen I, Lithell H (1994) The risk to develop NIDDM is related to the fatty acid composition of the serum cholesterol esters. Diabetes 43:1353–1357
Vessby B, Unsitupa M, Hermansen K et al (2001) Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: the KANWU Study. Diabetologia 44:312–319
van Dam RM, Willett WC, Rimm EB, Stampfer MJ, Hu FB (2002) Dietary fat and meat intake in relation to risk of type 2 diabetes in men. Diabetes Care 25:417–424
Luan J, Browne PO, Harding AH et al (2001) Evidence for gene–nutrient interaction at the PPARgamma locus. Diabetes 50:686–689
Lindi V, Schwab U, Louheranta A et al (2003) Impact of the Pro12Ala polymorphism of the PPAR-gamma2 gene on serum triacylglycerol response to n−3 fatty acid supplementation. Mol Genet Metab 79:52–60
Franks PW, Luan J, Browne PO et al (2004) Does peroxisome proliferator-activated receptor gamma genotype (Pro12ala) modify the association of physical activity and dietary fat with fasting insulin level? Metabolism 53:11–16
Browning LM (2003) n−3 Polyunsaturated fatty acids, inflammation and obesity-related disease. Proc Nutr Soc 62:447–453
Storlien LH, Kraegen EW, Chisholm DJ, Ford GL, Bruce DG, Pascoe WS (1987) Fish oil prevents insulin resistance induced by high-fat feeding in rats. Science 237:885–888
Storlien LH, Jenkins AB, Chisholm DJ, Pascoe WS, Khouri S, Kraegen EW (1991) Influence of dietary fat composition on development of insulin resistance in rats. Relationship to muscle triglyceride and omega-3 fatty acids in muscle phospholipid. Diabetes 40:280–289
Nawrocki AR, Scherer PE (2004) The delicate balance between fat and muscle: adipokines in metabolic disease and musculoskeletal inflammation. Curr Opin Pharmacol 4:281–289
Ruan H, Lodish HF (2004) Regulation of insulin sensitivity by adipose tissue-derived hormones and inflammatory cytokines. Curr Opin Lipidol 15:297–302
Fasching P, Ratheiser K, Waldhausl W et al (1991) Metabolic effects of fish-oil supplementation in patients with impaired glucose tolerance. Diabetes 40:583–589
Acknowledgements
We thank S. Molzer, P. Nowotny and E. Nowotny for technical assistance. This work was supported by CeMM (Center of Molecular Medicine), a basic research institute within the Austrian Academy of Sciences to T. M. Stulnig and W. Waldhäusl, the Austrian Science Fund (P16788-B13) and the Joseph Skoda Award of the Austrian Association of Internal Medicine (both to T. M. Stulnig).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material Table
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Todoric, J., Löffler, M., Huber, J. et al. Adipose tissue inflammation induced by high-fat diet in obese diabetic mice is prevented by n−3 polyunsaturated fatty acids. Diabetologia 49, 2109–2119 (2006). https://doi.org/10.1007/s00125-006-0300-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0300-x