Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2014; 23(4): 324-336
Published online December 31, 2014
https://doi.org/10.5607/en.2014.23.4.324
© The Korean Society for Brain and Neural Sciences
Protein Transmission, Seeding and Degradation: Key Steps for α-Synuclein Prion-Like Propagation
Abid Oueslati1#*, Methodios Ximerakis2# and Kostas Vekrellis2
1Centre de Recherche du Centre Hospitalier de Québec, Axe Neuroscience et Département de Médecine Moléculaire
de l'Université Laval, Québec G1V4G2, Canada, 2Center for Neurosciences, Biomedical Research Foundation,
Academy of Athens, Athens 11526, Greece
Correspondence to: #A. O. and M. X. contributed equally to this work.
*To whom correspondence should be addressed.
TEL: 14185254444, Ext 49119, FAX: 14186542125
e-mail: Abid.Oueslati@crchudequebec.ulaval.ca
Abstract
Converging lines of evidence suggest that cell-to-cell transmission and the self-propagation of pathogenic amyloidogenic proteins play a central role in the initiation and the progression of several neurodegenerative disorders. This "prion-like" hypothesis has been recently reported for α-synuclein, a presynaptic protein implicated in the pathogenesis of Parkinson's disease (PD) and related disorders. This review summarizes recent findings on α-synuclein prion-like propagation, focusing on its transmission, seeding and degradation and discusses some key questions that remain to be explored. Understanding how α-synuclein exits cells and propagates from one brain region to another will lead to the development of new therapeutic strategies for the treatment of PD, aiming at slowing or stopping the disease progression.
Keywords: secretion, seeding, turnover, extracellular proteins, disease propagation
INTRODUCTION
Abnormal accumulation of aggregated proteins is a common neuropathological feature of several neurodegenerative disorders, including Parkinson's disease (PD), Alzheimer's disease (AD), Huntington's disease (HD) and many other disorders [1]. Over the past few years, an increasing body of evidence suggests that the amyloidogenic proteins implicated in these neurological diseases can propagate from one brain region to another in a "prion-like" manner and contribute to the disease progression [2,3].
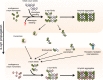
This concept of "prion-like propagation" was recently described for the presynaptic protein, α-synuclein (α-syn), which is the major component of Lewy bodies (LB) and a key player in the pathophysiology of PD and other diseases, collectively termed synucleinopathies [4,5]. This prion-like paradigm comprises a cascade of cellular events including: the secretion, the uptake and the protein seeding by the recipient cells. This process results on self-perpetuating states and the formation of pathogenic protein strains capable of recruiting endogenous soluble proteins (Fig. 1). Finally, the formed pathogenic species induce axonal degeneration and neuronal death.
Understanding how and where α-syn propagation is initiated and the characterization of the major factors playing a role in the modulation of intracerebral α-syn spreading will lead to the identification of new therapeutic targets aiming at slowing or stopping the disease progression. In this review, we explore the recent findings from cell-based assays and animal models of α-syn prion-like propagation and discuss how and where the pathological α-syn species could be initiated and the key mechanisms related to its intercellular transmission.
α-SYNUCLEIN TRANSMISSION
A crucial tilting point in the PD field was the discovery of α-syn secretion. α-syn was thought at first to be an exclusively intracellular protein [6], as it lacks a canonical endoplasmic reticulum signal peptide that would direct it to the secretory pathway. However, this notion was challenged when α-syn was detected in biological fluids, such as blood plasma and Cerebrospinal fluid [7,8]. Gradually, it became evident that α-syn secretion is a physiologic process in the protein's life cycle and that a proportion of α-syn is located in the extracellular milieu [8,9,10,11,12]. Although, the mechanism of α-syn release has not been fully elucidated, data point towards non-classic secretory pathways [13] that involve vesicle trafficking through exocytotic vesicles [12] and exosomes (Fig. 1) [9].
The discovery of α-syn secretion prompted the examination of the paracrine role of extracellular α-syn in brain homeostasis, which under pathological conditions could contribute to the cascade of events leading to neuronal degeneration [14,15]. It is therefore possible that certain abnormalities or deficits affecting α-syn proteostasis [16] could lead to increased extracellular α-syn accumulation and aggregation, and subsequently to its concomitant detrimental effects. In support of this, a growing number of reports have demonstrated that various species of α-syn when applied extracellularly are potent to induce multiple neurotoxic [9,11,17] and inflammatory responses [18,19,20,21,22,23,24], as well as, to cause deleterious seeding effects [25].
The hypothesis of α-syn prion-like propagation and its potential implication in the progression of PD was first proposed by two independent studies showing that intra-striatal transplants of fetal ventral mesencephalic progenitors in PD patients developed α-syn-positive LB-like inclusions, a decade after transplantation, suggesting a direct transfer of pathogenic α-syn from host-to-grafted tissue [26,27]. Subsequent
Although the precise mechanism(s) of α-syn exocytosis, the nature of extracellular α-syn in terms of its aggregation state, the physiologic role(s) of extracellular α-syn, as well as, its potential association with other molecules (e.g. chaperones, lipids) remain unresolved issues, these biological observations opened up new avenues for investigations to uncover the pathophysiological significance of extracellular α-syn in synucleinopathies.
The exact molecular mechanisms by which the newly synthesized α-syn amyloidogenic species are released from "diseased" (donor) cells and transmitted to "healthy" bystander (recipient) cells are still elusive. Over the last years, various species-specific and cell-type dependent mechanisms have been suggested and hypothesized [38]. α-syn can be actively secreted via non-classic exocytotic vesicles [12,39] and exosomes [9], but could also be directly released from injured/dying neurons. Once in the extracellular space, α-syn could either gain access to neighboring cells via active processes involving conventional endocytosis [17, 28, 40], exosomal transport [9, 41], or receptor-mediated internalization [17]. Alternatively, extracellular α-syn could be transmitted to neighboring cells via direct penetration of the plasma membrane (Fig. 1) [40, 42, 43].
Among these proposed mechanisms for α-syn cell-to-cell spreading, the transmission of pathogenic α-syn through exosomes necessitates further exploration. Exosomes enable the transfer of membrane and cytosolic components from donor cells to the extracellular matrix and recipient cells by various mechanisms, such as endocytosis, receptor-ligand binding, or fusion with the plasma membrane [44]. As such, these small vesicles could serve as a system to facilitate cell-to-cell communication
Another question that still remains to be answered is whether the PD-linked α-syn mutations affect the secretion of α-syn. Future studies need to explore the possibilities that these mutants might have higher affinities for exosome-association, thus altering the total amount of α-syn that is packed in these vesicles, or whether they affect the total number or composition of exosomes
Collectively, all these novel findings support the idea for common mechanisms of neurodegeneration in which the critical events are protein misfolding, aggregate deposition and propagation [2, 3]. As such, development of novel therapies that would prevent and/or reverse the protein's conformational changes and that would halt the onset and progression of several neurodegenerative diseases require a precise understanding not only of why proteins misfold and accumulate locally, but also of how this pathology spreads through-out the brain [2, 3]
α-SYNUCLEIN SEEDING AND DISEASE PROPAGATION
The initiation step of protein propagation consists of the formation of pathogenic species, which are able of self-amplification by the recruitment of soluble proteins. The resulting malicious strains are then transferred from one cell to another via different routes [4] and thus the process of self-amplification continues in an uninterrupted manner (Fig. 1).
This process, termed seeding, is a nucleation-dependent mechanism in which the seed provides a template for the assembly of other soluble monomers and leads to the formation of highly ordered protein aggregates defined by their insolubility and β-sheet structure. The seeding mechanism, that was first described for prion protein (PrP) and later on for β-amyloid (Aβ), provided an explanation on the conversion of soluble proteins to amyloid fibrils
In mammalian cell lines, the addition of a small amount of exogenous Pffs has been shown to seed the formation of intracellular α-syn aggregates that recapitulate the main features of LBs [53, 54]. Using tagged Pffs, Luk et al., showed that the exogenous fibrils form the core of the inclusions, which are surrounded by non-tagged endogenous proteins, thus demonstrating that the Pffs play the role of the template and recruit soluble monomers (Fig. 1) [53]. A similar phenomenon was also observed in primary neuronal cultures [33, 55, 56]. For instance, the addition of human or mouse Pffs in primary cultures of hippocampal neurons induced the formation of LB-like structures [33, 55]. Moreover, using microfluidics systems, Volpicelli-Daley et al., and Tran et al., demonstrated that the formed α-syn inclusions are able to propagate from one neuron to another and to induce axonal degeneration and neuronal toxicity [33, 55].
Thereafter, several independent groups, using various well-characterized
It is worth noting that the inoculation of tissue homogenate from brains of PD patient [30] or symptomatic α-syn-transgenic mice [31] are also able to seed the aggregation of the endogenous protein and to initiate the formation of α-syn prion-like strains. Importantly, a recent study by Schweighauser et al., reported that the injection of α-syn-transgenic brain extracts, either fresh or fixed with formaldehyde, are able to induce α-syn pathology in mice brains. This observation demonstrates that homogenate of α-syn extracted from transgenic animals share a common and important prion propriety, which is the resistance to inactivation by formaldehyde [60].
Together, these observations demonstrate that seeding is an important step in α-syn propagation and represents a rate-limiting factor for the initiation of this process. However, the question of whether α-syn Pffs alone are capable of self-seeding or whether other proteins/factors are also required to augment this process is still under debate.
Although the central role of the seeding process on the initiation of α-syn cell-to-cell propagation has been demonstrated, the identity of the seed(s) that initiates the formation of pathogenic α-syn species in PD-diseased brains remains elusive.
Up to date, the available experimental data demonstrate that the exogenous α-syn amyloid-like fibrils are able to induce the aggregation of counterpart cytosolic soluble proteins in cell culture [33, 53] and
Together these observations suggest that the sequence-specific templating is important for α-syn seeding
Moreover, the fact that other amyloidogenic proteins, such as tau, Huntingtin (Htt) and Aβ, are able to promote α-syn aggregation suggests that these proteins could also initiate the heterotypic seeding of pathogenic α-syn. Indeed, tau and α-syn synergistically promote the fibrillization of each other in vitro [63] and in cell culture systems [64]. Moreover, both proteins co-localize in brains of PD patients and in transgenic animal models [36, 63], thus suggesting that tau could promote the formation of pathogenic α-syn species. Htt, a protein that is implicated in Huntington's disease (HD), is also capable of seeding α-syn aggregation. Several studies showed that Htt and α-syn co-aggregate
The idea of a possible propagation of pathogenic α-syn between the brain regions during the progression of PD was first suggested in the seminal work by Braak et al. [71]. In this study, the authors performed several longitudinal analyses to evaluate the neuroanatomical changes in the brain of PD patients and proposed a model in which the disease stages are correlated with the regional distribution of Lewy neurites (LN) and LB in the central nervous system. According to the Braak's model, LB and LN formation starts early in the disease (even before the motor symptoms emerge) and originate in the olfactory bulb and in the brainstem, specifically at the dorsal motor nucleus of the vagus nerve [72]. In parallel to disease progression, LB and LN are detected in other brain regions and appear to propagate through brain structures, in a stereotypic pattern, to reach the other regions including the midbrain and, at later stages, the cerebral cortex [38, 72]. Moreover, subsequent neuroanatomical investigations reported the presence of α-syn-positive inclusions in other organs, including the gastro-intestinal track, notably the gastric myenteric plexus [73] and colon [74], at the early stages of the disease, thus suggesting that the formation of pathogenic α-syn species could start outside the central nervous system. Together, these neuropathological observations suggest that the process of α-syn prion-like spreading could start in the brain, as well as in the peripheral nervous system.
Although the Braak's model remains under debate [75, 76], recent
In the recently developed animal models of α-syn prion-like propagation, based on the inoculation of exogenous α-syn Pffs, several intracerebral injections sites have been tested. In 2012, Luk et al., showed that the intra-cerebral delivery of Pffs induced the formation of intra-neuronal inclusions in naïve mouse brains. In this study, the authors inoculated α-syn Pffs in different brain regions, including the striatum, the cortex and the hippocampus and in all cases the delivery of the exogenous fibrils induced the formation of pathogenic inclusions and promoted their spreading from the injected site to different interconnected brain structures [34]. Other groups reproduced these observations in wild type and in M20 transgenic animal model of PD and confirmed that the intracerebral inoculation of Pffs in the hippocampus [59, 63] as well as in the midbrain (substantia nigra) [58] induced α-syn inclusion formation and protein propagation. Together, these observations demonstrate that any region in the brain represents a potential area where α-syn propagation could start.
Interestingly, a recent study by Sacino et al., reported that hind limb intramuscular injection of α-syn Pffs was sufficient to induce pathology in brains of transgenic mouse models of PD [37]. Two to four months post-injection, the authors detected pathogenic α-syn aggregates in the brains as well as in the spinal cord. Transection of the sciatic nerve innervating the injected muscle significantly delayed the appearance of the pathology in the brain, thus suggesting the implication of long-distance retrograde transport from the periphery to the central nervous system.
Taken together, neuropathological and experimental data demonstrate that the initiation of α-syn seeding and spreading could start at any structure in both peripheral and central nervous system. However, the possibility that the transmission of α-syn pathology described in the above studies might be attributed to the simple diffusion of the initial injected α-syn strains cannot be excluded. Therefore, future studies need to determine the relative contribution of local versus trans-synaptic transmission of pathogenic α-syn and which mechanisms are responsible for their long-distance axonal transportation.
α-SYNUCLEIN DEGRADATION AND POTENTIAL THERAPEUTIC TARGETS
Although the extent to which α-syn is involved in synucleinopathies is not yet clear, it is of high importance to elucidate the underlying mechanisms that regulate its protein levels. Considering that the levels of α-syn depend on a dynamic balance between the rate of protein synthesis and the rate of clearance mechanisms [4, 79, 80], an imbalance between these mechanisms caused by dysfunction of one or more of these pathways can result in abnormal levels of α-syn that might favor the formation and/or accumulation of oligomeric and fibrillar species that, as mentioned above, promote the pathology [4, 81].
Consequently, strategies aspiring to reduce the total protein burden of α-syn could be promising and could be proven therapeutic and beneficial to patients suffering from PD or other synucleinopathies [4, 81, 82]. One of these potential strategies would be the triggering of proteins involved in the clearance of α-syn [79]. Therefore, elucidation of the exact mechanisms that regulate the protein levels of α-syn is of high importance.
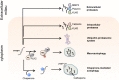
However, the simple question of how α-syn is degraded has caused much controversy and a body of conflicting results. Although the exact mechanisms are still unclear, overall, it has been demonstrated that intracellular α-syn monomers and aggregates are predominantly degraded by the ubiquitin-proteasome system (UPS) [83, 84, 85, 86] and the autophagy-lysosome pathway [85, 87, 88, 89], including chaperone-mediated autophagy (CMA) [90, 91]. Lysosomal cathepsins [92, 93, 94] and cytoplasmic calpains [95, 96, 97, 98] are considered to be major α-syn degrading enzymes, while kallikrein-related peptidase 6 (KLK6) has also been shown to play a pivotal role in proteolysis of intracellular α-syn (Fig. 2) [99, 100].
Over the recent years, an increasing body of evidence suggests that extracellular α-syn is quickly removed from the brain parenchyma by efficient cleaning and disposal mechanisms. Such clearance mechanisms include cell-mediated uptake and degradation, as well as proteolysis by extracellular proteases [79, 81]. In the context of cell-mediated uptake (endocytosis and phagocytosis), it has been suggested that adjacent cells, including neurons [40] and astrocytes [21], as well as, the innate (microglia) [101, 102] and adaptive immune system (infiltrating lymphocytes and autoantibodies) [103, 104, 105] are crucially involved. While, in the context of extracellular degradation, studies in cell-free systems and in cultured cells have implicated that certain matrix metalloproteinases (MMPs) - particularly MMP3 [10, 106], KLK6 [107, 108, 109] and plasmin [110] could degrade extracellular α-syn (Fig. 2).
Despite more than a decade of investigation, it is evident that the exact mechanisms and key players responsible for the clearance of α-syn are less studied. Accordingly, several questions need further investigation in the coming future. For example, only a small number of α-syn-degrading enzymes have been identified and our understanding of those characterized so far is incomplete. It is worth mentioning that there are still no studies available to ascertain whether the aforementioned α-syn-degrading enzymes are indeed able to catabolize extracellular α-syn
Little is known also about the ability of α-syn-degrading enzymes to cleave/degrade both monomeric and aggregated (oligomeric and fibrillar) α-syn forms that are increasingly assuming pathogenic significance promoting the protein spreading. For instance, studies in AD field showed the existence of conformation specific enzymes, as insulin-degrading enzyme (IDE), one of the major degrading enzymes of Aβ, selectively degrades the monomeric but not the oligomeric or fibrillar forms [111], thus suggesting that α-syn-degrading enzymes might also have differences in their substrate specificities. This is further supported by recent studies, which demonstrated the existence of diverse fibrillar α-syn strains with substantial differences in their proteolysis by proteinase K [36, 52]. Another critical query here is whether the produced proteolytic fragments are innocuous or even more toxic promoting further aggregation. For example, the proteolytic processing of α-syn by KLK6 [99] or plasmin [110] generates fragments that inhibit α-syn aggregation. On the other hand, the processing of α-syn by MMP3 [10, 112] or calpain I [95, 97] leads to fragments that promote α-syn aggregation, suggesting that these proteases may participate in the disease-linked α-syn aggregation in synucleinopathies and that cannot be promising therapeutic targets.
This is an area that merits more detailed exploration and in coming years the complete set of α-syn-degrading enzymes remains to be established and critically assessed. Although, up-regulation of these enzymes in well-characterized mouse models of PD could prove effective in the clearance of α-syn aberrant forms, complete elimination of extracellular α-syn is not desirable as it probably has a hitherto unidentified physiological role in brain parenchyma. In addition, another potential complication would be that the excessive protein degradation could be proven detrimental to neuronal integrity, since enzymes hydrolyze not only the targeted protein but also a range of other physiological substrates.
Another attractive direction of future research is to study human samples for differences in enzymatic activities between healthy and diseased individuals. However, a major obstacle in deciphering roles played by extracellular enzymes in the brain function and dysfunction is the apparent cellular and molecular complexity of the system. Indicatively, recent studies have revealed that certain proteases are also associated with exosomes, thus suggesting that these proteases may also contribute to extracellular proteolysis of their target substrates [113]. It is also known that many enzymes are often partnered with their specific inhibitors and act in very intricate cascades of enzymatic activities. In support of this, the degrading enzymes of extracellular α-syn, MMPs, KLK6 and plasmin, have been shown to cross talk each other in various systems [114, 115, 116]. Taking this into consideration, future studies should investigate the plausible assumption that α-syn-degrading enzymes work synergistically in complex proteolytic cascades [117, 118], along with other eliminative processes, in order to regulate the catabolism of extracellular α-syn and perhaps this of other amyloidogenic proteins, such as Aβ.
FUTURE DIRECTIONS
Studies of protein prion-like propagation have important implications in the understanding of the etiology and the progression of PD and other neurodegenerative disorders and the development of disease-modifying treatments aiming at stopping or slowing the disease progression. However, some important questions remain unexplored.
Over the recent years a broad spectrum of data have been accumulated pointing to an involvement of extracellular α-syn in PD and other synucleinopathies. The experimental models based on the inoculation of exogenous α-syn Pffs provide an important tool to explore the cellular and the molecular mechanisms of α-syn prion-like spreading. However, "Parkinson's disease is not a disorder in which somebody injects synuclein into your brain," (Prof. Ted Dawson, http://www.nature.com/news/misfolded-protein-transmits-parkinson-s-from-cell-to-cell-1.11838) and an important question remains to be elucidated concerning the origin and the identity of the endogenous seed(s).
Moreover, the majority of the existent evidence stems from experiments where primarily purified or synthetic forms of the proteins were used. Evidence from our laboratory for a differential susceptibility between recombinant and cell-secreted α-syn forms to KLK6-proteolysis raises new questions regarding their structural and functional properties [109]. The interaction of α-syn with lipids and other factors seems to alter its behavior and provide a biological context to understand that such modifications may force the protein to adopt multiple conformations and exert different functions. In support of this, it has been already demonstrated that cell-secreted α-syn forms are strong agonists of Toll-like receptor 2 (TLR2), which activates inflammatory responses in microglia, while the recombinant α-syn forms exhibited much less potency [102]. Given that the
In view of the importance of α-syn-degrading enzymes in α-syn-clearing mechanisms and in PD progression and given that α-syn may accumulate because of dysfunctional degradation, several genetic association studies should also be attempted to find potential links between defective proteases (e.g. polymorphisms or mutations) and PD pathology.
The future challenges for synucleionopathies are 1) to identify the origin of the pathogenic entities that initiate α-syn prion-like propagation; 2) to decipher the underlying propagation mechanisms, 3) to elucidate how and where this pathogenic process starts, and 4) to develop novel targeted strategies that facilitate the clearance of unwanted proteins but without disturbing the balance in protein homeostasis. Advanced understanding of the implications of protein degradation pathways will lead to diverse strategies for up-regulation of specific α-syn-degrading enzymes, either by means of gene therapy or by pharmacological modulation using small compounds that would modify the activity of the endogenous enzymes (or their inhibitors) at the transcriptional or protein level.
Figures
{kind=link}
{kind=link}
References
- Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 2003;4:49-60.

- Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013;501:45-51.
- Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 2014;20:130-138.
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 2013;14:38-48.
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 2006;52:33-38.
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995;14:467-475.
- Borghi R, Marchese R, Negro A, Marinelli L, Forloni G, Zaccheo D, Abbruzzese G, Tabaton M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson's disease and normal subjects. Neurosci Lett 2000;287:65-67.
- El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, Cookson MR, Hardy J, Allsop D. Alpha-synuclein implicated in Parkinson's disease is present in extracellular biological fluids, including human plasma. FASEB J 2003;17:1945-1947.
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Vekrellis EK. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 2010;30:6838-6851.
- Sung JY, Park SM, Lee CH, Um HJ, Lee JY, Kim J, Oh YJ, Lee ST, Paik SR, Chung KC. Proteolytic cleavage of extracellular secreted {alpha}-synuclein via matrix metalloproteinases. J Biol Chem 2005;280:25216-25224.
- Danzer KM, Ruf WP, Putcha P, Joyner D, Hashimoto T, Glabe C, Hyman BT, McLean PJ. Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J 2011;25:326-336.
- Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 2005;25:6016-6024.
- Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol 2009;10:148-155.
- Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of α-synuclein in neurological disorders. Lancet Neurol 2011;10:1015-1025.
- Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis 2012;3:e350.
- Roth DM, Balch WE. Modeling general proteostasis: proteome balance in health and disease. Curr Opin Cell Biol 2011;23:126-134.
- Sung JY, Kim J, Paik SR, Park JH, Ahn YS, Chung KC. Induction of neuronal cell death by Rab5A-dependent endocytosis of alpha-synuclein. J Biol Chem 2001;276:27441-27448.
- Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J 2005;19:533-542.
- Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson's disease. Neurobiol Aging 2008;29:1690-1701.
- Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B, Thomas MP, Mosley RL, Gendelman HE. Nitrated alpha-synuclein-activated microglial profiling for Parkinson's disease. J Neurochem 2008;104:1504-1525.
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 2010;285:9262-9272.
- Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Fernandez-Montesinos R, Caro M, Lachaud CC, Waudby CA, Delgado M, Dobson CM, Pozo D. Glial innate immunity generated by non-aggregated alpha-synuclein in mouse: differences between wild-type and Parkinson's disease-linked mutants. PLoS One 2010;5:e13481.
- Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res 2011;69:337-342.
- Couch Y, Alvarez-Erviti L, Sibson NR, Wood MJ, Anthony DC. The acute inflammatory response to intranigral α-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation 2011;8:166.
- Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci 2007;27:9220-9232.
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med 2008;14:504-506.
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med 2008;14:501-503.
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009;106:13010-13015.
- Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 2011;121:715-725.
- Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, Fariñas I, Obeso JA, Bezard E, Vila M. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 2014;75:351-362.
- Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L, Legastelois S, Baron T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 2012;33:2225-2228.
- Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B. Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J Neurochem 2009;111:192-203.
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011;72:57-71.
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012;338:949-953.
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 2012;209:975-986.
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 2013;154:103-117.
- Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, McGarvey NH, Ayers JI, Notterpek L, Borchelt DR, Golde TE, Giasson BI. Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci U S A 2014;111:10732-10737.
- Visanji NP, Brooks PL, Hazrati LN, Lang AE. The prion hypothesis in Parkinson's disease: braak to the future. Acta Neuropathol Commun 2013;1:2.
- Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem 2010;113:1263-1274.
- Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol 2008;40:1835-1849.
- Vella LJ, Sharples RA, Lawson VA, Masters CL, Cappai R, Hill AF. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J Pathol 2007;211:582-590.
- Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 2004;279:46363-46366.
- Park JY, Kim KS, Lee SB, Ryu JS, Chung KC, Choo YK, Jou I, Kim J, Park SM. On the mechanism of internalization of alpha-synuclein into microglia: roles of ganglioside GM1 and lipid raft. J Neurochem 2009;110:400-411.
- Simons M, Raposo G. Exosomes--vesicular carriers for intercellular communication. Curr Opin Cell Biol 2009;21:575-581.
- Chivet M, Hemming F, Pernet-Gallay K, Fraboulet S, Sadoul R. Emerging role of neuronal exosomes in the central nervous system. Front Physiol 2012;3:145.
- Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp Mol Med 2013;45:e22.
- Kondo K, Obitsu S, Teshima R. α-Synuclein aggregation and transmission are enhanced by leucine-rich repeat kinase 2 in human neuroblastoma SH-SY5Y cells. Biol Pharm Bull 2011;34:1078-1083.
- Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, Lee HJ, Kim S, Masliah E, Sardi SP, Lee SJ. Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat Commun 2014;5:4755.
- Come JH, Fraser PE, Lansbury PT. A kinetic model for amyloid formation in the prion diseases: importance of seeding. Proc Natl Acad Sci U S A 1993;90:5959-5963.
- Jarrett JT, Lansbury PT. Seeding "one-dimensional crystallization" of amyloid: a pathogenic mechanism in Alzheimers disease and scrapie?. Cell 1993;73:1055-1058.
- Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J Biol Chem 1999;274:19509-19512.
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R. Structural and functional characterization of two alpha-synuclein strains. Nat Commun 2013;4:2575.
- Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alphasynuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 2009;106:20051-20056.
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014;82:1271-1288.
- Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM. Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep 2014;7:2054-2065.
- Sacino AN, Thomas MA, Ceballos-Diaz C, Cruz PE, Rosario AM, Lewis J, Giasson BI, Golde TE. Conformational templating of α-synuclein aggregates in neuronal-glial cultures. Mol Neurodegener 2013;8:17.
- Sacino AN, Brooks M, McKinney AB, Thomas MA, Shaw G, Golde TE, Giasson BI. Brain injection of α-synuclein induces multiple proteinopathies, gliosis, and a neuronal injury marker. J Neurosci 2014;34:12368-12378.
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M. Prion-like spreading of pathological α-synuclein in brain. Brain 2013;136:1128-1138.
- Sacino AN, Brooks M, McGarvey NH, McKinney AB, Thomas MA, Levites Y, Ran Y, Golde TE, Giasson BI. Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol Commun 2013;1:38.
- Schweighauser M, Bacioglu M, Fritschi S, Shimshek D, Kahle P, Eisele Y, Jucker M. Formaldehyde-fixed brain tissue from spontaneously ill α-synuclein transgenic mice induces fatal α-synucleinopathy in transgenic hosts. Acta Neuropathol 2014
- Krammer C, Schätzl HM, Vorberg I. Prion-like propagation of cytosolic protein aggregates: insights from cell culture models. Prion 2009;3:206-212.
- Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem 2001;276:2380-2386.
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003;300:636-640.
- Badiola N, de Oliveira RM, Herrera F, Guardia-Laguarta C, Gonçalves SA, Pera M, Suárez-Calvet M, Clarimon J, Outeiro TF, Lleó A. Tau enhances α-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS One 2011;6:e26609.
- Pieri L, Madiona K, Bousset L, Melki R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys J 2012;102:2894-2905.
- Herrera F, Outeiro TF. α-Synuclein modifies huntingtin aggregation in living cells. FEBS Lett 2012;586:7-12.
- Charles V, Mezey E, Reddy PH, Dehejia A, Young TA, Polymeropoulos MH, Brownstein MJ, Tagle DA. Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington's disease patients and transgenic mouse models. Neurosci Lett 2000;289:29-32.
- Corrochano S, Renna M, Carter S, Chrobot N, Kent R, Stewart M, Cooper J, Brown SD, Rubinsztein DC, Acevedo-Arozena A. α-Synuclein levels modulate Huntingtons disease in mice. Hum Mol Genet 2012;21:485-494.
- Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci U S A 2001;98:12245-12250.
- Pletnikova O, West N, Lee MK, Rudow GL, Skolasky RL, Dawson TM, Marsh L, Troncoso JC. Abeta deposition is associated with enhanced cortical alpha-synuclein lesions in Lewy body diseases. Neurobiol Aging 2005;26:1183-1192.
- Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rüb U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (preclinical and clinical stages). J Neurol 2002;249:III/1-III/5.
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197-211.
- Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci Lett 2006;396:67-72.
- Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson's disease: frequency and pathophysiology. Neurology 1992;42:726-732.
- Burke RE, Dauer WT, Vonsattel JP. A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 2008;64:485-491.
- Jellinger KA. A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 2009;1792:730-740.
- Ulusoy A, Rusconi R, Pérez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, Di Monte DA. Caudorostral brain spreading of α-synuclein through vagal connections. EMBO Mol Med 2013;5:1051-1059.
- Rey NL, Petit GH, Bousset L, Melki R, Brundin P. Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathologica 2013;126:555-573.
- Park SM, Kim KS. Proteolytic clearance of extracellular α-synuclein as a new therapeutic approach against Parkinson disease. Prion 2013;7:121-126.
- Xilouri M, Brekk OR, Stefanis L. α-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol 2013;47:537-551.
- Lee HJ, Bae EJ, Lee SJ. Extracellular α--synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol 2014;10:92-98.
- Vekrellis K, Stefanis L. Targeting intracellular and extracellular alpha-synuclein as a therapeutic strategy in Parkinson's disease and other synucleinopathies. Expert Opin Ther Targets 2012;16:421-432.
- McNaught KS, Mytilineou C, Jnobaptiste R, Yabut J, Shashidharan P, Jennert P, Olanow CW. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J Neurochem 2002;81:301-306.
- McNaught KS, Björklund LM, Belizaire R, Isacson O, Jenner P, Olanow CW. Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport 2002;13:1437-1441.
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003;278:25009-25013.
- Emmanouilidou E, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging 2010;31:953-968.
- Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, Ischiropoulos H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci 2001;21:8053-8061.
- Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 2004;24:1888-1896.
- Oueslati A, Schneider BL, Aebischer P, Lashuel HA. Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc Natl Acad Sci U S A 2013;110:E3945-E3954.
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004;305:1292-1295.
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 2008;283:23542-23556.
- Sevlever D, Jiang P, Yen SH. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008;47:9678-9687.
- Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S, Xie ZL, Speake LD, Parks R, Crabtree D, Liang Q, Crimmins S, Schneider L, Uchiyama Y, Iwatsubo T, Zhou Y, Peng L, Lu Y, Standaert DG, Walls KC, Shacka JJ, Roth KA, Zhang J. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain 2008;1:17.
- Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, Boston H, Saftig P, Woulfe J, Feany MB, Myllykangas L, Schlossmacher MG, Tyynelä J. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain 2009;2:5.
- Mishizen-Eberz AJ, Guttmann RP, Giasson BI, Day GA, Hodara R, Ischiropoulos H, Lee VM, Trojanowski JQ, Lynch DR. Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro. J Neurochem 2003;86:836-847.
- Kim HJ, Lee D, Lee CH, Chung KC, Kim J, Paik SR. Calpain-resistant fragment(s) of alpha-synuclein regulates the synuclein-cleaving activity of 20S proteasome. Arch Biochem Biophys 2006;455:40-47.
- Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, Oxford JT, Feany MB, Masliah E, Rohn TT. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol 2007;170:1725-1738.
- Esteves AR, Arduíno DM, Swerdlow RH, Oliveira CR, Cardoso SM. Dysfunctional mitochondria uphold calpain activation: contribution to Parkinson's disease pathology. Neurobiol Dis 2010;37:723-730.
- Iwata A, Maruyama M, Akagi T, Hashikawa T, Kanazawa I, Tsuji S, Nukina N. Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum Mol Genet 2003;12:2625-2635.
- Spencer B, Michael S, Shen J, Kosberg K, Rockenstein E, Patrick C, Adame A, Masliah E. Lentivirus mediated delivery of neurosin promotes clearance of wild-type α-synuclein and reduces the pathology in an α-synuclein model of LBD. Mol Ther 2013;21:31-41.
- Lee HJ, Suk JE, Bae EJ, Lee SJ. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem Biophys Res Commun 2008;372:423-428.
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 2013;4:1562.
- Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron 2005;46:857-868.
- Reynolds AD, Stone DK, Mosley RL, Gendelman HE. Nitrated {alpha}-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J Immunol 2009;182:4137-4149.
- Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, Mueller-Steiner S, Seubert P, Barbour R, McConlogue L, Buttini M, Games D, Schenk D. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 2011;6:e19338.
- Levin J, Giese A, Boetzel K, Israel L, Högen T, Nübling G, Kretzschmar H, Lorenzl S. Increased alpha-synuclein aggregation following limited cleavage by certain matrix metalloproteinases. Exp Neurol 2009;215:201-208.
- Kasai T, Tokuda T, Yamaguchi N, Watanabe Y, Kametani F, Nakagawa M, Mizuno T. Cleavage of normal and pathological forms of alpha-synuclein by neurosin in vitro. Neurosci Lett 2008;436:52-56.
- Tatebe H, Watanabe Y, Kasai T, Mizuno T, Nakagawa M, Tanaka M, Tokuda T. Extracellular neurosin degrades α-synuclein in cultured cells. Neurosci Res 2010;67:341-346.
- Ximerakis M, Pampalakis G, Roumeliotis TI, Sykioti VS, Garbis SD, Stefanis L, Sotiropoulou G, Vekrellis K. Resistance of naturally secreted α-synuclein to proteolysis. FASEB J 2014;28:3146-3158.
- Kim KS, Choi YR, Park JY, Lee JH, Kim DK, Lee SJ, Paik SR, Jou I, Park SM. Proteolytic cleavage of extracellular α-synuclein by plasmin: implications for Parkinson disease. J Biol Chem 2012;287:24862-24872.
- Saido T, Leissring MA. Proteolytic degradation of amyloid β-protein. Cold Spring Harb Perspect Med 2012;2:a006379.
- Choi DH, Kim YJ, Kim YG, Joh TH, Beal MF, Kim YS. Role of matrix metalloproteinase 3-mediated alpha-synuclein cleavage in dopaminergic cell death. J Biol Chem 2011;286:14168-14177.
- Shimoda M, Khokha R. Proteolytic factors in exosomes. Proteomics 2013;13:1624-1636.
- Blaber SI, Yoon H, Scarisbrick IA, Juliano MA, Blaber M. The autolytic regulation of human kallikrein-related peptidase 6. Biochemistry 2007;46:5209-5217.
- Baramova EN, Bajou K, Remacle A, L'Hoir C, Krell HW, Weidle UH, Noel A, Foidart JM. Involvement of PA/plasmin system in the processing of pro-MMP-9 and in the second step of pro-MMP-2 activation. FEBS Lett 1997;405:157-162.
- Okumura Y, Sato H, Seiki M, Kido H. Proteolytic activation of the precursor of membrane type 1 matrix metalloproteinase by human plasmin. A possible cell surface activator. FEBS Lett 1997;402:181-184.
- Sotiropoulou G, Pampalakis G, Diamandis EP. Functional roles of human kallikrein-related peptidases. J Biol Chem 2009;284:32989-32994.
- Kessenbrock K, Plaks V, Werb Z. Matrix metallo-proteinases: regulators of the tumor microenvironment. Cell 2010;141:52-67.