The ribosomal protein Asc1/RACK1 is required for efficient translation of short mRNAs
- Massachusetts Institute of Technology, United States
Abstract
Translation is a core cellular process carried out by a highly conserved macromolecular machine, the ribosome. There has been remarkable evolutionary adaptation of this machine through the addition of eukaryote-specific ribosomal proteins whose individual effects on ribosome function are largely unknown. Here we show that eukaryote-specific Asc1/RACK1 is required for efficient translation of mRNAs with short open reading frames that show greater than average translational efficiency in diverse eukaryotes. ASC1 mutants in S. cerevisiae display compromised translation of specific functional groups, including cytoplasmic and mitochondrial ribosomal proteins, and display cellular phenotypes consistent with their gene-specific translation defects. Asc1-sensitive mRNAs are preferentially associated with the translational ‘closed loop’ complex comprised of eIF4E, eIF4G, and Pab1, and depletion of eIF4G mimics the translational defects of ASC1 mutants. Together our results reveal a role for Asc1/RACK1 in a length-dependent initiation mechanism optimized for efficient translation of genes with important housekeeping functions.
https://doi.org/10.7554/eLife.11154.001eLife digest
Ribosomes are structures within cells that are responsible for making proteins. Molecules called messenger RNAs (or mRNAs), which contain genetic information derived from the DNA of a gene, pass through ribosomes that then “translate” that information to build proteins. Although all living cells contain ribosomes, the protein building blocks that make up the structure of the ribosome are not the same in all species. Furthermore, the exact roles that each building block plays during translation are not known.
The ribosomes of plants, animals, and budding yeast contain the same protein, known as Asc1 in budding yeast and RACK1 in plants and animals. Thompson et al. have now explored the role of Asc1 in yeast cells by measuring translation in the absence of Asc1 using a technique called ribosome footprint profiling. This analysis revealed that cells lacking Asc1 translate fewer short mRNA molecules than normal cells. Short mRNAs encode small proteins that tend to play important ‘housekeeping’ roles in the cell — by forming the structural building blocks of ribosomes, for example.
It has been observed previously that short mRNAs are translated at a higher rate than longer mRNAs on average, although the reasons behind this bias are still mysterious. The findings of Thompson et al. suggest that the ribosome itself may discriminate between short and long mRNAs and that the Asc1 protein is involved in calibrating the ribosome’s preference for short mRNAs.
Cells need differing amounts of small proteins in different growth conditions. It will therefore be interesting to investigate whether mRNA length discrimination can be regulated by Asc1 and/or other components of the ribosome to tune gene expression to the environment.
https://doi.org/10.7554/eLife.11154.002Introduction
Ribosomes are universal protein-synthesizing machines that are highly conserved in their structure and function throughout all kingdoms of life. However, each domain of life has evolved unique ribosomal proteins that are added to the conserved core. The fundamental tasks of ribosomes — deciphering the genetic code and synthesizing peptide bonds — are the same in all organisms, so the functions of these ‘extra’ ribosomal proteins are intriguing, yet almost entirely unknown.
Eukaryotic ribosomes contain 13 domain-specific proteins that may play roles in translation initiation, which is both more complicated and more highly regulated in eukaryotes than in prokaryotes (Ban et al., 2014; Sonenberg and Hinnebusch, 2009). Recruitment of prokaryotic ribosomes to mRNAs requires only three initiation factors, IF1, 2, and 3, and relies on base-pairing between the RNA of the small ribosomal subunit and the anti-Shine-Delgarno sequence of the mRNA (Boelens and Gualerzi, 2002). In contrast, translation initiation in eukaryotes requires at least 12 initiation factors and proceeds by a complex series of steps beginning with recognition of the mRNA 5′ cap structure, followed by unwinding of mRNA secondary structure, recruitment of the small (40S) ribosomal subunit, scanning, recognition of the initiation codon, and finally, joining of the large (60S) ribosomal subunit to form a functional ribosome (Aitken and Lorsch, 2012). Although the eukaryotic ribosome is generally considered to be a passive player during canonical initiation, several of its proteins have been implicated in mRNA recruitment. For example, RPL38 is required for the translation of the Hox body-patterning genes during embryonic development, allowing spatiotemporal regulation of gene expression through translational control (Kondrashov et al., 2011). Other proteins including RPS25, RPL40, and RACK1 are essential for the translation of viral mRNAs that are recruited to the ribosome via alternative initiation pathways (Cherry et al., 2005; Landry et al., 2009; Lee et al., 2013; Majzoub et al., 2014).
The eukaryote-specific ribosomal protein RACK1 is a WD40-repeat β-propeller protein that binds the solvent-exposed face of the 40S subunit near the mRNA exit channel, in close proximity to proteins that contact the mRNA during translation initiation (Pisarev et al., 2008; Sengupta et al., 2004). In addition to its function as a core ribosomal protein, in mammalian cells, RACK1 has been found in complex with several proteins involved in signal transduction including protein kinase C, Src kinase, and cAMP phosphodiesterase, among many others (Adams et al., 2011). The location of RACK1 on the ribosome together with its interactions with signaling proteins suggests a possible role in conveying stimulus-dependent information to the translation machinery (Nilsson et al., 2004). However, signaling pathways in yeast and human have diverged significantly compared to genes required for ribosomal function (Stuart et al., 2003), suggesting that RACK1 might have another, more conserved function during translation.
Loss of RACK1 causes widespread and pleiotropic defects in many organisms. Deletion of the RACK1 homolog in budding yeast, ASC1, leads to slow growth, loss of invasive growth, loss of cell wall integrity, and decreased 60S subunit levels, among many described effects (Li et al., 2009; Melamed et al., 2010; Valerius et al., 2007; Yoshikawa et al., 2011). In metazoans, RACK1 is required for cell migration, neural tube closure, and control of post-synaptic excitation in the brain (Kiely et al., 2009; Ron et al., 1999; Wehner et al., 2011; Yaka et al., 2002). These cellular functions may explain why homozygous RACK1 loss-of-function mutations cause early developmental lethality in mouse and flies (Kadrmas et al., 2007; Volta et al., 2013). However, it is not known whether and how the effects of RACK1 on ribosome function contribute to the myriad cellular and organismal phenotypes observed in RACK1/ASC1 mutants (Gibson, 2012).
Here we have examined the translational functions of Asc1/RACK1 genome-wide by ribosome footprint profiling in yeast ASC1 mutants. We show that Asc1 is required for the efficient translation of short mRNAs, including those encoding cytoplasmic and mitochondrial ribosomal proteins. This requirement is specific as deletion of other ribosomal proteins does not cause similar translation defects. Using translational reporters we demonstrate that length per se determines translational sensitivity to Asc1, thus confirming a role for Asc1 in the translational privileging of short mRNAs, which is a dominant trend in genome-wide translational efficiency data from diverse eukaryotes. Remarkably, mRNA enrichment with proteins that mediate the formation of a ‘closed loop’ during translation — eIF4E, eIF4G, and Pab1 — is strongly biased towards short mRNAs and predicts Asc1-sensitivity, suggesting a role for Asc1 in closed-loop-dependent ribosome recruitment. Consistent with this prediction, we find that depletion of the central closed loop factor eIF4G mimics the translational effects of mutating ASC1. Finally, we show that loss of ASC1 reduces mitochondrial translation and renders cells unable to use alternative carbon sources that require enhanced mitochondrial function, demonstrating the functional significance of translational perturbation in ASC1 mutants. Together, our results reveal a role for Asc1 in the enhanced translation of short mRNAs and establish a direct connection between gene-specific effects of Asc1 on translation and defects in cellular physiology. Furthermore, because mitochondria are essential for energy generation and regulation of many cellular networks, our results suggest that the pleiotropic phenotypes associated with the Asc1/RACK1 protein should be re-examined in the context of mitochondrial health.
Results
Loss of the Asc1 protein perturbs global translation
The ASC1 locus encodes two distinct gene products — the Asc1 protein and an intronic small nucleolar RNA, snR24. Because snR24 directs 2′-O-methylation of 25S rRNA at positions C1437, C1449, and C1450, some of the reported phenotypes of ASC1 null mutants (asc1Δ) could be due to effects of deleting SNR24 on ribosome biogenesis or function. In addition, Asc1/RACK1 may have functions off the ribosome (Baum et al., 2004; Coyle et al., 2009; Warner and McIntosh, 2009). We therefore created an allelic series of yeast mutants with altered Asc1 function to enable direct comparison of cellular and translational effects of Asc1/RACK1 (Figure 1A). We created protein null alleles by mutating a codon early in the ASC1 ORF to a stop codon (asc1-M1X and asc1-E5X, where X denotes a stop codon), which abolished Asc1 protein expression but maintained wild type levels of SNR24 (Figure 1B,C). Although bulk polysomes appeared normal in these strains, both asc1∆ and asc1-M1X showed reduced levels of free 60S subunits (Figure 1D). This slight discrepancy between our results and the literature (Li et al., 2009) may stem from differences in strain backgrounds because the Sigma1278b strain used here has higher free 60S subunit levels than S288C. Restoring SNR24 expression rescued the temperature-sensitive polysome defect of the asc1∆ mutant in agreement with previous observations (Figure 1—figure supplement 1A–D) (Li et al., 2009). Both asc1-M1X and asc1∆ grow slowly under standard laboratory conditions, whereas a mutant lacking only snR24 grows as well as wild type, further demonstrating the importance of the Asc1 protein (Figure 1—figure supplement 1E).
Figure 1 with 1 supplement see all

Loss of the Asc1 protein causes widespread changes in translation efficiency.
(A) Gene model of ASC1, showing the SNR24 snoRNA and location of protein null (M1X and E5X) and ribosome binding (DE and D109Y) mutations. (B) Asc1 protein levels quantified by Western blot. Pgk1 blot on the same membrane is shown as a loading control. Dilutions of the WT sample are shown on the left. Data is representative of three biological replicates. (C) ASC1 mRNA and SNR24 snoRNA levels quantified by qRT-PCR. Levels were normalized to ACT1 mRNA levels. Error bars represent s.d. from three technical replicates. Data is representative of three biological replicates. (D) Polysome profiles of the ASC1 mutants at 30˚C. The polysome/monosome (P/M) ratio and 60S/40S (60/40) ratio are shown with s.d. from two biological replicates. (E) Calculation of translation efficiency as the ratio of ribosome-protected mRNA fragments to total mRNA abundance. (F) Distribution of changes in TE comparing two biological replicates from WT cells (i.e. replicate error) or asc1-M1X or asc1∆ to its corresponding WT comparison. #1 and #2 denote biological replicate experiments. (G) Scatterplot of TE changes between the two ASC1 null mutants. The Pearson correlation coefficient is shown.
To define the translational function of Asc1, we subjected the ASC1 mutants to ribosome footprint profiling and RNA-seq. Together, these techniques allow quantification of ribosome densities transcriptome-wide and can be used to infer changes in gene-specific translation activity (Ingolia et al., 2009). Loss of the Asc1 protein caused changes in translation activity for many mRNAs as measured by translational efficiency (TE) — the number of ribosomal footprints normalized by the number of total RNA fragments for each mRNA (Figure 1E–G). The magnitude and pervasiveness of translation changes in asc1-M1X and asc1Δ are notable given the normal appearance of bulk polysomes in ASC1 mutants (Figure 1D). Thus superficially normal polysomes can conceal significant perturbations of cellular translation. Together, these results demonstrate that the lack of Asc1 substantially alters the translational landscape of yeast cells.
ASC1 ‘ribosome-binding’ mutants associate with ribosomes and are largely functional
Next we examined isogenic yeast strains that express normal levels of Asc1 protein with perturbed association to the ribosome. Asc1 is a WD-repeat protein that interacts with helices 39 and 40 of the 18S rRNA primarily through its N-terminal blade (Sengupta et al., 2004). Directed mutation of several basic residues in this region interferes with the ribosome-binding capacity of the protein, with the strongest defect observed in the R38D K40E (DE) mutant (Coyle et al., 2009; Sengupta et al., 2004). Another Asc1 ribosome-binding mutant, D109Y, was discovered serendipitously in a forward genetic screen for mutants with defects in no-go decay, a ribosome-associated RNA quality control mechanism (Kuroha et al., 2010). These mutant proteins were expressed at near wild type levels (Figure 1B), and both mutations substantially decreased co-sedimentation of Asc1 with ribosomes in sucrose gradients, with D109Y having a markedly stronger effect (Figure 2A) that is consistent with previous reports (Kuroha et al., 2010).
Figure 2
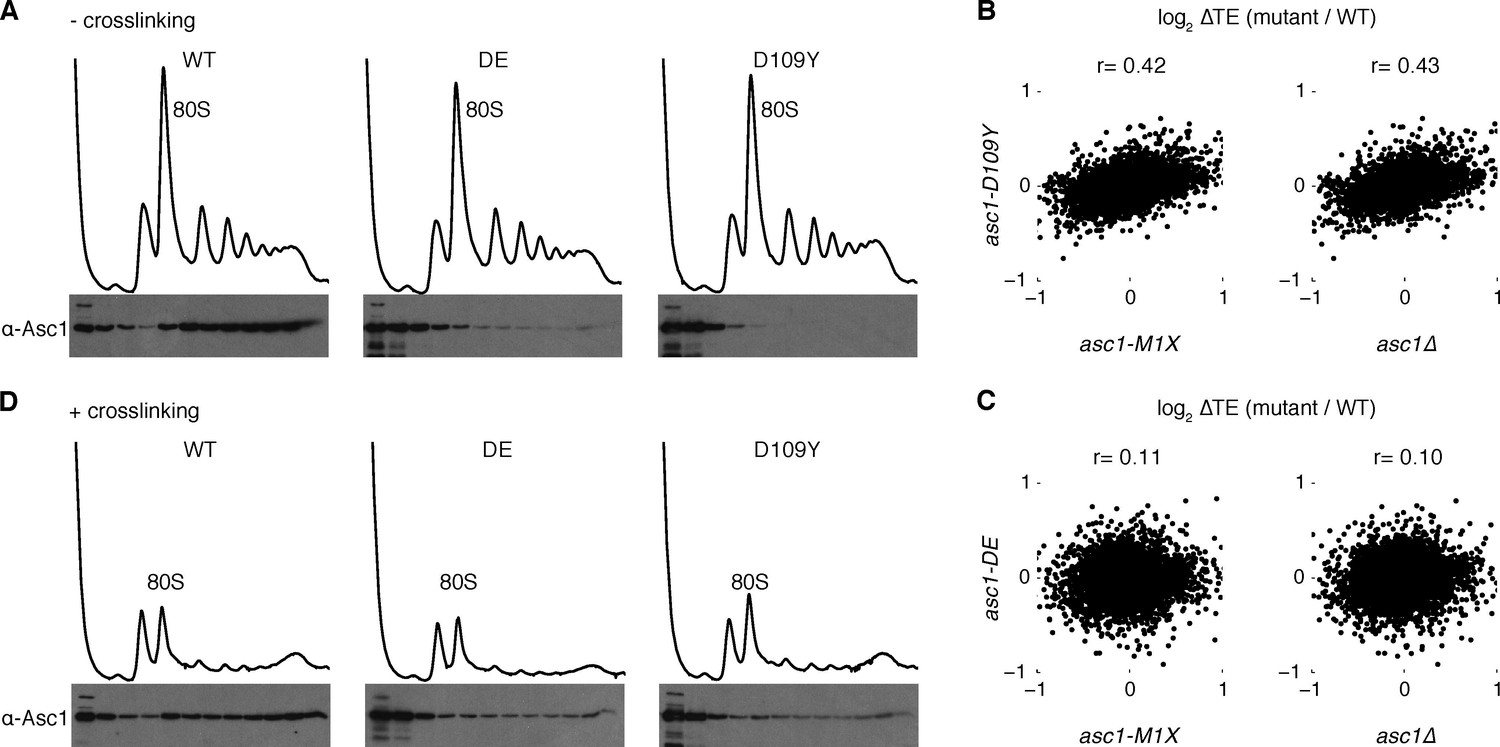
Asc1 ‘ribosome-binding’ mutants retain ribosomal association in vivo.
(A) Association of Asc1 mutant proteins with the ribosome assayed by Western blot of fractions isolated after velocity gradient sedimentation. (B, C) Scatterplot of TE changes between the two ASC1 null mutants and the asc1-D109Y and asc1-DE ribosome-binding mutants. The Pearson correlation coefficients are shown. (D) The same as (A) but proteins were crosslinked with formaldehyde in vivo before sample processing.
The D109Y strong ribosome-binding mutant showed translational defects that, although correlated with those observed in the ASC1 null mutants (r=0.43, p=10–221 for asc1∆; r=0.42, p=10–204 for asc1-M1X), were much smaller in magnitude (Figure 2B), while the DE mutant showed almost negligible effects on translation (Figure 2C). These findings suggest that either Asc1 primarily affects translation from a location off the ribosome, or that the ribosome-binding assay underestimates the extent of in vivo association of the D109Y and DE mutant proteins because ribosome-bound factors can dissociate during ultracentrifugation (Valásek et al., 2007). To test this second possibility, we performed formaldehyde crosslinking before ultracentrifugation. In the presence of crosslinking, we observed significant co-sedimentation of the DE and D109Y proteins with polysomes (Figure 2D). Crosslinking is not quantitative (Orlando, 2000); thus this assay underestimates the extent of ribosome binding by the mutant Asc1 proteins in vivo, which are likely much closer to wild type than previously appreciated.
An important implication of these findings is that phenotypic differences between ‘ribosome-binding’ alleles and ASC1 null mutants likely reflect different degrees of perturbing ribosome function and do not constitute strong evidence for ‘extra-ribosomal’ activity by Asc1/RACK1. We attempted to generate stronger ribosome-binding-defective alleles by combining multiple mutations, but these proteins were expressed at very low levels potentially due to misfolding (data not shown). Given the overall correlation between asc1-D109Y and ASC1 null alleles for translation changes transcriptome-wide, we infer that many of the translational changes in asc1-M1X and asc1Δ are likely to be caused by direct effects of Asc1 on ribosome function.
Asc1 promotes translation of mRNAs with short open reading frames
To probe the mechanism by which Asc1 promotes translation of specific mRNAs, we searched for shared attributes among mRNAs with decreased TE in the asc1-M1X mutant. Motif analysis of 5′ UTRs revealed the presence of a U-rich sequence in mRNAs sensitive to loss of Asc1 (Figure 3—figure supplement 1), but not found in mRNAs resistant to loss of Asc1. However, this motif was present in only 11% of Asc1-sensitive mRNAs and so cannot be generally required for translational enhancement by Asc1. We next examined various physical properties of Asc1-sensitive mRNAs (Table 1). Among the tested attributes, ORF length was notably well-correlated with ∆TE in asc1-M1X (r=0.27, p=10–84, Table 1) and ORFs <500 nts were the most strongly affected (Figure 3A). Short ORFs are highly translated in wild type cells (Figure 3B and [Arava et al., 2003]), an effect that has been hypothesized to reflect a higher rate of translation initiation on short mRNAs for reasons that are mechanistically mysterious (Figure 3C and Arava et al., 2005; Shah et al., 2013). Because short ORFs are among the most highly expressed, the loss of Asc1/RACK1 significantly alters the gene expression landscape of the cell.
Table 1
Properties of Asc1-sensitive mRNAs. Gene or mRNA attributes were correlated with ∆TE in the asc1-M1X mutant. The spearman correlation coefficients and p-values are shown.
| attribute | Spearman r (∆TE asc1-M1X vs. attribute) | p-value |
|---|---|---|
| wild type protein level | 0.103 | 1.49e-2 |
| wild type translation efficiency | -0.091 | 1.55e-10 |
| tRNA adaptation index (tAI) | 0.023 | 1.17e-1 |
| 5′ UTR length | -0.004 | 7.73e-1 |
| 3′ UTR length | 0.079 | 9.62e-8 |
| ORF length | 0.272 | 3.05e-84 |
| 5′ folding energy (MFE) | 0.030 | 3.97e-2 |
| 3′ folding energy (MFE) | -0.077 | 1.83e-7 |
| poly(A) tail length | 0.029 | 7.38e-2 |
Figure 3 with 4 supplements see all
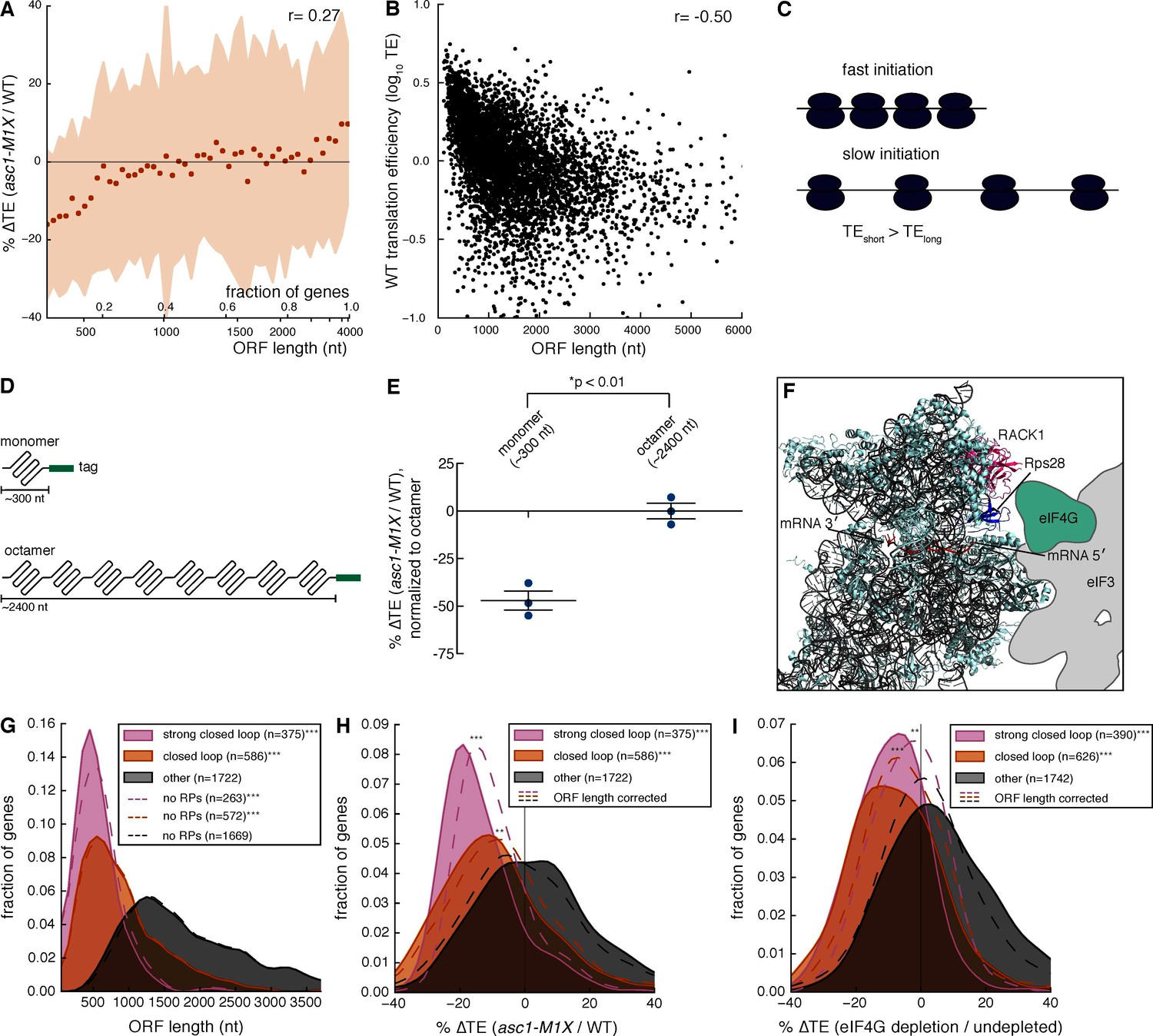
Asc1 is required for efficient translation of short ORFs that form closed loop complexes.
(A) Relationship between ORF length and TE changes in asc1-M1X. The values shown represent the average percent change in TE for bins of 100 genes arranged by length. The ORF lengths shown correspond to the point at which the average ORF length of the bin exceeds the indicated value. Shaded areas represent +/- 1 s.d. from the average change. The ASC1 gene is excluded from the plot. (B) Relationship between ORF length and translational efficiency in WT yeast cells (data from this study). The Spearman correlation coefficient is shown. (C) Model showing the expected effect of a higher initiation rate on short mRNAs compared to long mRNAs on translation efficiency measurements. (D) Diagram of ORF length reporter constructs. The I27 monomer was repeated to make the octamer and each ORF was fused to a C-terminal V5 epitope tag. (E) Result of ORF length reporter experiment. TE is calculated as the normalized protein (V5 tag/Pgk1) to mRNA ratio (V5 mRNA/18S) and the ∆TE (ratio between mutant and WT) is shown. Relative protein concentration was obtained from quantitative Western blotting and mRNA concentration from qRT-PCR. *p=0.002, two-tailed Student’s t-test (monomer vs. octamer). Error bars are SEM from 3 biological replicates derived from independent genetic isolates of asc1-M1X. (F) The structure of the mammalian 48S pre-initiation complex is shown (Lomakin and Steitz, 2013) with the mRNA, RACK1, and Rps28, which crosslinks to the -7 and -10 positions of the mRNA relative to the AUG (Pisarev et al., 2008), indicated. The outline of eIF3 from Hashem et al. (2013) is shown. eIF4G is placed on the left arm of eIF3 based on electron microscopy data from Siridechadilok et al. (2005). (G, H, I) The relationship between closed loop complex association and ORF length (p=10–172and 10–135 for strong closed loop and closed loop groups vs. other mRNAs, respectively) (G), ∆TE in asc1-M1X (p=10–71and 10–42 for strong closed loop and closed loop vs. other mRNAs, respectively) (H), and ∆TE after eIF4G depletion (p=10–70and 10–73 for strong closed loop and closed loop vs. other mRNAs, respectively. Data from Park et al., 2011) (I). In (H) and (I), the dotted lines show the results after accounting for the relationship between ORF length and ∆TE using linear regression. For asc1-M1X, ORF length corrected p-values are 10–30 and 10–14 for strong closed loop and closed loop groups, respectively. For eIF4G depletion, ORF length corrected p-values are 10–17 and 10–28 for strong closed loop and closed loop groups, respectively. p-values are from the one-sided Mann-Whitney U test. Closed loop association groups are from Costello et al. (2015). For G-I, ***p<10–18, **p<10–9, *p<10–3
We then sought to determine whether ORF length or transcript length is more predictive of translational efficiency. ORF length was slightly more predictive of wild type translation efficiency than transcript length, (Figure 3—figure supplement 2A–E, partial correlation r=-0.09 (p=10–8) vs. r=0.03 (p=10–1)). For simplicity, and because transcript boundary annotations are not available for all yeast genes, we have used the ORF length metric in subsequent analyses.
To test whether length per se, and not some other feature common to short mRNAs, is responsible for Asc1-sensitive translation, we generated two constructs with identical regulatory regions (promoter, 5′ UTR, 3′ UTR) that differed only in the length of the ORF (Figure 3D). These ORF length reporters contain either one or eight repeats of the I27 domain from the human cardiac protein titin, which has been used extensively in studies of protein folding because the small globular domains fold and unfold independently of each other (Hoffmann and Dougan, 2012). This modular architecture allows the construction of proteins of different lengths that resemble linear chains and minimizes the potential for differential protein folding or stability to impact the abundance of the reporter proteins. We performed quantitative Western blotting by fluorescent detection of a common C-terminal epitope tag in combination with qRT-PCR measurements of mRNA levels to determine the translational efficiency (protein/mRNA) of each construct (Figure 3—figure supplement 3A). Remarkably, the translational efficiency of the short ORF (~300 nt) was two-fold lower in the asc1-M1X mutant compared to the long ORF (~2400 nt) (p=0.002, Figure 3E). Together with the genome-wide trend, these reporter results demonstrate a role for Asc1 in the translational advantage of short mRNAs. Given that ORF length is strongly anti-correlated with translational efficiency in diverse eukaryotes (Figure 3—figure supplement 3B–D, data from Guo et al., 2010; Stadler and Fire, 2011), this function of Asc1/RACK1 may be conserved.
How might short ORFs be translationally privileged and sensitive to loss of Asc1? Asc1’s position near the mRNA exit channel places it in close proximity to translation initiation factors that interact with the 5′ end of the mRNA during initiation, including eIF3 and eIF4G (Kouba et al., 2012) (Figure 3F, note that the structure shown is the mammalian ribosome, for which structural information regarding the orientation of eIF3 and eIF4G has been reported [Hashem et al., 2013; Lomakin and Steitz, 2013; Siridechadilok et al., 2005]). eIF4G has a well-characterized role in promoting a circularized form of the mRNA in which the 5′ and 3′ regions of the mRNA are bundled together via the interaction between the eIF4G protein, associated with the mRNA 5’ cap through the eIF4F complex, and Pab1, an RNA-binding protein that binds the poly(A) tail. The mRNA in this conformation is known as the closed loop, and closed loop formation is thought to enhance translation (Kahvejian et al., 2001). We hypothesized that mRNAs with short ORFs might form closed loop structures more efficiently than mRNAs with longer ORFs, and that Asc1 could promote the function of the closed loop in translation.
According to this model, mRNAs with short ORFs should be more highly associated with the closed loop factors — eIF4E, eIF4G, and Pab1 — than other mRNAs. To test this prediction, we analyzed data quantifying the association of specific mRNAs with the closed loop factors and the eIF4E-binding proteins (4E-BPs) by RNA immunoprecipitation and sequencing (Costello et al., 2015). We grouped mRNAs into ‘closed loop’, ‘strong closed loop’, and ‘other’ categories based on the following enrichment profiles: ‘Strong closed loop’ mRNAs are enriched in immunoprecipitations of eIF4E, eIF4G, and Pab1, and de-enriched in immunoprecipitations of the 4E-BPs, which should not be associated with mRNAs in closed loops because 4E-BPs and eIF4G compete for binding to eIF4E (Haghighat et al., 1995). ‘Closed loop’ mRNAs have similar enrichment profiles to ‘strong closed loop’ mRNAs, but are not de-enriched for association with the 4E-BPs. Remarkably, we found that both ‘closed loop’ and ‘strong closed loop’ mRNAs were dramatically shorter than other mRNAs (median ORF lengths= 489, 774, and 1694 nt for ‘strong closed loop’, ‘closed loop’, and ‘other’ mRNAs, respectively). This association between ORF length and closed loop association was observed regardless of whether mRNAs encoding ribosomal proteins were included in the analysis (Figure 3G and Figure 3—figure supplement 3E). Thus, although ~30% of the ‘strong closed loop’ mRNAs encode ribosomal proteins, a specialized mechanism for enhancing the translation of ribosomal protein mRNAs cannot explain the ORF length bias of the ‘strong closed loop’ group. This discovery — that closed-loop-associated mRNAs are much shorter than other mRNAs — provides a plausible biochemical explanation for the preference for higher translation efficiency of mRNAs with short ORFs observed here and previously (Arava et al., 2003). Remarkably, loss of Asc1 or eIF4G depletion (data from Park et al., 2011) similarly decreased the translation of closed-loop-associated mRNAs (Figure 3H and I).
Although ORF length, closed loop enrichment, and ∆TE in ASC1 and eIF4G mutants are correlated, some longer RNAs are strongly associated with the closed loop and require Asc1 for efficient translation while some short mRNAs are neither enriched with closed loop factors nor particularly dependent on Asc1 for their translation. Accounting for the global relationship between ORF length and ∆TE by linear regression showed that closed loop association has additional explanatory power for translational sensitivity to Asc1 and eIF4G: the observed reductions in translation efficiency for closed-loop-enriched mRNAs were significantly more than would be expected if ORF length alone determined their translation efficiencies (p=10–14 and 10–30 for ‘closed loop’ and ‘strong closed loop’ groups, respectively, Figure 3H and I). These results suggest that Asc1 is important for closed loop formation and/or stability or for closed-loop-dependent ribosome recruitment, a process that is apparently biased towards short ORFs.
What are the potential consequences of impairing translation of short mRNAs? Using gene ontology analysis, we found that transcripts annotated to the category ‘ribosomal subunit’ had significantly decreased TE in the ASC1 null mutants (asc1-M1X, p=10–35, Figure 4A and Figure 4—source data 1). This category is composed of short mRNAs encoding both cytoplasmic and mitochondrial ribosomal proteins (RPs, MRPs), which both displayed ~20% decreased TE in ASC1 null mutants (asc1-M1X, p=10–37 and p=10–10; asc1∆, p=10–35 and p=10–10, respectively, Figure 4B). As the median RP and MRP ORF lengths are 434 and 716 nt, respectively, their translational defects are within the range predicted by their length. Indeed, removing RP and MRP genes does not significantly alter the global relationship between ∆TE and ORF length in asc1-M1X (r=0.23, p=10–58, Figure 4—figure supplement 1A) indicating that all classes of genes with short ORFs have decreased TE in asc1-M1X. Although these GO categories were the clear outliers, most GO categories with short median ORF lengths also displayed decreased TE in the ASC1 null mutants, including several additional groups of genes whose protein products function in mitochondria (Figure 4—figure supplement 1B). Because short ORF length is associated with specific functional categories, loss of Asc1 — and, potentially, modulation of its activity — leads to coherent changes in gene expression.
Figure 4 with 2 supplements see all
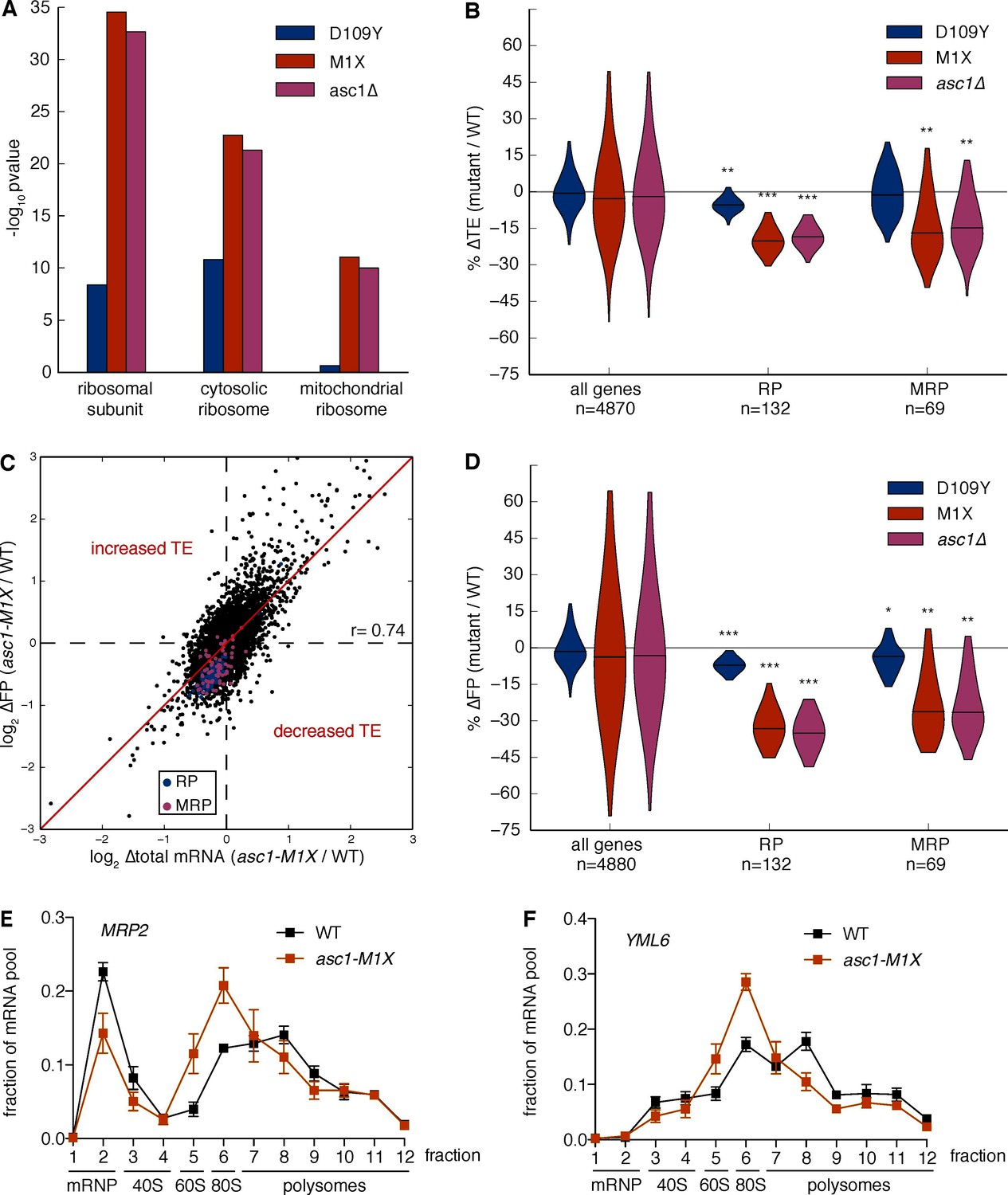
Loss of Asc1 causes decreased translational efficiency of cytoplasmic and mitochondrial ribosomal protein mRNAs.
(A) GO Component category enrichments for mRNAs with decreased TE in the ASC1 mutants. GO categories related to the top category ‘ribosomal subunit’ for the asc1-M1X mutant are displayed. (B) Violin plot showing the decreases in TE for the cytosolic ribosomal protein (RP) and mitochondrial ribosomal protein (MRP) gene sets in the ASC1 mutants. The violin shape represents a kernel density estimation and the top and bottom of the plot extend to the most extreme data point within 1.5x of the inner quartile range. Midlines represent the medians. ***p<10–18, **p<10–9, *p<10–3. (C) Scatterplot showing the decrease in both the footprint (FP) and total RNA pool for RP and MRP mRNAs. The Pearson correlation coefficient in shown. (D) As in (B), but with the change in ribosome association (FP) shown. (E, F) Polysome qRT-PCR showing decreased association of MRP genes with heavy polysomes. Values are normalized to an RNA spike-in control in each fraction and then set so that the sum of all fractions=1.
-
Figure 4—source data 1
GO category enrichments for mRNAs with changes in FP, total, or TE in ASC1 mutants.
- https://doi.org/10.7554/eLife.11154.013
We noted that RP and MRP mRNAs decreased in both the total RNA pool and the ribosome-protected footprint (FP) pool (Figure 4C,D). The additional decrease in the FP pool shows that these mRNA substrates are translationally disadvantaged in the ASC1 mutants. In support of this interpretation, qRT-PCR analysis of polysome gradient fractions demonstrated that representative MRP mRNAs associated with fewer ribosomes in asc1-M1X (Figure 4E,F), which specifically indicates a defect in translation initiation. Because inhibiting translation initiation can induce mRNA degradation (Coller and Parker, 2005; LaGrandeur and Parker, 1999; Schwartz and Parker, 1999), decreased translation may account for the reduction in total mRNA levels although we cannot exclude the possibility of transcriptional effects or translation-independent effects of Asc1 on mRNA stability. We note that our translation efficiency measurements are correlated with steady-state mRNA half-life estimates using non-invasive metabolic labeling approaches (r=0.43, p=10–194, Figure 4—figure supplement 1C, data from Miller et al., 2011 and r=0.39, p=10–168, Figure 4—figure supplement 1D, data from Neymotin et al., 2014), consistent with the hypothesis that the decay rates of mRNAs are coupled to their translational status. The same trends of decreased TE for the RP and MRP genes were observed using an rRNA-depletion strategy instead of poly(A) selection (Figure 4—figure supplement 1E,F), ruling out a significant effect of poly(A) tail length on our ∆TE calculations (Subtelny et al., 2014). Thus, Asc1 is required for efficient translation of short ORFs, which includes most ORFs encoding cytosolic and mitochondrial ribosomal proteins.
Although Asc1 has been implicated in the ribosome-dependent no-go decay pathway (Kuroha et al., 2010), the observed co-directional changes in mRNA abundance and translational efficiency are not consistent with widespread defects in no-go decay as a driver of changes in translation efficiency. If decreases in translation efficiency were caused by defects in no-go decay stabilizing mRNAs, thus inflating the denominator in the footprint RNA/total RNA calculation, then the levels of affected mRNAs should increase in the total RNA pool. However, the overall trend was for the levels of total mRNA for genes with decreased TE in the asc1-M1X mutant to go down or remain constant rather than increase (Figure 4C, Figure 4—figure supplement 2A–D).
The translational defects of ASC1 mutants are not a general consequence of perturbing the ribosome
The mRNAs that are sensitive to the loss of Asc1 are among the most efficiently translated in a cell. We therefore considered the possibility that reduced translation of these mRNAs might be a general consequence of perturbing the ribosome. To assess the specificity of the translational phenotypes of ASC1 null mutants, we tested four additional ribosomal protein mutants, rpl23b∆ rpp1a∆, rps0b∆, and rps16b∆, each of which deletes one paralog encoding a core ribosomal protein. Like asc1-M1X, rpl23b∆ and rpp1a∆ show reduced growth on glucose and decreased 60S subunit levels (Figure 5—figure supplement 1A,B). RPS0B and RPS16B encode small ribosomal subunit proteins that bind the ribosome near the mRNA exit channel in the vicinity of Asc1/RACK1 and deletion of these loci results in increased 60S subunit levels relative to 40S levels (Figure 5—figure supplement 2A and B). However, none of the tested ribosomal protein mutants showed notable similarity to asc1-M1X in their translational dysregulation genome-wide (r= -0.06 to 0.18, Figure 5A and B), and they did not display decreased translation efficieny of ‘closed loop’ mRNAs (Figure 5C). Because the growth and bulk translation phenotypes of these other ribosomal protein mutants are more severe than asc1-M1X, any shared defects in gene-specific translation should have been readily detected. Thus, decreased translation of RP genes is not a general feature of slow-growing mutants, ribosomal subunit imbalance, or perturbations in the vicinity of the mRNA exit channel near RACK1.
Figure 5 with 2 supplements see all
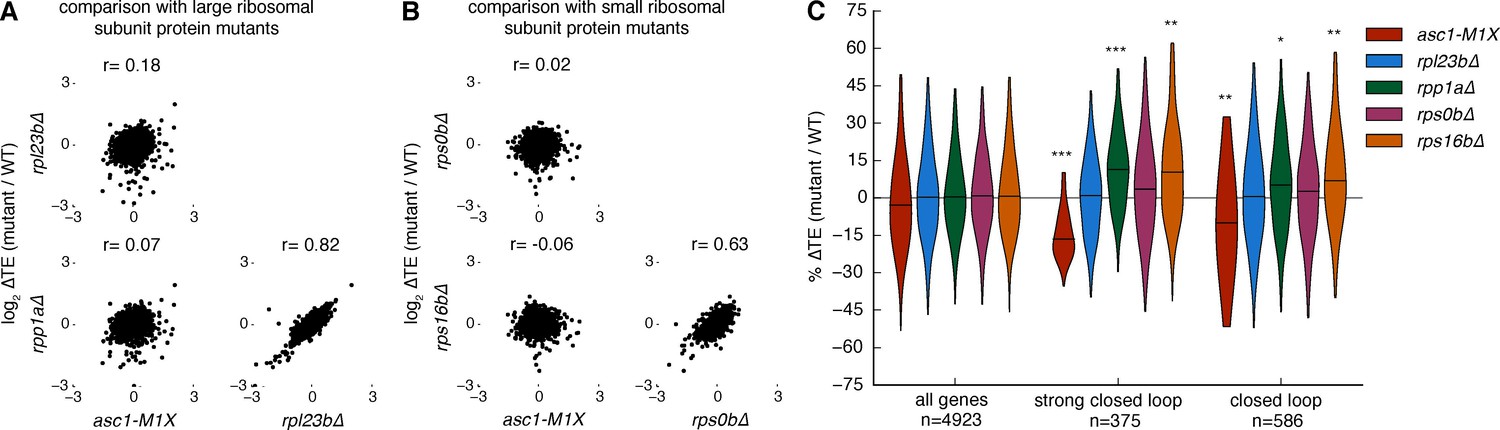
Other ribosomal protein mutants do not share translational phenotypes with the ASC1 mutants.
(A) Correlations between ∆TE among asc1-M1X and mutants with reduced expression of large ribosomal subunit proteins, rpl23b∆ and rpp1a∆. The Pearson correlation coefficient is shown. (B) Correlations between ∆TE among asc1-M1X and mutants with reduced expression of small ribosomal proteins in the vicinity of Asc1, rps0b∆ and rps16b∆. The Pearson correlation coefficient is shown. (C) Violin plots showing the change in TE for the ‘strong closed loop’ and ‘closed loop’ mRNAs in asc1-M1X and the other ribosomal protein mutants. Violin plot parameters are described in Figure 4B.
Loss of Asc1 impairs mitochondrial function in yeast
To assess the physiological significance of gene-specific translation defects in ASC1 mutants, we looked for phenotypes related to gene categories with significantly impaired translation. In particular, the requirement of Asc1 for efficient MRP translation suggested the possibility of impaired mitochondrial function in ASC1 mutants. To assess mitochondrial health, we measured growth on the non-fermentable carbon source glycerol, which requires the activity of the mitochondrial respiratory chain to generate energy (Dimmer et al., 2002). When shifted to glycerol-containing media, wild type yeast resumed rapid growth after an initial adaptation phase, but the asc1-M1X mutant completed only ~3 doublings before ceasing growth (Figure 6A). In contrast, the rpl23b∆ and rpp1a∆ mutants grew better in glycerol than asc1-M1X, demonstrating the specificity of this phenotype (Figure 6—figure supplement 1A). Consistent with our results, a proteomic survey of asc1∆ cells showed a shift away from respiration and towards fermentative metabolism (Rachfall et al., 2012). Because mitochondrial ribosomes are required for mitochondrial biogenesis and function, it is plausible that the growth and metabolic defects of ASC1 mutants are consequences of the translation defects observed for MRP genes.
Figure 6 with 1 supplement see all
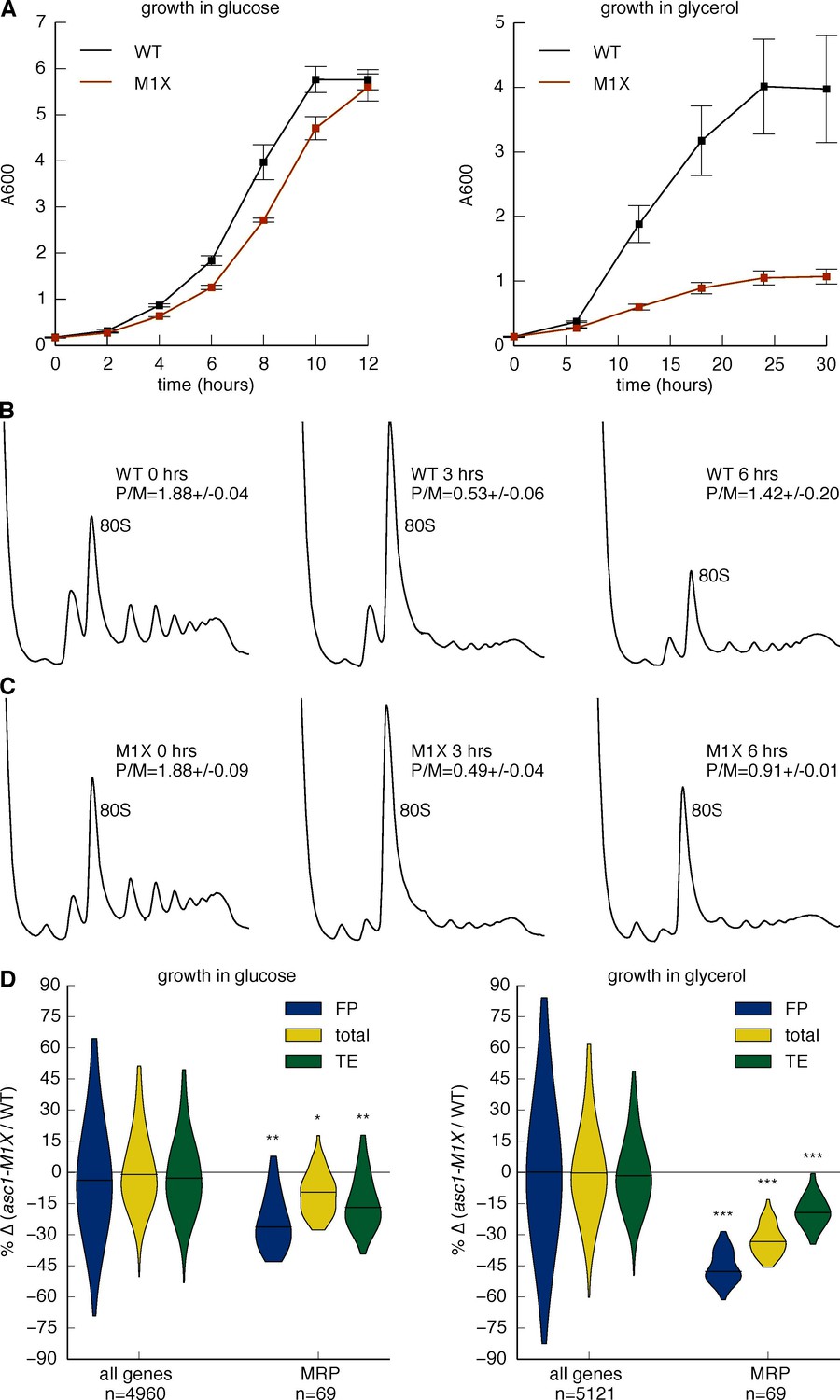
Asc1 is required for adaptation to a non-fermentable carbon source.
(A) Growth curves of WT and asc1-M1X cells after a shift from YPAD to fresh media containing either glucose (left) or glycerol (right). Curves are averages of two biological replicates, error bars are s.d. (B,C) Polysome profiles of WT (B) and asc1-M1X (C) yeast after a shift from glucose- to glycerol-containing media. Yeast were shifted at OD600=0.5. The polysome/monosome (P/M) and 60S/40S (60/40) ratios are shown with s.d. from two biological replicates. (D) Violin plot of the FP, total mRNA, and TE changes in asc1-M1X for MRP transcripts during growth in glucose (left, FP and TE data is also represented in Figure 4B,D) or after 6 hr of growth in glycerol (right). Violin plot parameters are as described for Figure 4B.
To directly determine the impact of the MRP translation defects on mitochondrial translation activity, we performed 35S metabolic labeling assays in the asc1-M1X mutant in the presence of cycloheximide, which inhibits cytosolic but not mitochondrial ribosomes. Synthesis of all mitochondrially-translated proteins was reduced >two-fold in asc1-M1X compared to wild type (Figure 6—figure supplement 1B and C). Thus pervasive, moderate impairment of MRP translation is associated with substantial defects in mitochondrial protein synthesis.
Given the severe growth defects of the asc1-M1X mutant in glycerol, we wondered whether the moderate impairment of MRP translation observed in glucose would worsen under conditions of increased MRP expression. Adaptation to growth in non-fermentable carbon sources or low glucose is accompanied by a rewiring of the transcriptional network in yeast (Galdieri et al., 2010) and widespread reprogramming of translation (Vaidyanathan et al., 2014). To investigate whether the glycerol growth defect of asc1-M1X is linked to inadequate translational adaptation, we examined translation genome-wide 6 hr after transfer from glucose to glycerol, a point just before the resumption of rapid growth in wild type cells (Figure 6A) and coincident with the recovery of polysomes after the initial collapse upon glucose withdrawal (Figure 6B). In the asc1-M1X mutant, polysomes recovered only partially (Figure 6C). Moreover, the ribosome-associated pool was strongly depleted of MRP mRNAs compared to wild type (Figure 6D). However, the magnitude of translational defect (ΔTE) for this class of mRNAs was similar in both glucose and glycerol, supporting a constitutive rather than regulatory role for Asc1 in translation of MRP mRNAs (Figure 6D). The fact that asc1-M1X shows a bulk translation defect in glycerol but not in glucose may reflect the fact that MRP mRNAs make up a larger portion of the translatome in glycerol. Thus, the cellular context is an important factor in determining the phenotypic consequences of translational perturbations in asc1-M1X and likely in other ribosomal protein mutants as well. Taken together, our results suggest an important role for Asc1 in supporting cellular respiration by promoting synthesis of mitochondrial ribosomal proteins.
Discussion
Here we have demonstrated that the eukaryote-specific ribosomal protein Asc1/RACK1 is required for efficient translation of short mRNAs, a category that includes functionally related groups of genes required for vital cellular processes (e.g. cytoplasmic and mitochondrial ribosomal proteins). A correlation between ORF length and translation efficiency or ribosome density has been observed since the advent of genome-wide translation profiling (Arava et al., 2003; Ingolia et al., 2009) and we observed this relationship in data collected from diverse eukaryotes including yeast, nematodes, mice, and humans (Guo et al., 2010; Stadler and Fire, 2011). To account for this trend, it was proposed that the rate of translation initiation is higher for short ORFs (Arava et al., 2005; Shah et al., 2013), but the mechanism(s) underlying length-dependent initiation rate differences were unknown.
It has been suggested that the increased probability of mRNA circularization by diffusion could make initiation more efficient on short mRNAs (Chou, 2003; Guo et al., 2015). Our results add an additional nuance to these physical models — the presence of a ribosome-dependent regulatory mechanism that specifically enhances the translation of short mRNAs by promoting the formation and/or function of the closed loop. Our analyses reveal a clear trend that short mRNAs preferentially associate with closed loop factors in vivo, and consistent with these observations, short mRNAs form more stable closed loop complexes than longer mRNAs in vitro (Amrani et al., 2008). A challenge for the future will be to determine how the mRNA, the closed loop factors, and the ribosome cooperate to privilege the translation of short mRNAs.
How might Asc1/RACK1 promote closed loop formation? RACK1’s placement on the solvent exposed side of the head of the small subunit puts it in close proximity to the mRNA exit channel, in a position with the potential to interact with the mRNA-bound closed loop factors during initiation. Intriguingly, eIF4G co-purifies with Asc1 from yeast lysates under stringent conditions in which most other initiation factors do not (Gavin et al., 2002; 2006), suggesting that Asc1 may interact directly with the closed loop via eIF4G. Our results also raise the possibility that translation of many mRNAs could be co-regulated by mechanism(s) that target Asc1/RACK1’s function in closed-loop-dependent initiation. Moving forward, it will be important to determine how the many signaling pathways that have been linked to Asc1/RACK1 impact the translation of closed-loop-dependent mRNAs.
More generally, our study highlights the fact that individual ribosomal proteins can contribute to efficient translation of subsets of mRNAs with important consequences for cellular physiology. In particular we show that loss of the non-essential ribosomal protein Asc1/RACK1 causes a concerted decrease in MRP expression that leads to mitochondrial insufficiency. Given the central role of mitochondria in energy and metabolite production in eukaryotic cells, it is not surprising that mitochondrial defects elicit pleiotropic consequences (Calvo and Mootha, 2010; Fleming et al., 2001; Kushnir et al., 2001; Shoffner et al., 1990; Vafai and Mootha, 2012). In light of our findings, many of Asc1/RACK1’s ascribed cellular functions should be re-evaluated for potential connections to mitochondrial dysfunction. Finally, it is intriguing that several distinct mutations in human ribosomal proteins and ribosome biogenesis factors result in anemia, the cause of which is currently the source of much debate (Freed et al., 2010; Narla et al., 2011). Given that many forms of heritable anemia have been traced to defects in mitochondrial iron metabolism (Dailey and Meissner, 2013; Huang et al., 2011), it will be interesting to see whether translation of nuclear-encoded mitochondrial proteins is affected in these diseases and whether these defects contribute to pathogenesis.
Materials and methods
Plasmid construction
Request a detailed protocolThe cDNA encoding the I27 domain monomer from human cardiac titin was a generous gift from Julio Fernandez. The I27 monomer was fused to a serine-glycine linker (SGGGGG) followed by the V5 epitope tag. The I27 octamer was made using the iterative subcloning method that relies upon the compatible cohesive ends of BamHI and BglII and results in an arginine-serine linker added between individual domains (Hoffmann and Dougan, 2012). I27 proteins were expressed under the GAL1 promoter and followed by the CYC1 terminator in the pRS415 low-copy yeast vector.
Yeast strain construction
Request a detailed protocolDeletion strains of the ASC1, RPL23B, and RPP1A loci were obtained by homologous recombination using the pFA6a-kanMX6 plasmid as a template and PCR product adding 40 nt of homology to each side of the kanMX6 cassette (Longtine et al., 1998). Isolates were confirmed by PCR. Deletion strains of RPS0B, RPS16B, and their isogenic wild type were obtained from the Sigma1278b deletion collection (Dowell et al., 2010). To make the ASC1 protein null alleles, a codon early in the ASC1 open reading frame was mutated to a stop codon, denoted as X (i.e. M1X, E5X). Integration of mutant ASC1 alleles was performed using the two-step gene replacement strategy. First, the URA3 marker was integrated at the ASC1 locus. Subsequently, ASC1 mutant alleles were amplified by PCR from plasmid templates and integrated into the asc1::URA3 strain at the ASC1 locus. Isolates were identified by 5-FOA resistance and correct integration was confirmed by sequencing. All strains were constructed in the Sigma1278b strain background.
Yeast growth
Request a detailed protocolYeast were cultivated in liquid or on solid (2% agar) YPAD media (yeast extract, peptone, dextrose (2% w/v) supplemented with adenine hemisulfate). Liquid cultures were grown with rapid agitation at 30˚C, unless otherwise noted, and harvested at OD 0.6–0.9 (0.6-0.7 for ribosome footprint profiling experiments in YPAD). For glycerol shift polysome experiments, yeast were grown to mid log phase (OD 0.5–0.6) in YPAD and then media was removed by brief centrifugation and replaced with YPAG (YPA + 3% (w/v) glycerol). For the yeast growth curves, yeast were diluted from saturated cultures into fresh media and allowed to double 1–2 times before rediluting to an OD of 0.1 in glucose- or glycerol-containing media.
Polysome analysis
Request a detailed protocolCycloheximide (CHX, Sigma-Aldrich, St. Louis, Missouri) was added to a final concentration of 0.1 mg/ml to cells and incubated an additional 2 min at the growth temperature with shaking. Cells were rapidly cooled and washed with polysome lysis buffer (PLB: 20 mM Hepes-KOH, pH 7.4, 2 mM Mg acetate, 100 mM K acetate, 3 mM DTT, 0.1 mg/ml CHX + 1% Triton X-100). Formaldehyde crosslinking experiments were performed as described (Valásek et al., 2007). 10–15 OD260 units were loaded on 10–50% sucrose gradients in polysome gradient buffer (PGB: PLB –Triton) and centrifuged in an SW 41 rotor (Beckman Coulter, Brea, California) at 35,000 rpm for 3 hr. Fractions were collected from the top using a BioComp Gradient Station (Biocomp Instruments, Canada). To calculate the ratio of free 60S/40S subunits, A254 traces of the native polysome profiles (without dissociation into free subunits) were quantified with a custom script, available on github: https://github.com/marykthompson/Thompson_eLife_2016/. Minima were identified and used as boundaries for each peak. Values are the integral under the curve to the baseline, which was set as a line connecting the lowest minimum in the first half of the trace with the lowest minimum in the second half of the trace.
Ribosome footprint profiling
Request a detailed protocolRibosome footprint profiling was performed essentially as described (Ingolia et al., 2009) with the following modifications: monosomes were isolated manually from 10–50% sucrose gradients. 50 A260 units were digested with 750 U of RNAse I (Ambion, Waltham, Massachusetts). Selective precipitation was used to enrich for small RNA fragments prior to size selection of 28mers on denaturing gels. In brief, RNA samples were resuspended in GuHCl buffer (8 M guanidine HCl, 20 mM MES hydrate, 20 mM EDTA) and brought to 33% ethanol before binding to a silica-based column (Zymoprep-V, Zymoresearch, Irvine, California) to precipitate and remove large RNAs. The eluate was brought to 70% ethanol to precipitate small RNAs. Total RNA for accompanying RNA-seq samples was isolated from the same cell extracts used for footprint library generation using the hot acid phenol method. Poly(A) selection was performed using oligo-dT cellulose (Sigma-Aldrich or NEB, Ipswich, Massachusetts) as previously described (Sambrook et al., 2001). For experiments using rRNA-depletion to enrich for coding transcripts, the Ribo-Zero kit (Epicentre, Madison, Wisconsin) was used. The asc1-DE and matched WT libraries were constructed using an earlier version of the protocol that used Microcon YM-100 (EMD Millipore, Billerica, Massachusetts) filters to enrich for small RNA fragments, poly(A) tailing to capture the small RNA fragments, and downstream library construction steps as previously described (Ingolia et al., 2009). For all other libraries, we ligated a pre-adenylated 3’ adaptor (5Phos/TGGAATTCTCGGGTGCCAAGG/3ddC/) to the fragments using T4 RNA Ligase 1 (NEB). First strand synthesis was performed with Superscript III (Life Technologies, Carlsbad, California) or AMV (Promega, Madison, Wisconsin) using primer OJA225 (/5Phos/GATCGTCGGACTGTAGAACTCTGAACCTGTCGGTGGTCGCCGTATCATT/iSp18/CACTCA/iSp18/GCCTTGGCACCCGAGAATTCCA). cDNA was amplified using primer oNTI230 (AATGATACGGCGACCACCGA) and (CAAGCAGAAGACGGCATACGAGATXXXXXXGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA), where XXXXXX denotes a six nucleotide barcode used to distinguish samples run in the same lane. Samples were run on an Illumina HiSeq 2000 instrument or an Illumnina GAIIx.
qRT-PCR
Request a detailed protocolRNA was extracted using the hot acid phenol method. RNA was treated with TURBO DNase (Life Technologies). First strand synthesis was performed with AMV Reverse Transcriptase (Promega) using an anchored oligo-dT primer (for coding transcripts) or a random hexamer primer (for SNR24). Quantitative PCR was performed with SYBR Fast reagents (Kapa Biosystems, Wilmington, Massachusetts) using a Lightcycler 480 (Roche, Switzerland). Gene-specific primer sequences are: ACT1: (TTCTGAGGTTGCTGCTTTGG, CTTGGTGTCTTGGTCTACCG), ASC1: (ATGTTTGGCCACTTTGTTGG, GTTACCGGCAGAAATGATGG), MRP2: (AATAGGTGCGTGGACTCTGG, CTGGCAAATTACCCTTCAGAGC), SNR24: (TTGCTACTTCAGATGGAACTTTG, TCAGAGATCTTGGTGATAATTGG), V5: (AGATCTTCCGGAGGCGGG, GGATCTATTACGTAGAATCGAGACC), YML6: (AGAGTAGGCGCCTCAAATCC, TTGGAGAGTTAGCATCCCCG), 18S: (TGGCGAACCAGGACTTTTAC, CCGACCGTCCCTATTAATCAT), FLUC: ( GTACCAGAGTCCTTTGATCGTGA, ACCCAGTAGATCCAGAGGAATTC).
Western blotting
Request a detailed protocolTotal protein levels were determined using the BCA assay (Thermo Scientific, Waltham, Massachusetts). For total Asc1 level quantification, 1 μg of total protein obtained by TCA precipitation followed by cell lysis was loaded onto 12% SDS-PAGE gels. For polysome Westerns, the same volume of each fraction was loaded per well. Blots were overexposed to show the remaining ribosome-associated protein for the ribosome-binding mutants. Membranes were blotted with α-Asc1 (Coyle et al., 2009) and α-Pgk1 (Life Technologies 22C5D8). After secondary antibody incubation, blots were incubated with ECL (GE Healthcare Life Sciences, United Kingdom) and exposed to X-ray film.
ORF length reporter assays
Request a detailed protocolYeast grown overnight in SC-Leu (synthetic complete media lacking leucine) were diluted to OD 0.2 in YPA + 2% galactose and grown for 8 hr before harvest. Cells were lysed in PBS pH 7.4 supplemented with protease inhibitors (1X Roche complete mini EDTA-free, 1 mM PMSF) with glass beads. Total protein was quantified by the BCA assay (Thermo Scientific) and 1 ug (octamer) or 2 ug (monomer) total protein was loaded per lane with each sample loaded in 4 different lanes as technical replicates for each of three biological replicates. A standard curve encompassing 2X, 1X, 0.5X and 0.25X of the WT extract concentration was loaded on each gel. Western blotting was performed using the ECL Plex kit (GE) according to the manufacturer’s instructions and blots were scanned with a Typhoon imager (FLA 9500, GE). Primary antibodies were α-Pgk1 (Life Technologies 22C5D8) and α-V5 (Sigma-Aldrich V8137). Images were quantified with ImageStudio (LI-COR Biosciences, Lincoln, Nebraska) and the values of each sample were calculated relative to the standard curve. Although all standards were in linear range (linear fit of signal vs. concentration r2 ≥ 0.95 for all blots), we used a quadratic fit as it fit the standards slightly better. RNA was extracted from the extracts in parallel, and the mRNA levels of each reporter were quantified by qRT-PCR using primers recognizing the region encoding the V5 tag and normalized to 18S levels also determined by qRT-PCR. For each sample, a translation efficiency was calculated from the ratio of the normalized protein levels of the reporter (V5 protein/Pgk1 protein) to the normalized mRNA levels of the reporter (V5 mRNA/18S rRNA).
Mitochondrial translation
Request a detailed protocolMitochondrial translation products were labeled with 35S-methionine as previously described (Funes and Herrmann, 2007). In brief, cells were grown overnight in SC-Met (with glucose) to OD 0.4 then transferred to SC-Met with glycerol for 3 hr. Equal OD units of cells were then incubated with 35S-methionine (EasyTag L-[35S]-Methionine, PerkinElmer, Waltham, Massachusetts) and cycloheximide to inhibit cytoplasmic protein synthesis. After 30 min, total protein synthesis was halted by the addition of puromycin. TCA-precipitated protein was visualized by Coomassie staining (total protein normalization) and autoradiography on a Typhoon imager (mitochondrial proteins). Total protein in each sample was quantified with ImageJ using Coomassie signal across the whole lane. Six bands corresponding to mitochondrial translation products were quantified with ImageQuant (GE).
Read mapping and positional alignment
Request a detailed protocolYeast reads were aligned to the Sigma1278b (Dowell et al., 2010) genome downloaded from the Saccharomyces Genome Database on June 29, 2014. We used Tophat to map first to annotated splice junctions and then to the genome. We used only uniquely-mapping reads for all downstream analyses. Because ribosome footprint reads generally start 12 nt upstream of start codons and end 18 nt upstream of stop codons (Ingolia et al., 2009), we used only reads mapping within these boundaries. Additionally, to avoid potential variability that can be present at the 5’ end of mRNAs, we excluded the first 30 codons from counting for quantification of gene expression.
Gene expression analysis
Request a detailed protocolFor comparisons between libraries, we used normalized values obtained from running count data through the DE-Seq package (Anders and Huber, 2010) because RPKM values are strongly biased by the transcript lengths of the RNA pool (Wagner et al., 2012). For gene expression measurements, we only included genes for which there were at least 128 mapping reads total among the libraries used for the analysis (Ingolia et al., 2009). All analyses were performed with custom Bash and Python scripts written in-house, available on github: https://github.com/marykthompson/Thompson_eLife_2016/. Data in figures represent the average of two biological replicates. Figures were constructed using Matplotlib (Hunter, 2007).
ORF length correction of ∆TE values for closed loop groups
Request a detailed protocolTo determine whether the decrease in translation efficiency of the ‘closed loop’ mRNAs in asc1-M1X could be accounted for completely by the relationship between ∆TE and ORF length, we first regressed ∆TE against ORF length. We then took the residuals from this correlation (i.e. the part of ∆TE that cannot be accounted for by the global correlation between ∆TE and ORF length) and plotted these values among the ‘strong closed loop’, ‘closed loop’ and ‘other’ mRNAs, as shown by the dashed lines in Figure 3H and I. Note that this analysis assumes linear relationships between ∆TE and ORF length. These results demonstrate that the decrease in translation efficiency of the ‘closed loop’ mRNAs in asc1-M1X is more than would be expected if ∆TE was determined entirely by ORF length, thus suggesting that ‘closed loop’ enrichment may be more important. However, as with all correlative analyses, the results cannot assign causality.
Analysis of TE in other organisms
Request a detailed protocolFor correlations of TE with ORF length shown in Figure 3—figure supplement 3, processed data files were downloaded from NCBI GEO and used to calculate TE. Only genes in which the pooled the reads or scaled reads (for Stadler and Fire, 2013) from footprint and total RNA libraries reached 128 reads were included.
Pathway analysis
Request a detailed protocolTo identify groups of genes with significantly altered TE in yeast mutants, we used the Mann Whitney U test and report one-sided p-values for groups of genes with significantly altered TE each condition. For this analysis, we included all genes without filtering for read cutoff and added a pseudocount of one read in cases where >1 read was detected in some but not all libraries.
Motif finding
Request a detailed protocolWe used MEME (Bailey and Elkan, 1994) to identify motifs present in 5′ UTRs of the selected groups of mRNAs. 5′ UTR boundaries were taken from the median UTR length reported in Pelechano et al. (2014). UTRs <8 nt were excluded from the motif analysis. WebLogo (Crooks et al., 2004) was used to generate sequence logos.
Data sources for mRNA attributes
Request a detailed protocol5′ and 3′ UTR lengths were taken as the median length from Pelechano et al. (2014). MFEs were calculated by running these sequences through RNAfold (Gruber et al., 2008) with temperature set to 30˚C and otherwise default parameters. Translation adaptation index values per gene were calculated by Eckhard Jankowsky and colleagues using values from Tuller et al. (2010). Poly(A) tail length was taken from Subtelny et al. (2014). Wild type protein levels were taken from de Godoy et al., 2008.
Data availability
-
The ribosomal protein Asc1/RACK1 is required for efficient translation of short mRNAsPublicly available at NCBI Gene Expression Omnibus (accession no. GSE61753).
-
Mammalian microRNAs predominantly act to decrease target mRNA levelsPublicly available at NCBI Gene Expression Omnibus (accession no. GSE22004).
-
mRNA and Ribosome Profiling in Four Nematode Species Traversing a Shared Developmental TransitionPublicly available at NCBI Gene Expression Omnibus (accession no. GSE48140).
-
Depletion of eIF4G from yeast cells narrows the range of translational efficiencies genome-widePublicly available at NCBI Gene Expression Omnibus (accession no. GSE25721).
-
Global assessment of the closed loop components (eIF4E, eIF4G and PABP) and the translational repressors (4E-BPs) in mRNA recognition for translation initiation.Publicly available at ArrayExpress (accession no. E-MTAB-2464).
References
-
RACK1, a multifaceted scaffolding protein: Structure and functionCell Communication and Signaling 9:.https://doi.org/10.1186/1478-811X-9-22
-
A mechanistic overview of translation initiation in eukaryotesNature Structural & Molecular Biology 19:568–576.https://doi.org/10.1038/nsmb.2303
-
Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiaeProceedings of the National Academy of Sciences of the United States of America 100:3889–3894.https://doi.org/10.1073/pnas.0635171100
-
Dissecting eukaryotic translation and its control by ribosome density mappingNucleic Acids Research 33:2421–2432.https://doi.org/10.1093/nar/gki331
-
Fitting a mixture model by expectation maximization to discover motifs in biopolymersProceedings International Conference on Intelligent Systems for Molecular Biology 2:28–36.
-
A new system for naming ribosomal proteinsCurrent Opinion in Structural Biology 24:165–169.https://doi.org/10.1016/j.sbi.2014.01.002
-
Structure and function of bacterial initiation factorsCurrent Protein & Peptide Science 3:107–119.https://doi.org/10.2174/1389203023380765
-
The mitochondrial proteome and human diseaseAnnual Review of Genomics and Human Genetics 11:25–44.https://doi.org/10.1146/annurev-genom-082509-141720
-
Direct link between RACK1 function and localization at the ribosome in vivoMolecular and Cellular Biology 29:1626–1634.https://doi.org/10.1128/MCB.01718-08
-
Erythroid heme biosynthesis and its disordersCold Spring Harbor Perspectives in Medicine 3:a011676.https://doi.org/10.1101/cshperspect.a011676
-
Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiaeMolecular Biology of the Cell 13:847–853.https://doi.org/10.1091/mbc.01-12-0588
-
When ribosomes go bad: diseases of ribosome biogenesisMolecular bioSystems 6:481–493.https://doi.org/10.1039/b919670f
-
Analysis of mitochondrial protein synthesis in yeastMethods in Molecular Biology 372:255–263.https://doi.org/10.1007/978-1-59745-365-3_18
-
RACK1 research - ships passing in the night?FEBS Letters 586:2787–2789.https://doi.org/10.1016/j.febslet.2012.04.048
-
Length-dependent translation initiation benefits the functional proteome of human cellsMolecular bioSystems 11:370–378.https://doi.org/10.1039/c4mb00462k
-
Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4EThe EMBO Journal 14:5701–5709.
-
Single molecule force spectroscopy using polyproteinsChemical Society Reviews 41:4781.https://doi.org/10.1039/c2cs35033e
-
Mitochondrial mayhem: the mitochondrion as a modulator of iron metabolism and its role in diseaseAntioxidants & Redox Signaling 15:3003–3019.https://doi.org/10.1089/ars.2011.3921
-
Matplotlib: A 2D Graphics EnvironmentComputing in Science & Engineering 9:90–95.https://doi.org/10.1109/MCSE.2007.55
-
Characterization of RACK1 function in Drosophila developmentDevelopmental Dynamics 236:2207–2215.https://doi.org/10.1002/dvdy.21217
-
The mRNA Closed-loop Model: The Function of PABP and PABP-interacting Proteins in mRNA TranslationCold Spring Harbor Symposia on Quantitative Biology 66:293–300.https://doi.org/10.1101/sqb.2001.66.293
-
Phosphorylation of RACK1 on tyrosine 52 by c-Abl is required for insulin-like growth factor I-mediated regulation of focal adhesion kinaseThe Journal of Biological Chemistry 284:20263–20274.https://doi.org/10.1074/jbc.M109.017640
-
A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAsProceedings of the National Academy of Sciences of the United States of America 110:324–329.https://doi.org/10.1073/pnas.1216454109
-
Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeastMolecular Systems Biology 7:458.https://doi.org/10.1038/msb.2010.112
-
Ribosome defects in disorders of erythropoiesisInternational Journal of Hematology 93:144–149.https://doi.org/10.1007/s12185-011-0776-0
-
Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitationTrends in Biochemical Sciences 25:99–104.https://doi.org/10.1016/s0968-0004(99)01535-2
-
RACK1/Asc1p, a ribosomal node in cellular signalingMolecular & Cellular Proteomics 12:.https://doi.org/10.1074/mcp.M112.017277
-
Coordinated movement of RACK1 with activated betaIIPKCThe Journal of Biological Chemistry 274:27039–27046.https://doi.org/10.1074/jbc.274.38.27039
-
Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiaeMolecular and Cellular Biology 19:5247–5256.https://doi.org/10.1128/mcb.19.8.5247
-
Identification of the versatile scaffold protein RACK1 on the eukaryotic ribosome by cryo-EMNature Structural & Molecular Biology 11:957–962.https://doi.org/10.1038/nsmb822
-
Translation efficiency is determined by both codon bias and folding energyProceedings of the National Academy of Sciences of the United States of America 107:3645–3650.https://doi.org/10.1073/pnas.0909910107
-
The Saccharomyces homolog of mammalian RACK1, Cpc2/Asc1p, is required for FLO11-dependent adhesive growth and dimorphismMolecular & Cellular Proteomics 6:1968–1979.https://doi.org/10.1074/mcp.M700184-MCP200
-
In vivo stabilization of preinitiation complexes by formaldehyde cross-linkingMethods in Enzymology 429:163–183.https://doi.org/10.1016/S0076-6879(07)29008-1
-
RACK1 depletion in a mouse model causes lethality, pigmentation deficits and reduction in protein synthesis efficiencyCellular and Molecular Life Sciences 70:1439–1450.https://doi.org/10.1007/s00018-012-1215-y
-
Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samplesTheory in Biosciences 131:281–285.https://doi.org/10.1007/s12064-012-0162-3
-
NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1Proceedings of the National Academy of Sciences of the United States of America 99:5710–5715.https://doi.org/10.1073/pnas.062046299
Article and author information
Author details
Paritosh Gangaramani

Funding
National Institutes of Health (GM094303)
- Mary Katherine Thompson
- Maria Fernanda Rojas-Duran
- Paritosh Gangaramani
- Wendy V Gilbert
National Institutes of Health (T32GM007287)
- Mary Katherine Thompson
- Paritosh Gangaramani
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank N Ingolia, J Weissman, D Bartel, and members of the Gilbert lab for discussion; and B Zinshteyn and P Vaidyanathan for discussion and scripts. The sequencing was performed at the BioMicro Center under the direction of S Levine. This work was supported by the National Institutes of Health (GM094303) to WVG. This work was supported in part by the NIH Pre-Doctoral Training Grant T32GM007287.
Version history
- Received: August 26, 2015
- Accepted: March 21, 2016
- Version of Record published: April 27, 2016 (version 1)
Copyright
© 2016, Thompson et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,768
- views
-
- 915
- downloads
-
- 89
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The ribosomal protein Asc1/RACK1 is required for efficient translation of short mRNAs
eLife 5:e11154.
https://doi.org/10.7554/eLife.11154
Further reading
-
- Biochemistry and Chemical Biology
- Cell Biology
Mediator of ERBB2-driven Cell Motility 1 (MEMO1) is an evolutionary conserved protein implicated in many biological processes; however, its primary molecular function remains unknown. Importantly, MEMO1 is overexpressed in many types of cancer and was shown to modulate breast cancer metastasis through altered cell motility. To better understand the function of MEMO1 in cancer cells, we analyzed genetic interactions of MEMO1 using gene essentiality data from 1028 cancer cell lines and found multiple iron-related genes exhibiting genetic relationships with MEMO1. We experimentally confirmed several interactions between MEMO1 and iron-related proteins in living cells, most notably, transferrin receptor 2 (TFR2), mitoferrin-2 (SLC25A28), and the global iron response regulator IRP1 (ACO1). These interactions indicate that cells with high MEMO1 expression levels are hypersensitive to the disruptions in iron distribution. Our data also indicate that MEMO1 is involved in ferroptosis and is linked to iron supply to mitochondria. We have found that purified MEMO1 binds iron with high affinity under redox conditions mimicking intracellular environment and solved MEMO1 structures in complex with iron and copper. Our work reveals that the iron coordination mode in MEMO1 is very similar to that of iron-containing extradiol dioxygenases, which also display a similar structural fold. We conclude that MEMO1 is an iron-binding protein that modulates iron homeostasis in cancer cells.
-
- Biochemistry and Chemical Biology
- Structural Biology and Molecular Biophysics
NADPH oxidases (NOX) are transmembrane proteins, widely spread in eukaryotes and prokaryotes, that produce reactive oxygen species (ROS). Eukaryotes use the ROS products for innate immune defense and signaling in critical (patho)physiological processes. Despite the recent structures of human NOX isoforms, the activation of electron transfer remains incompletely understood. SpNOX, a homolog from Streptococcus pneumoniae, can serves as a robust model for exploring electron transfers in the NOX family thanks to its constitutive activity. Crystal structures of SpNOX full-length and dehydrogenase (DH) domain constructs are revealed here. The isolated DH domain acts as a flavin reductase, and both constructs use either NADPH or NADH as substrate. Our findings suggest that hydride transfer from NAD(P)H to FAD is the rate-limiting step in electron transfer. We identify significance of F397 in nicotinamide access to flavin isoalloxazine and confirm flavin binding contributions from both DH and Transmembrane (TM) domains. Comparison with related enzymes suggests that distal access to heme may influence the final electron acceptor, while the relative position of DH and TM does not necessarily correlate with activity, contrary to previous suggestions. It rather suggests requirement of an internal rearrangement, within the DH domain, to switch from a resting to an active state. Thus, SpNOX appears to be a good model of active NOX2, which allows us to propose an explanation for NOX2’s requirement for activation.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}