PUMILIO, but not RBMX, binding is required for regulation of genomic stability by noncoding RNA NORAD
- University of Texas Southwestern Medical Center, United States
- Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, United States
Abstract
NORAD is a conserved long noncoding RNA (lncRNA) that is required for genome stability in mammals. NORAD acts as a negative regulator of PUMILIO (PUM) proteins in the cytoplasm, and we previously showed that loss of NORAD or PUM hyperactivity results in genome instability and premature aging in mice (Kopp et al., 2019). Recently, however, it was reported that NORAD regulates genome stability through an interaction with the RNA binding protein RBMX in the nucleus. Here, we addressed the contributions of NORAD:PUM and NORAD:RBMX interactions to genome maintenance by this lncRNA in human cells. Extensive RNA FISH and fractionation experiments established that NORAD localizes predominantly to the cytoplasm with or without DNA damage. Moreover, genetic rescue experiments demonstrated that PUM binding is required for maintenance of genomic stability by NORAD whereas binding of RBMX is dispensable for this function. These data provide an important foundation for further mechanistic dissection of the NORAD-PUMILIO axis in genome maintenance.
https://doi.org/10.7554/eLife.48625.001Introduction
Long noncoding RNAs (lncRNAs) have emerged as regulators of diverse biological processes. Among these transcripts, Noncoding RNA activated by DNA damage (NORAD) is particularly noteworthy, due to its unusually abundant expression in mammalian cells and tissues and strong evolutionary conservation across mammalian species. Initial studies of NORAD revealed that this lncRNA is required to maintain genomic stability in mammalian cells (Lee et al., 2016), and provided strong evidence that this function is mediated through the ability of NORAD to bind to and negatively regulate PUMILIO RNA binding proteins (PUM1 and PUM2) in the cytoplasm (Lee et al., 2016; Tichon et al., 2016). PUM proteins bind with high specificity to the eight nucleotide (nt) PUMILIO response element (PRE) (UGUANAUA or UGUANAUN) on target messenger RNAs (mRNAs), triggering their deadenylation, decapping, and eventual degradation (Miller and Olivas, 2011; Quenault et al., 2011). Notably, NORAD contains 18 conserved PREs and has the capacity to bind a large fraction of PUM1/2 within the cell, although it is not yet known whether NORAD limits PUM activity through a simple sequestration model or whether additional mechanisms contribute to PUM inhibition. Regardless, loss of NORAD results in PUM hyperactivity and increased repression of PUM targets that include important regulators of mitosis, DNA repair, and DNA replication, resulting in a dramatic genomic instability phenotype in NORAD-deficient cells and mouse tissues (Kopp et al., 2019; Lee et al., 2016). Accordingly, PUM1/2 overexpression is sufficient to phenocopy loss of NORAD in both human cells and mice (Kopp et al., 2019; Lee et al., 2016), while PUM1/2 loss-of-function suppresses the genomic instability phenotype in NORAD knockout cells (Lee et al., 2016).
Recently, an alternative mechanism for the regulation of genomic stability by NORAD was proposed (Munschauer et al., 2018). Proteomic analysis of the NORAD interactome revealed an interaction with RBMX, an RNA binding protein that contributes to the DNA damage response (Adamson et al., 2012). Subsequent experiments suggested that the NORAD:RBMX interaction facilitates the assembly of a ribonucleoprotein (RNP) complex in the nucleus that includes Topoisomerase I (TOP1) and other proteins that are critical for genome maintenance. Importantly, PUM and RBMX interact with different sites on NORAD and function in distinct subcellular compartments. Thus, while it remains to be determined whether the NORAD:RBMX interaction is necessary for regulation of genomic stability, both PUM and RBMX may play important, non-mutually exclusive roles in the genome maintenance functions of NORAD.
Here, we further examined the mechanism by which NORAD functions to maintain genome stability in human cells and directly tested the requirement for PUM and RBMX interactions in this activity. RNA fluorescent in situ hybridization (FISH) using a panel of probes spanning the entire length of NORAD, as well as cellular fractionation studies, definitively demonstrated that this lncRNA localizes predominantly to the cytoplasm and does not detectably redistribute to the nucleus upon induction of DNA damage. Genetic rescue experiments in NORAD knockdown cells established that PUM binding is essential for maintenance of genomic stability whereas interaction with RBMX is completely dispensable for this function. Further experiments demonstrated that RBMX is not required for induction of NORAD following DNA damage nor its cytoplasmic localization. Together, these studies establish the importance of the NORAD:PUM axis in regulating genomic stability in mammalian cells and provide a foundation for further dissection of the mechanism and physiologic role of this pathway.
Results and discussion
NORAD localizes predominantly to the cytoplasm with or without DNA damage
Initial studies of NORAD reported a predominantly cytoplasmic localization of this lncRNA in human cell lines, based on RNA FISH using pools of fluorescently-labeled oligonucleotide probes and subcellular fractionation experiments (Lee et al., 2016; Tichon et al., 2016). Recently, however, a distinct localization pattern was reported based upon RNA FISH performed using a commercially-available kit with a proprietary set of oligonucleotide probes that hybridize to an unknown segment of NORAD (Munschauer et al., 2018). In these more recent experiments, NORAD was reported to localize equally between the nucleus and cytoplasm and appeared to redistribute almost entirely to the nuclear compartment upon treatment of cells with the DNA damaging agents camptothecin and doxorubicin. Importantly, a single cell line (human colon cancer cell line HCT116) was used in both the previous (Lee et al., 2016) and more recent studies (Munschauer et al., 2018), arguing against a cell-type specific difference in NORAD trafficking as the cause of these discordant results.
We considered the possibility that the disparate observed localization patterns could be due to unrecognized processing of the NORAD transcript, such that different segments of the RNA that are recognized by the different FISH probes accumulate in distinct subcellular compartments. To investigate this possibility, and to reliably establish the localization of the entire NORAD transcript, we generated a panel of 11 in vitro transcribed RNA FISH probes spanning the complete NORAD sequence (Figure 1A) (Mito et al., 2016). One probe, that recognized a segment of NORAD containing an Alu repeat element (probe 7), gave rise to a nonspecific nuclear signal that was present in both NORAD+/+ and NORAD–/– HCT116 cells. The remaining 10 probes produced a highly consistent, predominantly cytoplasmic, punctate localization pattern in wild-type cells that was absent in NORAD–/– cells (Figure 1A–B). These results were confirmed using subcellular fractionation followed by quantitative reverse transcriptase-PCR (qRT-PCR) using primers located at the 3′ or 5′ ends of NORAD, which revealed that 80–90% of the transcript is localized to the cytoplasmic compartment (Figure 1C).
Figure 1
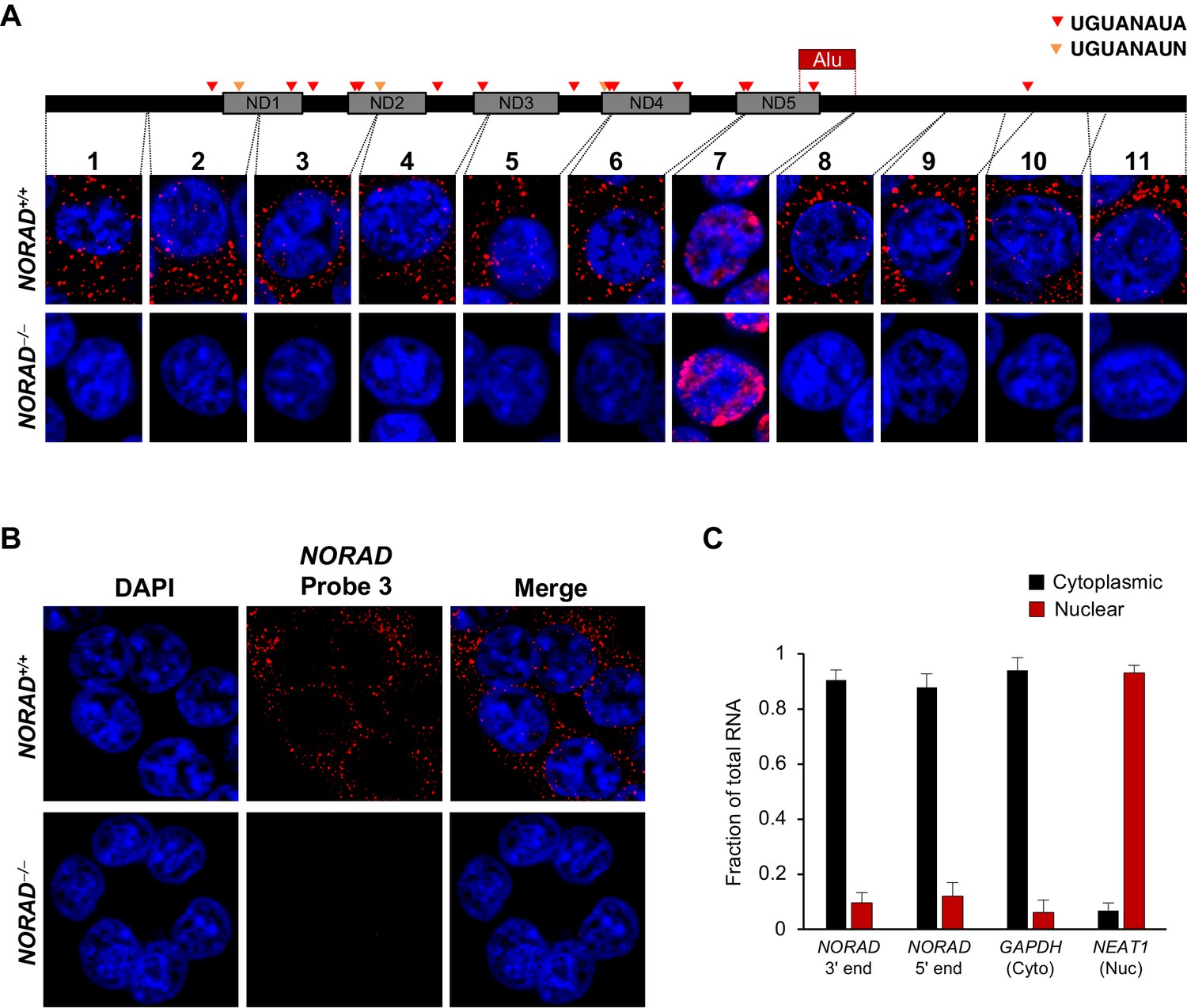
NORAD localizes predominantly to the cytoplasm.
(A) RNA FISH in HCT116 cells using a panel of 11 probes tiling the entire NORAD transcript reveals a predominantly cytoplasmic signal that is absent in NORAD–/– cells with all probes except probe 7, which produces a nonspecific signal likely due to the presence of an Alu repeat element. NORAD FISH signal in red, DAPI counterstain in blue. Locations of PREs indicated by arrowheads. ND1-ND5 represent repetitive NORAD domains, as previously described (Lee et al., 2016). (B) RNA FISH image using probe 3 showing a wider field of cells. (C) Subcellular fractionation followed by qRT-PCR in HCT116 cells using primers located at the 3′ or 5′ end of NORAD, in GAPDH (cytoplasmic control), or in NEAT1 (nuclear control). n = 3 biological replicates each with three technical replicates.
Next, we examined NORAD localization following treatment of cells with agents that induce DNA damage (Figure 2—figure supplement 1A–B). RNA FISH using the panel of probes spanning NORAD revealed clear cytoplasmic localization after treatment with doxorubicin or camptothecin, without a significant increase in nuclear signal compared to untreated cells (Figure 2A–B). Consistent with the previously reported induction of NORAD after DNA damage (Lee et al., 2016), a clear increase in cytoplasmic NORAD signal was apparent in treated cells (Figure 2B). These findings were further corroborated by subcellular fractionation experiments following treatment with DNA damaging agents, which confirmed that NORAD remained predominantly in the cytoplasmic compartment at all time points (Figure 2C and Figure 2—figure supplement 1C). Interestingly, we observed a modest increase in nuclear NORAD levels that peaked after 12 hr of camptothecin or doxorubicin treatment. We speculated that this might represent a burst of NORAD transcription in response to accumulating DNA damage. To test this hypothesis, we co-treated cells with DNA damaging agents and the transcriptional inhibitor actinomycin D. As expected, this completely abrogated any detectable increase in nuclear NORAD abundance in treated cells (Figure 2D and Figure 2—figure supplement 1D).
Figure 2 with 1 supplement see all

NORAD remains predominantly in the cytoplasm after treatment with DNA damaging agents.
(A) RNA FISH in HCT116 cells using the indicated NORAD probes following a 12 hr treatment with doxorubicin or camptothecin. (B) NORAD RNA FISH (probe 5) after the indicated drug treatments. Images captured with identical microscope settings. (C–D) Subcellular fractionation followed by qRT-PCR in HCT116 cells after treatment with camptothecin (C) or camptothecin plus actinomycin D (D) for the indicated number of hours. n = 3 biological replicates each with three technical replicates. ns, not significant; *p<0.05; **p<0.01; ***p<0.001; one-tailed t-test comparing each sample to the 0 hr time-point.
These comprehensive RNA FISH and subcellular fractionation experiments provide definitive evidence that NORAD is a predominantly cytoplasmic RNA in HCT116 cells and does not detectably redistribute to the nucleus upon DNA damage. These findings are consistent with the reported localization of NORAD in other human and mouse cell lines (Kopp et al., 2019; Lee et al., 2016; Tichon et al., 2016). We speculate that the disparate localization pattern observed using a commercially-available RNA FISH probe set (Munschauer et al., 2018) most likely represented a non-specific signal.
PUM1, PUM2, and RBMX are components of the NORAD interactome
Previous crosslinking-immunoprecipitation coupled with high throughput sequencing (CLIP-seq) studies demonstrated that NORAD is the preferred binding partner of PUM2 in both human cells (Lee et al., 2016) and mouse brain (Kopp et al., 2019). In light of these findings, it was surprising that PUM1/2 were not reported among the most enriched NORAD-bound proteins in the recent RNA antisense purification with quantitative mass spectrometry (RAP-MS) experiments performed in HCT116 cells that identified the NORAD:RBMX interaction (Munschauer et al., 2018). Since these RAP-MS experiments utilized pulse labeling with 4-thiouridine to crosslink NORAD to protein interactors, a bias towards detection of proteins that bind to newly synthesized NORAD would be expected, potentially explaining the enrichment of nuclear interactors observed. Nevertheless, we reanalyzed the published RAP-MS dataset to determine whether PUM1 or PUM2 were enriched in NORAD pull-downs compared to control RMRP pull-downs. Peptides were identified and scored using a combined algorithm that employed three search engines (Sequest HT, Mascot, and MS Amanda). Isoforms of PUM1 and PUM2, along with RBMX, were indeed identified as significantly-enriched interacting partners of NORAD compared to RMRP (Figure 3—figure supplement 1). Notably, PUM1 was more enriched than PUM2 in our analysis, which may reflect its higher abundance in HCT116 cells (Lee et al., 2016). These results confirmed that both PUM proteins and RBMX are identified by RAP-MS as significant NORAD-interacting partners.
Binding of PUMILIO, but not RBMX, to NORAD is necessary for genome stability
Genetic epistasis experiments have strongly implicated a role for PUM1/2 in the regulation of genomic stability by NORAD, with PUM2 overexpression phenocopying, and PUM1/2 knockdown suppressing, the effects of NORAD deletion (Kopp et al., 2019; Lee et al., 2016). Nevertheless, it has not yet been directly tested whether binding of PUM1/2 is required for NORAD function. Similarly, a requirement for RBMX binding in genome maintenance by NORAD has not been established. Therefore, to directly interrogate the importance of PUM and RBMX binding for NORAD function, we generated a series of mutant NORAD constructs lacking either PUM or RBMX binding sites (Figure 3A). For each of the 18 PREs within NORAD, the UGU sequence, which is essential for PUM binding (Bohn et al., 2018; Van Etten et al., 2012), was mutated to ACA to abolish PUM interaction (PREmut construct). To remove the RBMX binding site, the first 898 nucleotides (nt) of NORAD were deleted (Munschauer et al., 2018) (5′ deletion construct). We also sought to determine whether PUM or RBMX binding regions of NORAD could represent minimal functional domains that are sufficient for maintaining genomic stability. To this end, we generated a fragment comprising NORAD domain 4 (ND4), which represents the most conserved of five repeated segments within this lncRNA termed NORAD domains (Lee et al., 2016) and contains 4 PREs (nt 2494–3156). An RBMX binding site fragment, representing the 5′ end of NORAD (nt 33–898), which also harbors one PRE, was also generated (5′ fragment).
Figure 3 with 1 supplement see all
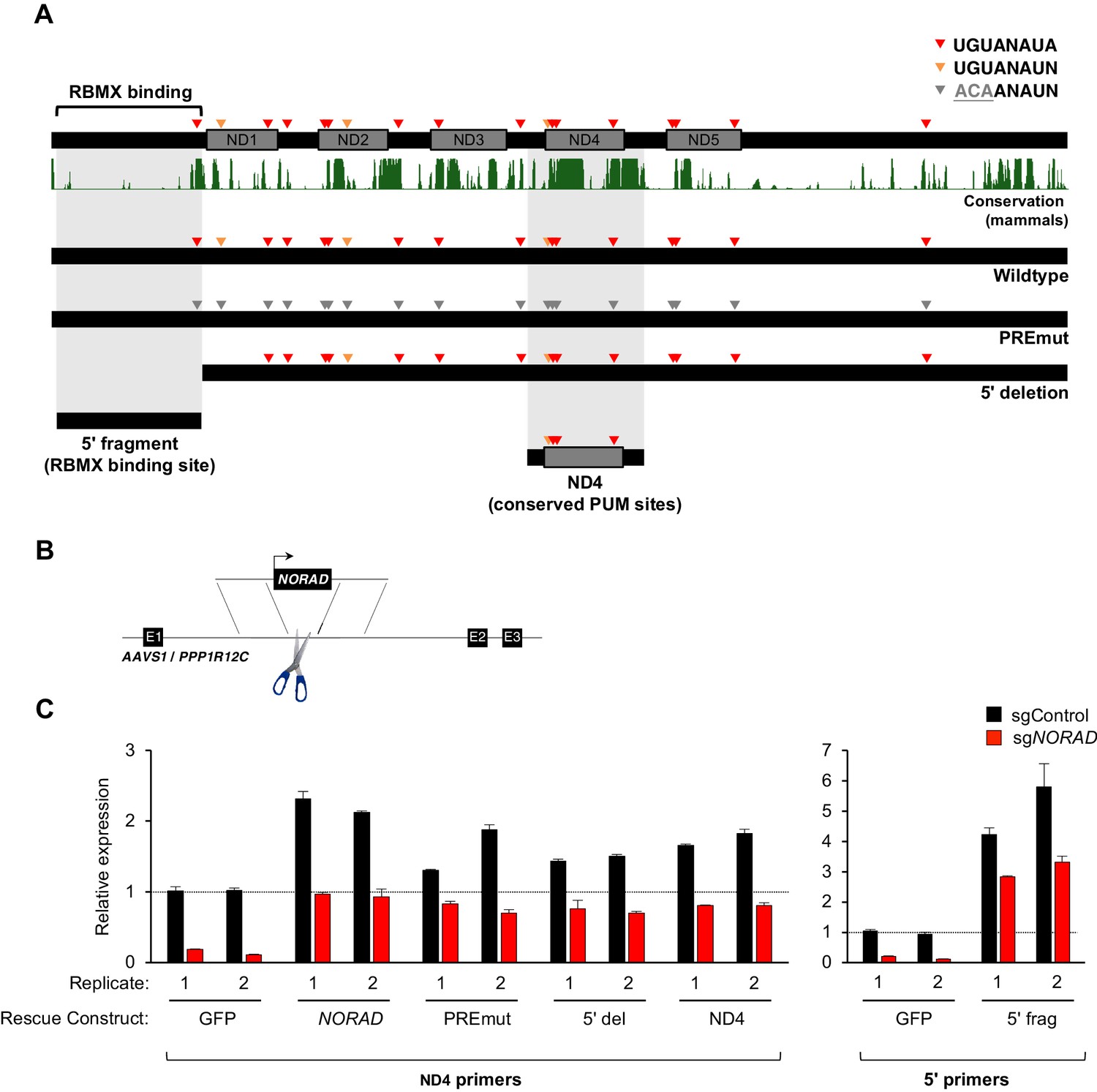
Generation and stable expression of NORAD constructs.
(A) Schematic depicting wild-type or mutant NORAD constructs. NORAD sequence conservation in mammals (UCSC Genome Browser Hg38 PhastCons track) highlights the strong conservation of the region of NORAD harboring PREs (arrowheads). PREmut contains 18 UGU to ACA mutations in PREs (gray arrowheads); 5′ deletion (5′ del) lacks the RBMX binding site (nt 1–898) (Munschauer et al., 2018); 5′ fragment (5′ frag) spans the RBMX binding site (nt 33–898); ND4 construct represents the most conserved segment of NORAD (nt 2494–3156). (B) Schematic depicting insertion of constructs into the AAVS1/PPP1R12C locus using TALENs. (C) qRT-PCR analysis of expression of each NORAD construct in HCT116 CRISPRi cells after infection with control or endogenous NORAD-targeting sgRNAs. Expression was normalized to endogenous NORAD level, represented by expression in AAVS1-GFP cells infected with sgControl (replicate 1 samples normalized to sgControl AAVS1-GFP replicate 1; replicate 2 samples normalized to sgControl AAVS1-GFP replicate 2). The data in the left graph were generated with a primer pair in ND4 that does not amplify the 5′ fragment, while the right graph used primers at the NORAD 5′ end. Replicates represent two independently-derived AAVS1 knock-in and sgRNA-infected cell lines. Values normalized to GAPDH expression. n = 3 technical replicates per sample.
Wild-type or mutant NORAD constructs, as well as a control GFP sequence, under the control of a constitutive promoter were introduced into the AAVS1/PPP1R12C locus of HCT116 cells using a previously published TALEN pair (Sanjana et al., 2012) (Figure 3B). Endogenous NORAD was then depleted using CRISPR interference (CRISPRi) with a single-guide RNA (sgRNA) targeting the endogenous NORAD promoter. As expected, in cells infected with a non-targeting control sgRNA (sgControl), increased total levels of NORAD were observed upon expression of NORAD rescue constructs in trans (~1.5–6 fold overexpression, depending on the construct) (Figure 3C). Upon silencing of the endogenous NORAD locus, however, near physiologic expression levels for all constructs were achieved, with the exception of the 5′ fragment which exhibited ~3 fold overexpression, perhaps indicating increased stability of this transcript segment when expressed in isolation.
We next used UV crosslinking and RNA immunoprecipitation (RIP) to assess binding of wild-type and mutant NORAD transcripts to endogenous PUM1, PUM2, and RBMX. Pull-downs of each of these proteins resulted in the expected enrichment of wild-type NORAD, but not GAPDH, relative to immunoprecipitation with control IgG (Figure 4A and Figure 4—figure supplement 1). The PREmut transcript as well as the 5′ fragment did not bind to PUM1/2 but were recovered in RBMX RIP samples as efficiently as wild-type NORAD. In contrast, the 5′ deletion construct and ND4 fragment retained PUM1 and PUM2 binding activity, but interaction with RBMX was not detectable above background. Furthermore, RNA FISH documented a predominantly cytoplasmic localization pattern of each construct (Figure 4B).
Figure 4 with 1 supplement see all
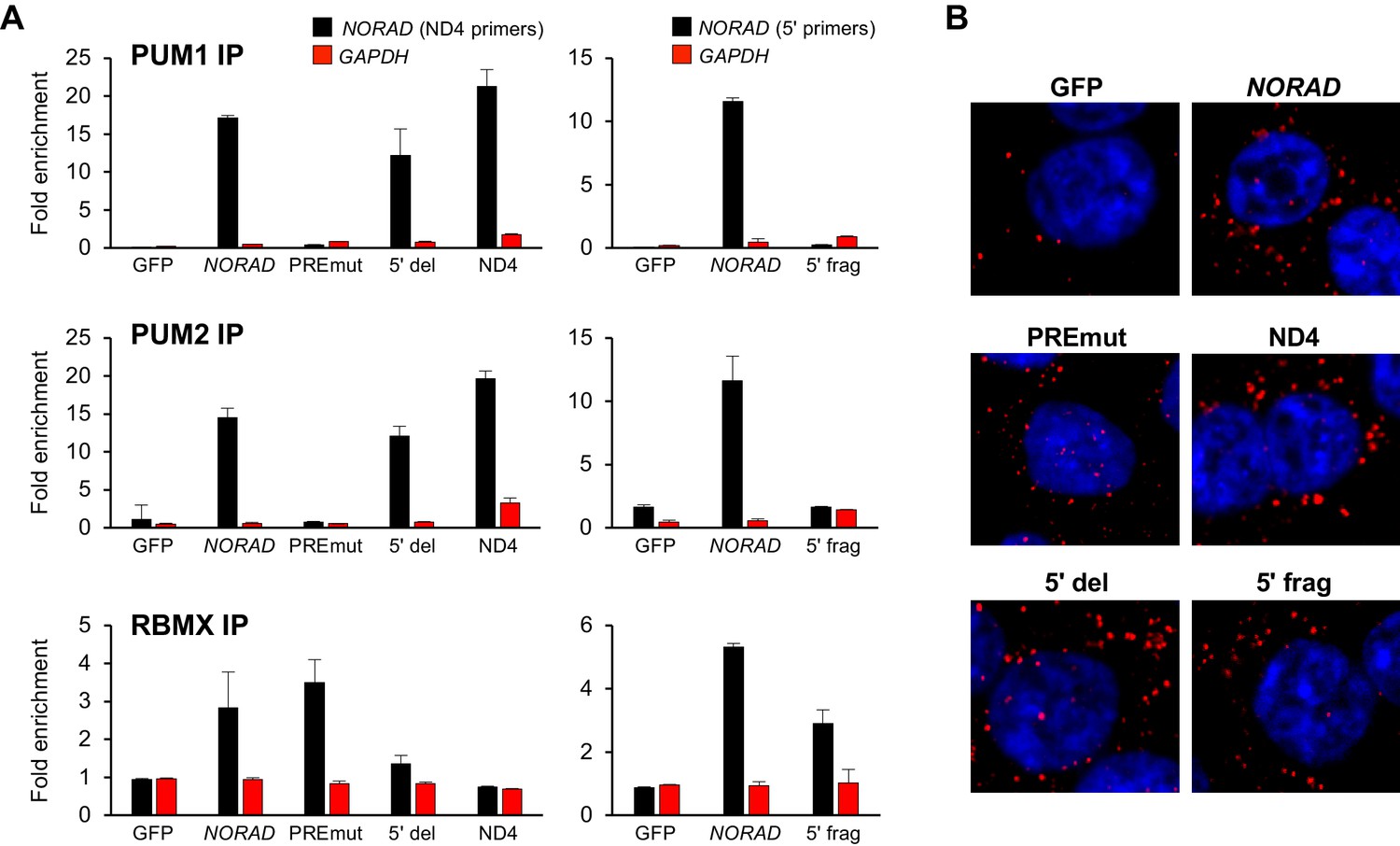
RNA immunoprecipitation and localization of NORAD constructs.
(A) UV crosslinking and RNA immunoprecipitation (RIP) was used to assess PUM1, PUM2, and RBMX interactions with GFP mRNA or the indicated NORAD constructs. After knock-in of the indicated constructs to the AAVS1 locus in HCT116 CRISPRi cells, endogenous NORAD was silenced with a lentivirally-expressed sgRNA. qRT-PCR was used to assess NORAD or GAPDH recovery in each RIP sample, expressed as fold-enrichment over pull-down with IgG. The data in the left graphs were generated with a primer pair in ND4 that does not amplify the 5′ fragment, while the right graphs used primers at the NORAD 5′ end. n = 2 biological replicates, each measured with three technical replicates. (B) Representative RNA FISH images of wild-type or mutant NORAD transcripts expressed from the AAVS1 locus in HCT116 CRISPRi cells after knockdown of endogenous NORAD. Probe 10 was used for full-length NORAD, PREmut, and 5′ del constructs; probe 1 was used for 5′ frag; and probe 6 was used for ND4.
Genome stability was first assessed in cell populations expressing wild-type or mutant NORAD constructs by quantifying the number of aneuploid cells in each population using DNA FISH for marker chromosomes 7 and 20, as described previously (Lee et al., 2016) (Figure 5A–B). Importantly, no significant increase in aneuploidy was observed in any of the NORAD rescue populations after infection with non-target sgRNA (sgControl), indicating that overexpression of the NORAD constructs in trans did not trigger genomic instability. Furthermore, as expected, knockdown of endogenous NORAD in GFP-control cells resulted in a significant accumulation of aneuploid cells. The frequency of aneuploidy observed under these conditions was very similar to that observed previously in NORAD–/– HCT116 cells (Lee et al., 2016). Expression of wild-type NORAD in trans was sufficient to fully suppress the accumulation of aneuploid cells after silencing endogenous NORAD. Cells expressing the PREmut transcript, however, exhibited high levels of aneuploidy, demonstrating that loss of PUM binding abrogated the ability of NORAD to maintain genomic stability. Moreover, the 5′ deletion construct that lacks the RBMX binding site, but preserves the PUM interaction, was fully functional in this assay and completely prevented the accumulation of aneuploid cells. Thus, RBMX binding to NORAD is dispensable for genome maintenance. Remarkably, we observed a strong suppression of aneuploidy in cells expressing the minimal ND4 fragment, further supporting the centrality of the PUM interaction for NORAD function, while the 5′ fragment of NORAD had no activity in this assay.
Figure 5
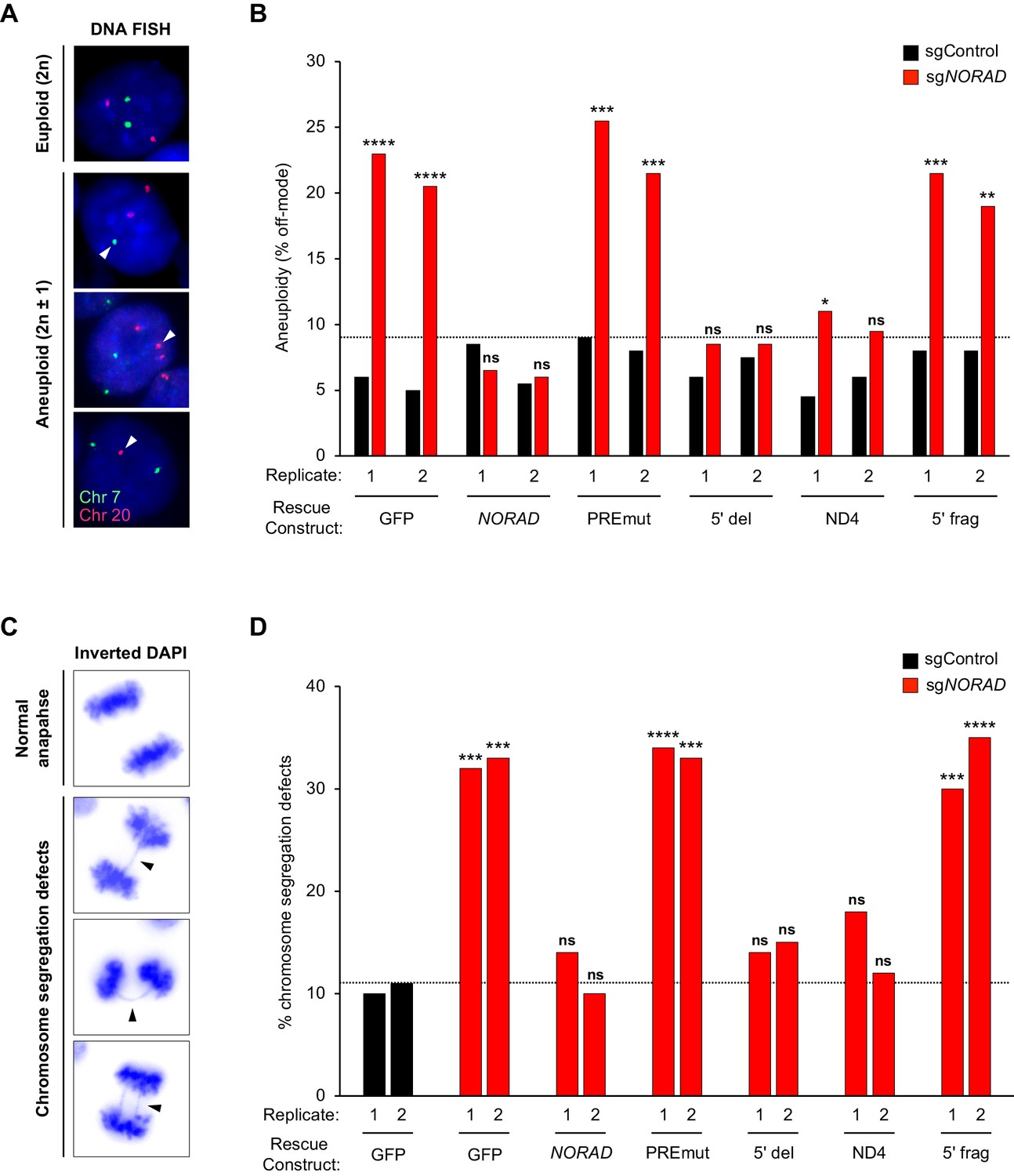
PUMILIO, but not RBMX, binding to NORAD is necessary for genome stability.
(A) Representative DNA FISH images for chromosome 7 (green) and 20 (red) showing examples of cells with modal (2 n) and non-modal (2n ± 1) chromosome numbers. Arrowheads indicate chromosome gain or loss. (B) HCT116 CRISPRi cells stably expressing the indicated AAVS1 knock-in construct were infected with lentivirus expressing control or endogenous NORAD-targeting sgRNA. Aneuploidy was assayed 18–21 days later using DNA FISH for chromosome 7 and 20, and the frequency of interphase cells exhibiting a non-modal (2 n) chromosome number was scored. Replicates represent two independently-derived AAVS1 knock-in and sgRNA-infected cell lines. 200 nuclei were scored per sample. The dotted line denotes the highest level of background aneuploidy observed in sgControl-infected cells. ns, not significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001, chi-square test comparing sgNORAD to sgControl for each replicate. (C) Representative images of anaphase cells with normal or abnormal (arrowheads) chromosome segregation in DAPI-stained HCT116 CRISPRi cells. (D) The frequency of mitotic cells exhibiting chromosome segregation defects was determined in both biological replicates of each cell population (100 anaphase cells assayed per sample). The dotted line denotes the highest percentage of chromosome segregation defects observed in sgControl-infected cells. ns, not significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001, chi-square test comparing all sgNORAD replicate 1 samples to sgControl GFP replicate 1 and all sgNORAD replicate 2 samples to sgControl GFP replicate 2.
To further assess the role of RBMX and PUM binding in genome maintenance by NORAD, we quantified the frequency of chromosomal segregation defects in HCT116 cell populations expressing the various NORAD rescue constructs. We have previously demonstrated that NORAD knockout cells exhibit a significant increase in mitotic errors (Lee et al., 2016) and the same phenotypic assay was later used by Munschauer et al. to confirm genomic instability in NORAD and RBMX knockdown cells (Munschauer et al., 2018). Examination of DAPI-stained anaphase nuclei revealed the expected increase in chromosomal segregation defects in GFP-expressing control cells following NORAD knockdown (Figure 5C–D). Consistent with our analyses of aneuploidy using DNA FISH, expression of full-length NORAD, or the 5′ deletion construct lacking the RBMX binding site, reversed this phenotype and suppressed the increase in mitotic errors. We further documented that mutation of the PUM binding sites (PREmut) abolished NORAD function in this assay. Finally, we found that the ND4 segment containing several highly conserved PUM binding sites, but not the 5′ fragment encompassing the RBMX binding site, exhibited significant rescue activity in this assay. Overall, these data provide compelling evidence that PUM, but not RBMX, binding to NORAD is necessary for the maintenance of genomic stability by this lncRNA.
RBMX is not required for NORAD expression or localization
Although RBMX is not required for maintenance of genomic stability by NORAD, we were able to confirm binding of this protein to the 5′ end of NORAD, as reported (Munschauer et al., 2018) (Figure 3 and Figure 4A). Thus, we investigated whether RBMX functions as an upstream regulator of NORAD expression or localization. Depletion of RBMX using CRISPRi resulted in an increase in NORAD expression that was further augmented by doxorubicin treatment (Figure 6A). We speculate that this increase in NORAD levels may be an indirect effect of the previously reported accumulation of DNA damage caused by RBMX loss of function (Adamson et al., 2012). Additionally, RBMX knockdown did not alter the predominantly cytoplasmic localization of NORAD, as indicated by subcellular fractionation experiments (Figure 6B). We conclude that RBMX is not an essential co-factor for NORAD expression or localization.
Figure 6
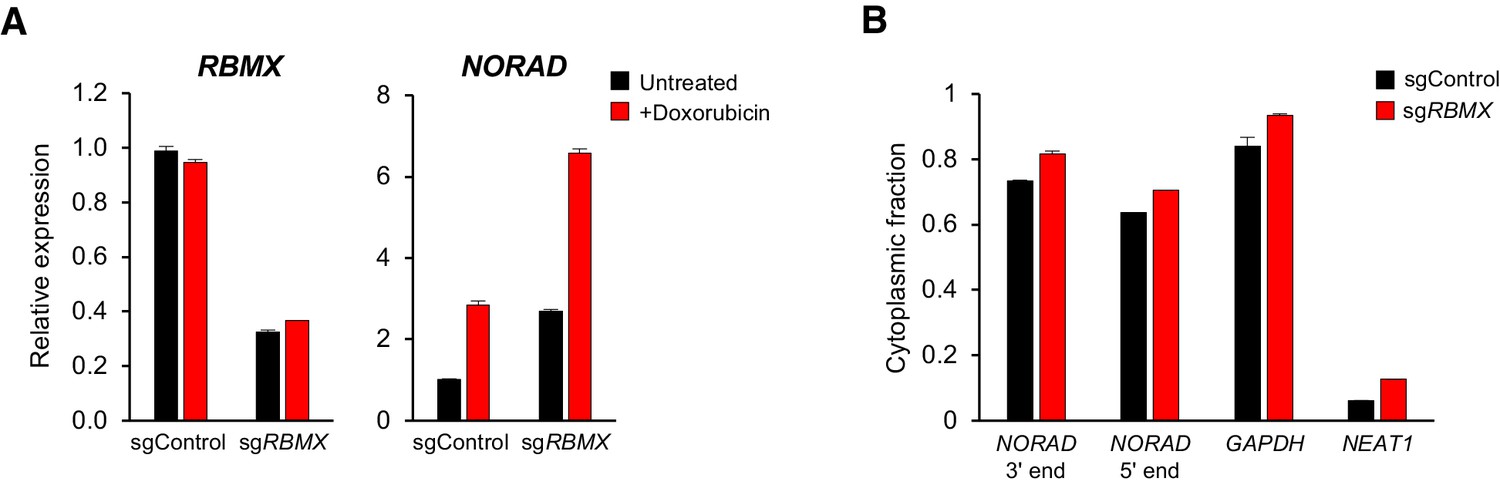
RBMX is not required for NORAD expression or localization.
(A) qRT-PCR analysis of RBMX and NORAD transcript levels in HCT116 CRISPRi cells after introduction of the indicated lentivirally-expressed sgRNA with or without doxorubicin treatment (1 μM for 24 hr). Quantification relative to GAPDH. n = 3 technical replicates. (B) Subcellular fractionation and qRT-PCR of NORAD, GAPDH (cytoplasmic control), or NEAT1 (nuclear control) following introduction of control or RBMX-targeting sgRNAs. n = 3 biological replicates each with three technical replicates.
In sum, these results establish the essential role of PUM binding for the regulation of genomic stability by NORAD. A systematic examination of the subcellular localization of this lncRNA unequivocally established its predominantly cytoplasmic localization under baseline conditions as well as after treatment with DNA-damaging agents. Moreover, genetic complementation experiments demonstrated that PUM binding is essential, whereas RBMX interaction is dispensable, for the genome maintenance function of NORAD. These results further define and clarify the NORAD molecular mechanism of action and direct future investigation towards elucidation of the regulation and physiologic roles of the NORAD:PUM axis.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Homo sapiens) | NORAD (LINC00657) | NA | Ensembl:ENSG00000260032 | |
| Cell line (Homo sapiens) | HCT116 | ATCC | CCL-247, RRID:CVCL_0291 | |
| Cell line (Homo sapiens) | NORAD-/- HCT116 | Lee et al., 2016 | ||
| Cell line (Homo sapiens) | HCT116 CRISPRi | this paper; see Materials and methods section Cell culture and generation of HCT116 CRISPRi cell line | ||
| Antibody | Anti-PUM1 (polyclonal rabbit) | Santa Cruz | sc-135049, RRID:AB_10610604 | RIP |
| Antibody | Anti-PUM2 (polyclonal goat) | Santa Cruz | sc-31535, RRID:AB_654939 | RIP |
| Antibody | Anti-RBMX (monoclonal rabbit) | Cell Signaling | #14794, RRID:AB_2798614 | RIP, WB (1:1000) |
| Antibody | Anti-PUM2 (monoclonal rabbit) | Abcam | ab92390, RRID:AB_10563318 | WB (1:1000) |
| Antibody | Anti-PUM1 (monoclonal rabbit) | Abcam | ab92545, RRID:AB_10563695 | WB (1:1000) |
| Antibody | Anti-GAPDH (monoclonal rabbit) | Cell Signaling | #2118, RRID:AB_561053 | WB (1:5000) |
| Antibody | Anti-phospho-histone H2A.X (Ser139) | Cell Signaling | #2577, RRID:AB_2118010 | IF (1:500), WB (1:1000) |
| Antibody | IRDye 800CW anti-rabbit (donkey) | Licor | 925–32213, RRID:AB_2715510 | WB (1:10000) |
| Antibody | Anti-Digoxigenin (monoclonal mouse) | Roche | 11333062910, RRID:AB_514495 | RNA FISH |
| Antibody | Anti-Mouse IgG, Cy3 (polyclonal goat) | EMD Millipore | AP124C | RNA FISH |
| Recombinant DNA reagent | AAVS1/PPP1R12C targeting vector | Addgene | #22072, RRID:Addgene_22072 | |
| Recombinant DNA reagent | hAAVS1 1L TALEN; hAAVS1 1R TALEN | Addgene | #35431, RRID:Addgene_35431; #35432, RRID:Addgene_35432 | |
| Recombinant DNA reagent | pU6-sgRNA EF1a-PuroR-T2A-BFP | Addgene | #60955, RRID:Addgene_60955 | CRISPRi-mediated knockdown |
| Sequence-based reagent | Chromosome enumeration Probe Chr. 7 (green) | Empire Genomics | CHR07-10-GR | DNA FISH |
| Sequence-based reagent | Chromosome enumeration Probe Chr. 20 (red) | Empire Genomics | CHR20-10-RE | DNA FISH |
| Software, algorithm | Prism 7 | GraphPad Software | ||
| Software, algorithm | Proteome Discoverer | Thermo Fisher | ||
| Software, algorithm | Limma package for R | Smyth, 2004 |
Cell culture and generation of HCT116 CRISPRi cell line
Request a detailed protocolHCT116 cells (ATCC) were cultured in McCoy’s 5a media (Thermo Fisher Scientific) supplemented with 10% FBS (Gibco, Sigma-Aldrich) and 1X AA (Gibco). The cell line was authenticated by ATCC using short tandem repeat (STR) analysis in November 2017. All cell lines were confirmed to be free of mycoplasma contamination. HCT116 NORAD–/– cells were generated previously (Lee et al., 2016).
To generate the HCT116 CRISPRi cell line, lentivirus expressing dCas9/BFP/KRAB was produced by first seeding 6 × 105 HEK293T cells per well in a six-well plate. The following day, cells were transfected with 1.4 μg of pHR-SFFV-dCas9-BFP-KRAB (Addgene plasmid #46911), 0.84 μg of psPAX2 (Addgene plasmid #12260), 0.56 μg of pMD2.G (Addgene plasmid #12259), 8.4 μl of FuGENE HD (Promega), and 165 μl Opti-MEM (Thermo Fisher) according to the manufacturer’s instructions. Medium was changed the next day. Two days after transfection, medium was collected and passed through a 0.45 μm SFCA sterile filter. Recipient HCT116 cells were transduced overnight using medium supplemented with 8 μg/ml polybrene (EMD Millipore). Cells expressing BFP were enriched by FACS and single-cell clonal lines were derived.
RNA Fluorescent in situ hybridization (RNA FISH)
Request a detailed protocolRNA FISH was performed as described previously (Mito et al., 2016) with the following modifications. DIG-labeled RNA probes for human NORAD were synthesized by in vitro transcription using a DIG-labeling mix (Roche). Primers used for amplification of the DNA template for each probe are provided in Supplementary file 1. 2 × 105 cells were grown on poly-L-lysine coated coverslips for 24 to 36 hr. For RNA FISH experiments with DNA damage treatment, cells were grown for 24 hr and treated with either 1 μM doxorubicin or 200 nM camptothecin for an additional 12 hr. Samples were rinsed twice in phosphate buffered saline (PBS), fixed in 4% paraformaldehyde for 10 min, washed again in PBS, and permeabilized in 0.5% Triton X-100 for 10 min. Samples were then washed twice with PBS and rinsed with DEPC-treated water prior to incubation in prehybridization buffer (50% formamide, 2X SSC, 1X Denhardt’s solution, 10 mM EDTA, 0.1 mg/ml yeast tRNA, 0.01% Tween-20) for 1 hr. 10 ng/μl DIG-labeled RNA probe was diluted in hybridization buffer (prehybridization buffer with 5% dextran sulfate) and used for hybridization at 55°C for 16 to 20 hr. Following hybridization, samples were washed, treated with RNase A, and blocked using Blocking Reagent (Roche). DIG-labeled probes were detected using mouse monoclonal anti-DIG primary antibody (Roche; 1:100 dilution) and a Cy3-labeled goat anti-mouse IgG secondary antibody (Roche; 1:100 dilution). Immunofluorescence and western blot analysis of the DNA damage marker γ-H2AX was performed using anti-γ-H2AX (Ser139) antibody (2577, Cell Signaling). Samples were mounted using SlowFade Diamond Antifade with DAPI mounting media (Invitrogen) and imaging was performed using a Zeiss LSM700 confocal microscope. ImageJ was used for further image analysis.
Subcellular fractionation
Request a detailed protocolCells were seeded in triplicate and 1 × 106 cells were collected for subcellular fractionation, which was performed as previously described (Kopp et al., 2019; Lee et al., 2016). Briefly, cell pellets were lysed in RLN1 buffer (50 mM Tris-HCl pH 8.0, 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, RNAse inhibitor) on ice for 5 min and centrifuged at 500g × 2 min. The supernatant containing the cytoplasmic fraction was separated from the pelleted nuclear fraction. RNA was then isolated from both fractions using the Qiagen RNeasy kit and equal cell equivalents of nuclear and cytoplasmic RNA were used in subsequent qRT-PCR reactions. All samples were tested for NORAD as well as NEAT1 (nuclear control) and GAPDH (cytoplasmic control). The sum of the nuclear and cytoplasmic expression level of each transcript was set to 100%, and the percentage of each transcript localized to each compartment was determined. NEAT1 and GAPDH, respectively, showed the expected nuclear and cytoplasmic localization in each experiment, confirming successful fractionation.
Reanalysis of NORAD RAP-MS data
Request a detailed protocolThe raw mass spectra files from iTRAQ-labeled NORAD and RMRP RAP-MS experiments (Munschauer et al., 2018) were downloaded from MassIVE (https://massive.ucsd.edu) using the identifier: MSV000082561. Peptide identification and quantification was performed using Proteome Discoverer (Thermo Fisher) with three search engines combined (Sequest HT, Mascot, and MS Amanda). MS/MS spectra were searched against the human Uniprot database. Search parameters included: trypsin enzyme specificity with a maximum of 2 missed cleavages tolerated, False Discovery Rate (FDR) set to 0.01 at both peptide and protein level, ±10 ppm for precursor mass tolerance with a shorter window for fragment mass tolerance for the first search, and carbamidomethylation of cysteine modification and iTRAQ labels on N-termini and lysine residues as fixed modifications and oxidation of methionine and N-termini acetylation as variable modifications. All peptide and protein identifications had scores surpassing the combined search engine significance threshold for identification. Protein abundance was calculated as the intensity given from precursor quantification and was then normalized to the total peptide amount. To correct for total abundance differences between samples, protein and peptide abundance values in each sample were corrected by a constant factor such that the end total abundance was equivalent across all samples. Fold change was calculated as the log2 difference of average scaled protein abundance in NORAD samples and RMRP sample. For statistical analysis, we used the limma package (Smyth, 2004) in R (https://www.r-project.org/) to calculate the adjusted p-value using a moderated t-test and Benjamini Hochberg method to control the FDR.
RNA isolation and quantitative reverse transcription PCR (qRT-PCR)
Request a detailed protocolRNA was isolated from cells using the RNeasy Mini Kit (Qiagen), or, for RIP experiments, Trizol (Invitrogen), and treated with RNase-free DNase (Qiagen). RNA was reverse transcribed with PrimeScript RT-PCR mix (Clonetech), and Power SYBR Green PCR Master Mix (Applied Biosystems) was used for qPCR. Biological replicates represent independently grown and processed cells. Technical replicates represent multiple measurements of the same biological sample. Primer sequences are provided in Supplementary file 1.
Generation and AAVS1 knock-in of NORAD constructs
Request a detailed protocolFull-length wild-type NORAD was amplified from a modified pcDNA3.1 vector containing the NORAD cDNA (Lee et al., 2016), along with an additional 115 base pairs downstream of the endogenous NORAD polyadenylation site. The PRE-mutant (PREmut) construct containing 18 PRE mutations (TGT to ACA) was synthesized by GENEWIZ. The 5′ deletion construct (∆1–898), 5′ fragment (nt 33–898), and ND4 were amplified from the full-length NORAD construct using primers provided in Supplementary file 1. Constructs were cloned into a AAVS1/PPP1R12C targeting vector (AAVS1 hPGK-PuroR-pA donor, Addgene plasmid #22072) modified by replacing the puromycin resistance gene with a hygromycin resistance gene and digested with KpnI and MfeI to remove the GFP cassette. These vectors, as well as a control GFP vector, were then inserted into the AAVS1 locus of HCT116 CRISPRi cells using a previously described TALEN pair targeting the AAVS1/PPP1R12C locus (Sanjana et al., 2012) (hAAVS1 1L TALEN, Addgene plasmid #35431; hAAVS1 1R TALEN, Addgene plasmid #35432). Transfection of these plasmids was performed using FugeneHD (Promega) at a 1:1:8 ratio of L-TALEN:R-TALEN:Donor as previously described (Lee et al., 2016). 48 hr after transfection, cells were selected with hygromycin (500 μg/ml) for at least 10 days prior to introducing sgRNAs for CRISPRi-mediated knockdown.
CRISPRi-mediated knockdown
Request a detailed protocolSingle guide RNAs (sgRNAs) targeting a sequence upstream of the endogenous NORAD transcription start site or targeting RBMX were cloned into a pU6-sgRNA EF1a-PuroR-T2A-BFP vector (Addgene plasmid #60955). sgRNA sequences are provided in Supplementary file 1. pU6-sgRNA vectors were then packaged into lentivirus by transfecting HEK293T cells using a 4:2:1 ratio of pU6-sgRNA:psPAX2:pMD2.G with FuGENE HD. Medium was changed the next day. Media containing the virus was collected and filtered at 48 hr and 72 hr after transfection. Virus was then diluted 1:3 with fresh media and used to transduce HCT116 CRISPRi cell lines overnight in a final polybrene concentration of 8 μg/ml. 48 hr after transduction, selection with 1 μg/ml puromycin was initiated. For HCT116 CRISPRi cells with AAVS1/NORAD construct insertion and sgRNA expression, cells were grown in 1 μg/ml puromycin and 500 μg/ml hygromycin.
UV crosslinking and RNA immunoprecipitation (RIP)
Request a detailed protocolPUM1, PUM2, and RBMX RIP experiments were performed in HCT116 CRISPRi cells stably expressing AAVS1/NORAD constructs and depleted of endogenous NORAD with CRISPRi as described above. 20 × 106 cells were washed in cold PBS and UV crosslinked on ice in a Spectrolinker XL-1500 (Spectronics) at 254 nm (400 mJ/cm2). Cells were then scraped, centrifuged, snap-frozen in liquid nitrogen, and stored at −80°C. RIP was performed following a modified eCLIP protocol (Van Nostrand et al., 2016) as follows: Cells were lysed in 1 mL cold iCLIP lysis buffer (50 mM Tris-HCl, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1:200 Protease Inhibitor Cocktail III, RNAse inhibitor) for 25 min on ice. Lysed cells were then centrifuged at 14,000 g for 15 min at 4°C and the supernatant was added to pre-washed and antibody-coupled Protein G Dynabeads (Invitrogen). For each RIP, 5 μg of antibody (anti-PUM1, Santa Cruz sc-135049; anti-PUM2, Santa Cruz sc-31535; anti-RBMX, Cell Signaling #14794; Goat IgG control, Santa Cruz sc-2028; Rabbit IgG control Cell Signaling #2729) was coupled to 3.75 mg of beads at room temperature for 45 min, after which unbound antibody was removed. Sample and beads were incubated at 4°C overnight. The next day, beads were washed three times with 900 μL cold High Salt Wash Buffer #1 (50mM Tris-HCl, 1M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate) and three times with 500 μL Wash Buffer #2 (20mM Tris-HCl, 10 mM MgCl2, 0.2% Tween-20). Beads were then resuspended in 100 μL Wash Buffer #2, and 70 μL was used for RNA extraction and the remainder for western blotting. Proteins were extracted by incubation in Laemmli buffer for 10 min at 70°C. Antibodies used for western blotting were anti-PUM1 (ab92545, Abcam), anti-PUM2 (ab92390, Abcam), and anti-RBMX (14794, Cell Signaling).
DNA fluorescence in situ hybridization (DNA FISH)
Request a detailed protocolAneuploidy in NORAD construct rescue experiments was assessed 18 to 21 days after knockdown of endogenous NORAD. DNA FISH was performed as described previously (Kopp et al., 2019; Lee et al., 2016). Chromosome enumeration probes for chromosome 7 (CHR7-10-GR) and chromosome 20 (CHR20-10-RE) were purchased from Empire Genomics. Cells were trypsinized, washed in PBS, and incubated in hypotonic 0.4% KCl solution for 5 min at room temperature. Cells were then fixed in 3:1 methanol:glacial acetic acid and dropped onto slides. DNA FISH hybridizations were performed by the Veripath Cytogenetics laboratory at UT Southwestern. Slides were analyzed using an AxioObserver Z1 microscope (Zeiss). For each sample, 200 nuclei were counted and aneuploidy was defined as a chromosome count that differed from 2 n for at least one of the two tested chromosomes. Samples were prepared and counted in an experimenter-blinded manner. Two independent HCT116 CRISPRi cell lines stably expressing each AAVS1 knock-in construct were generated, and each was independently tested for aneuploidy using this method.
Quantification of chromosome segregation defects
Request a detailed protocolFor anaphase nuclei imaging, cells were plated on poly-L-lysine coated coverslips and grown for 24 hr. Samples were then fixed with 4% PFA in PBS for 10 min at room temperature, carefully washed with PBS, rinsed with DEPC-treated water, and mounted using SlowFade Diamond Antifade with DAPI mounting media (Invitrogen). Slides were analyzed using an AxioObserver Z1 microscope. For each sample, 100 anaphase nuclei were imaged and assessed for the presence of chromosome segregation defects. Samples were counted in an experimenter-blinded manner.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
-
MassIVEThe NORAD lncRNA assembles a topoisomerase complex critical for DNA replication and genome stability.
References
-
Identification of diverse target RNAs that are functionally regulated by human pumilio proteinsNucleic Acids Research 46:362–386.https://doi.org/10.1093/nar/gkx1120
-
Roles of Puf proteins in mRNA degradation and translationWiley Interdisciplinary Reviews: RNA 2:471–492.https://doi.org/10.1002/wrna.69
-
PUF proteins: repression, activation and mRNA localizationTrends in Cell Biology 21:104–112.https://doi.org/10.1016/j.tcb.2010.09.013
-
Linear models and empirical bayes methods for assessing differential expression in microarray experimentsStatistical Applications in Genetics and Molecular Biology 3:1–25.https://doi.org/10.2202/1544-6115.1027
-
Human pumilio proteins recruit multiple deadenylases to efficiently repress messenger RNAsJournal of Biological Chemistry 287:36370–36383.https://doi.org/10.1074/jbc.M112.373522
Article and author information
Author details
Mahmoud M Elguindy

Funding
Cancer Prevention and Research Institute of Texas (RP160249)
- Joshua T Mendell
National Institutes of Health (R35CA197311)
- Joshua T Mendell
National Institutes of Health (P30CA142543)
- Joshua T Mendell
National Institutes of Health (P50CA196516)
- Joshua T Mendell
Welch Foundation (I-1961-20180324)
- Joshua T Mendell
Howard Hughes Medical Institute
- Joshua T Mendell
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Rudolf Jaenisch, Didier Trono, Stanley Qi, Jonathan Weissman, and Feng Zhang for plasmids; Shinichi Nakagawa for technical assistance with RNA FISH; Sushama Sivakumar and Hongtao Yu for assistance with quantification of chromosome segregation defects; Sangeeta Patel in the Veripath Cytogenetics laboratory at UT Southwestern for DNA FISH; and Sungyul Lee, Kathryn O’Donnell, and members of the Mendell laboratory for helpful discussions and comments on the manuscript. This work was supported by grants from CPRIT (RP160249 to JTM), NIH (R35CA197311 to JTM; P30CA142543 to JTM; and P50CA196516 to JTM), and the Welch Foundation (I-1961–20180324 to JTM). JTM is an investigator of the Howard Hughes Medical Institute.
Version history
- Received: May 22, 2019
- Accepted: July 22, 2019
- Accepted Manuscript published: July 25, 2019 (version 1)
- Version of Record published: August 2, 2019 (version 2)
Copyright
© 2019, Elguindy et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,966
- views
-
- 553
- downloads
-
- 53
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
PUMILIO, but not RBMX, binding is required for regulation of genomic stability by noncoding RNA NORAD
eLife 8:e48625.
https://doi.org/10.7554/eLife.48625
Further reading
-
- Chromosomes and Gene Expression
- Immunology and Inflammation
Ikaros is a transcriptional factor required for conventional T cell development, differentiation, and anergy. While the related factors Helios and Eos have defined roles in regulatory T cells (Treg), a role for Ikaros has not been established. To determine the function of Ikaros in the Treg lineage, we generated mice with Treg-specific deletion of the Ikaros gene (Ikzf1). We find that Ikaros cooperates with Foxp3 to establish a major portion of the Treg epigenome and transcriptome. Ikaros-deficient Treg exhibit Th1-like gene expression with abnormal production of IL-2, IFNg, TNFa, and factors involved in Wnt and Notch signaling. While Ikzf1-Treg-cko mice do not develop spontaneous autoimmunity, Ikaros-deficient Treg are unable to control conventional T cell-mediated immune pathology in response to TCR and inflammatory stimuli in models of IBD and organ transplantation. These studies establish Ikaros as a core factor required in Treg for tolerance and the control of inflammatory immune responses.
-
- Cell Biology
- Chromosomes and Gene Expression
Heat stress is a major threat to global crop production, and understanding its impact on plant fertility is crucial for developing climate-resilient crops. Despite the known negative effects of heat stress on plant reproduction, the underlying molecular mechanisms remain poorly understood. Here, we investigated the impact of elevated temperature on centromere structure and chromosome segregation during meiosis in Arabidopsis thaliana. Consistent with previous studies, heat stress leads to a decline in fertility and micronuclei formation in pollen mother cells. Our results reveal that elevated temperature causes a decrease in the amount of centromeric histone and the kinetochore protein BMF1 at meiotic centromeres with increasing temperature. Furthermore, we show that heat stress increases the duration of meiotic divisions and prolongs the activity of the spindle assembly checkpoint during meiosis I, indicating an impaired efficiency of the kinetochore attachments to spindle microtubules. Our analysis of mutants with reduced levels of centromeric histone suggests that weakened centromeres sensitize plants to elevated temperature, resulting in meiotic defects and reduced fertility even at moderate temperatures. These results indicate that the structure and functionality of meiotic centromeres in Arabidopsis are highly sensitive to heat stress, and suggest that centromeres and kinetochores may represent a critical bottleneck in plant adaptation to increasing temperatures.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}