Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach

 ,
,  ,
,
Abstract
:1. Introduction
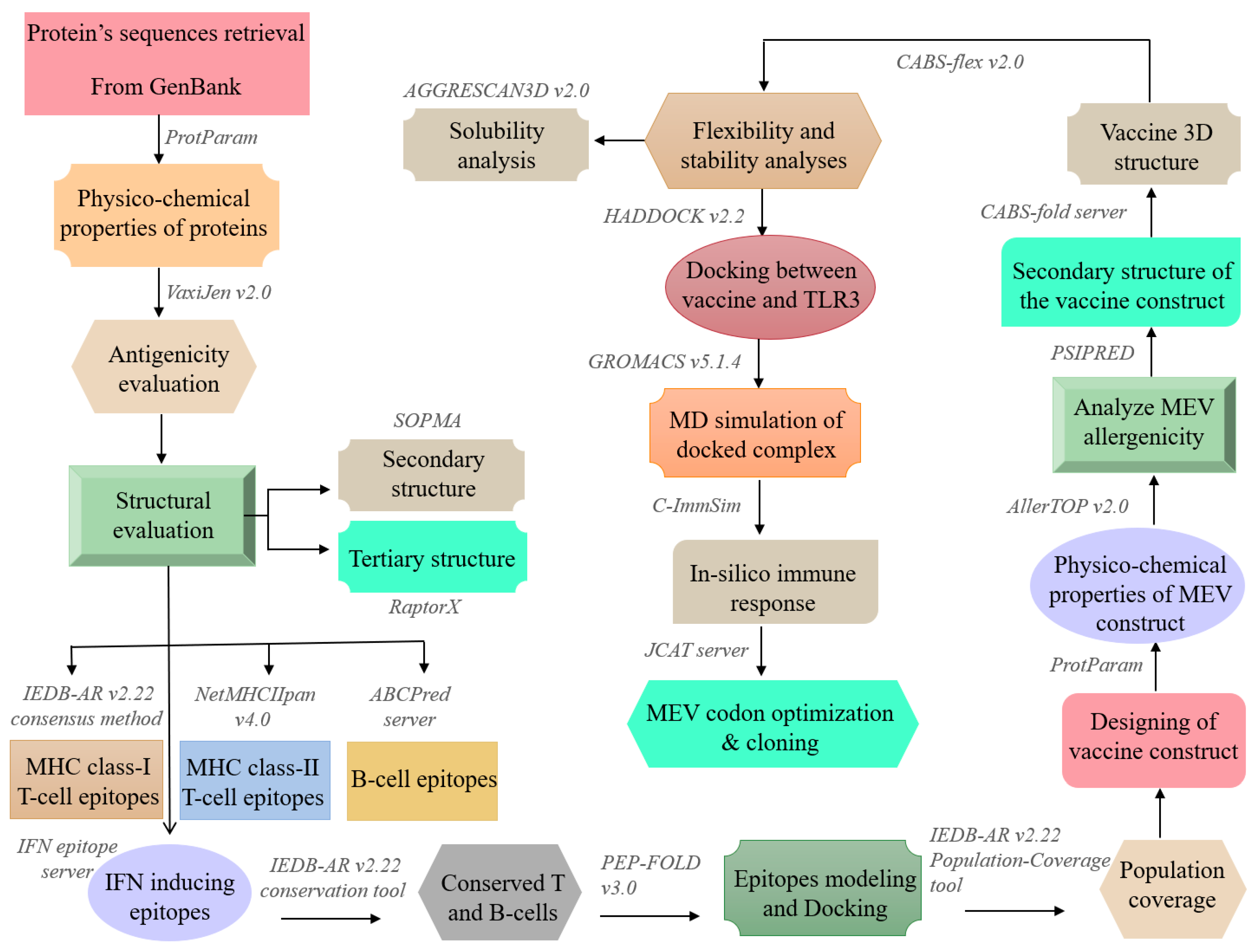
2. Materials and Methods
2.1. Immunoinformatic Analyses of the Antigen
2.1.1. Proteins Sequence Retrieval, Antigenicity Prediction, Physicochemical Properties, and Structural Evaluation
2.1.2. Prediction and Assessment of T-Cell Epitopes
2.1.3. Prediction and Assessment of B-Cell Epitopes
2.1.4. Conservation Analysis and Shortlisting of Predicted Epitopes
2.1.5. Epitopes Modeling and Molecular Docking
2.1.6. Assessment of Population Coverage
2.2. Multiple-Epitope Vaccines Designing and Evaluation
2.2.1. Designing of Vaccine Construct
2.2.2. Primary and Secondary Structural Analyses
2.2.3. Tertiary Structure Prediction, Refinement and Validation
2.2.4. Screening for B-Cell Epitopes
2.2.5. Molecular Docking between Vaccine and TLR3
2.2.6. Molecular Dynamics Simulation Analysis
2.2.7. MM/PBSA Binding Free Energy Calculation
2.3. Immunogenicity Evaluation of the Vaccine Construct
2.4. Codon Optimization and In Silico Cloning
2.5. Data Availability
3. Results
3.1. Pre-Vaccine Design Analyses
3.1.1. Target Proteins Sequence and Structural Analyses
3.1.2. Evaluation and Selection of Epitope
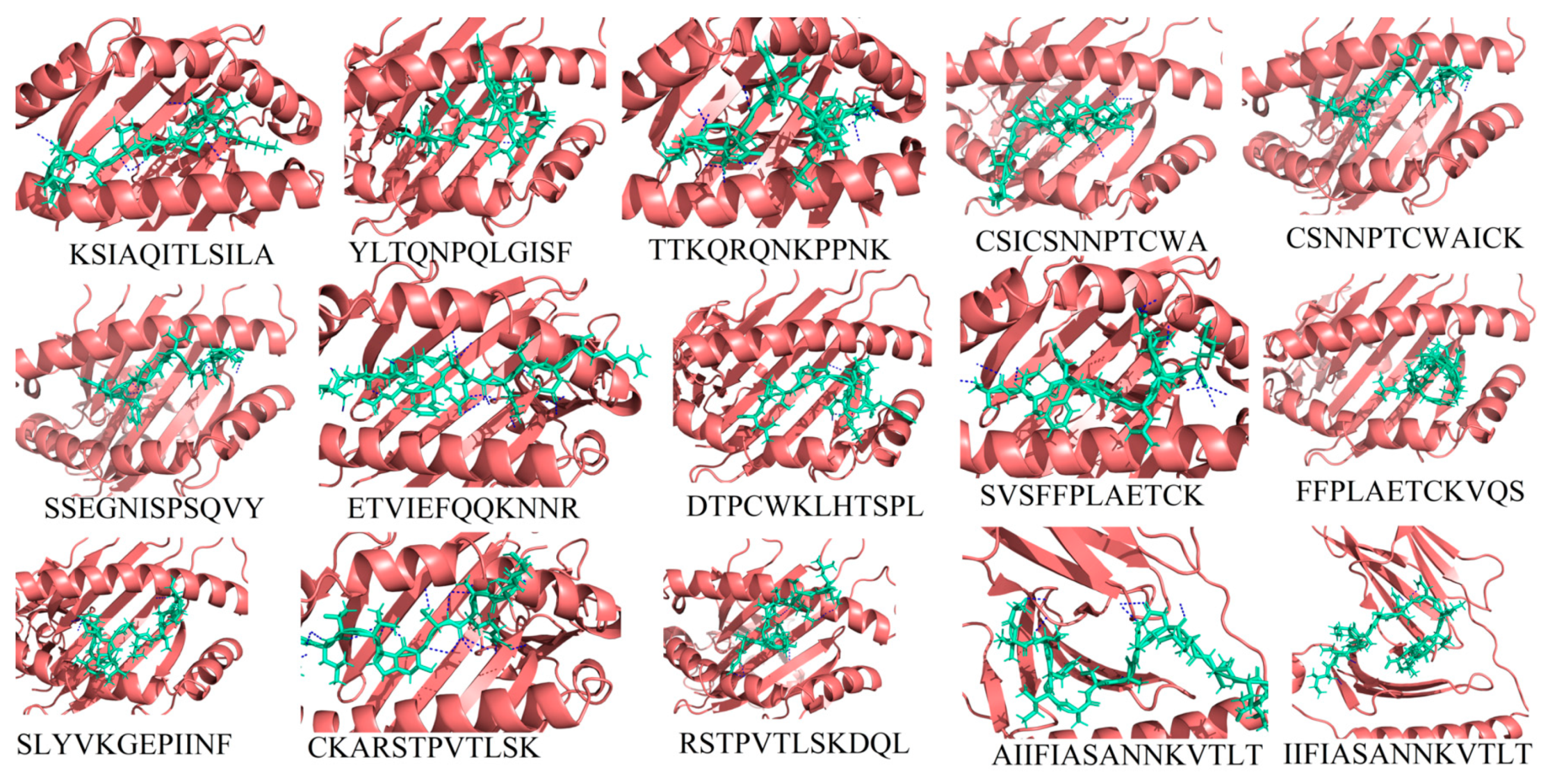
3.1.3. Molecular Docking between Epitopes and HLA Alleles
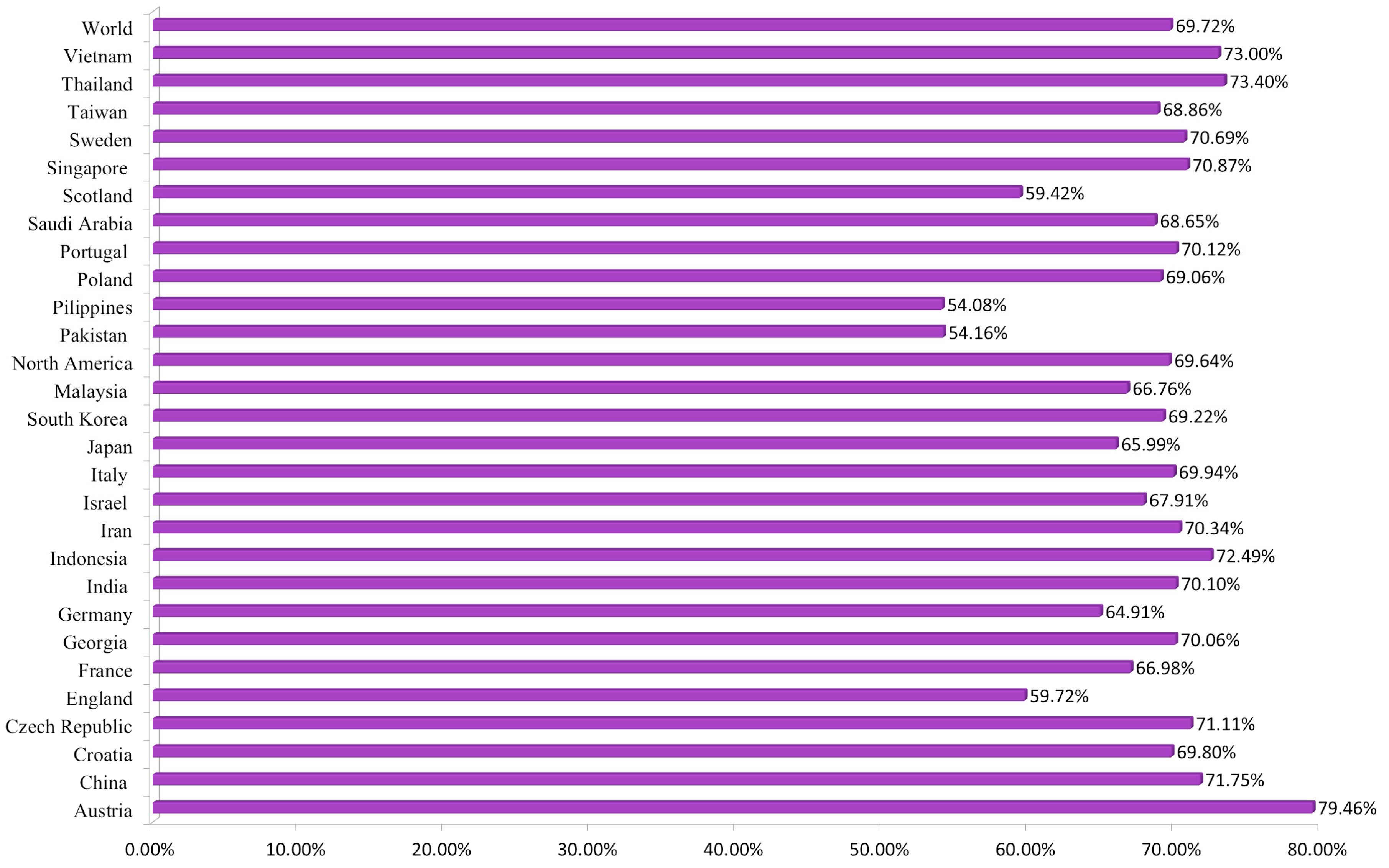
3.1.4. Population Coverage Analysis
3.2. Multiepitope Based Sub-Unit Vaccine Design and Validation
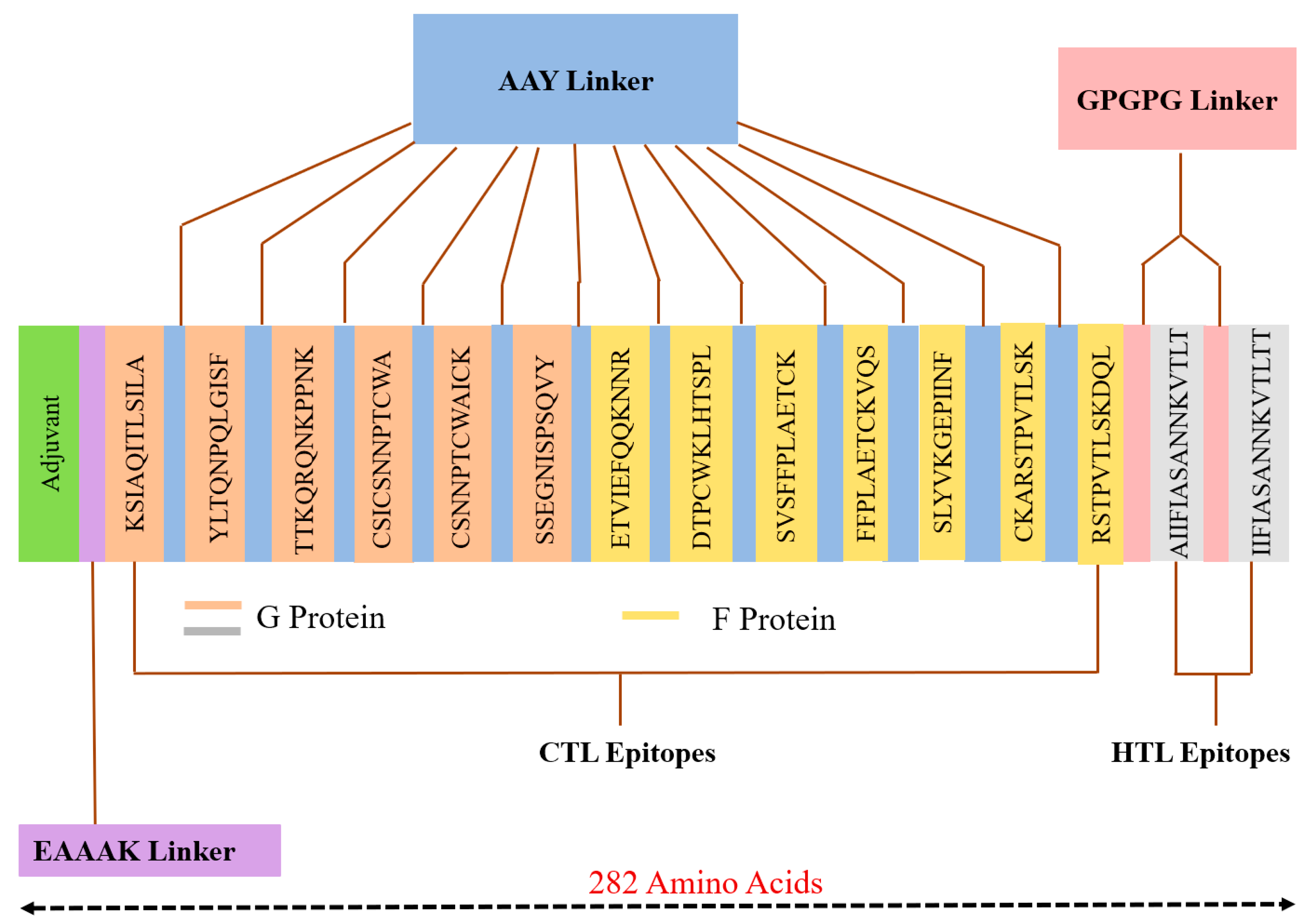
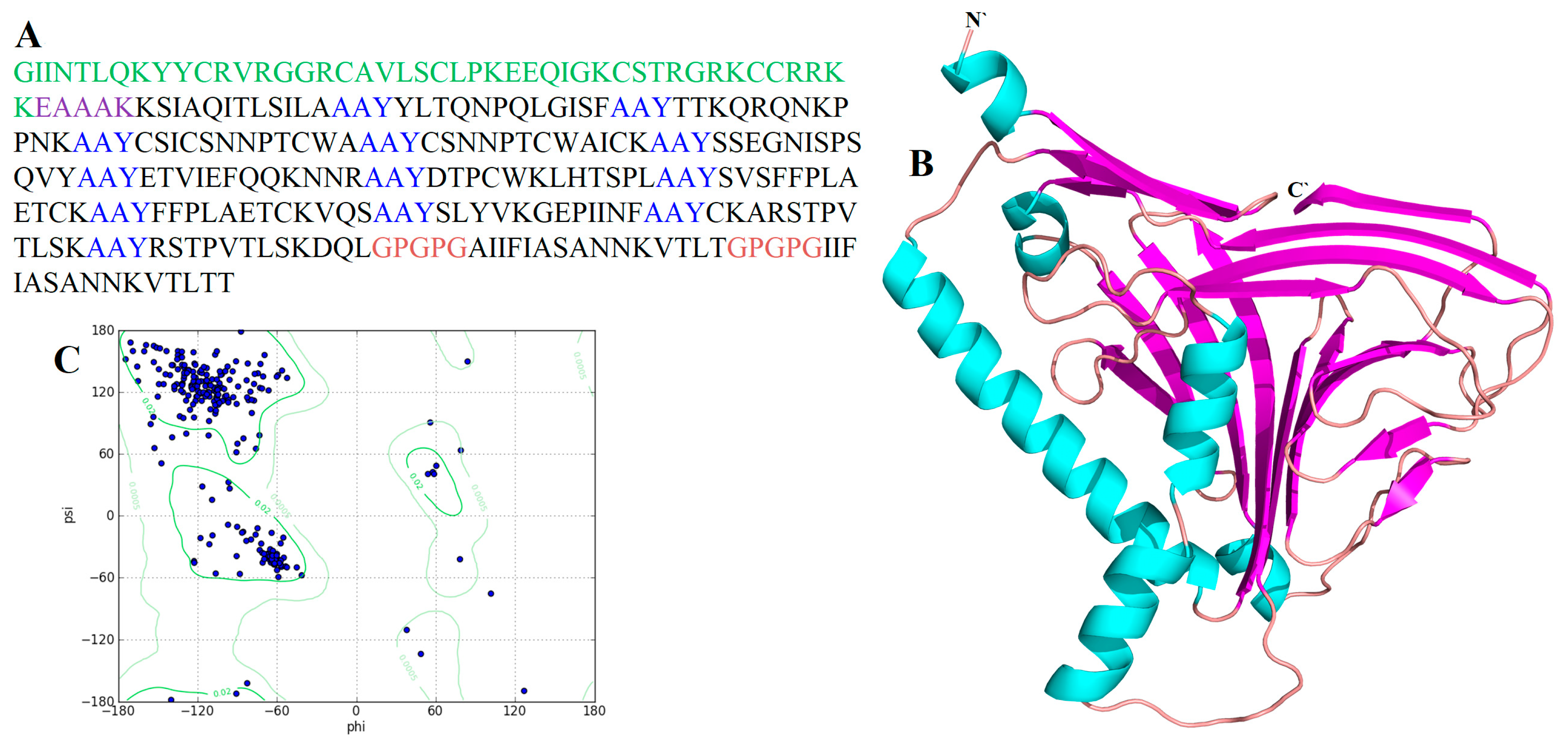
3.2.1. Construction of MEV
3.2.2. Physicochemical and Immunogenic Profiling
3.2.3. Secondary Structure Analysis
3.2.4. Tertiary Structure Prediction, Refinement and Validation
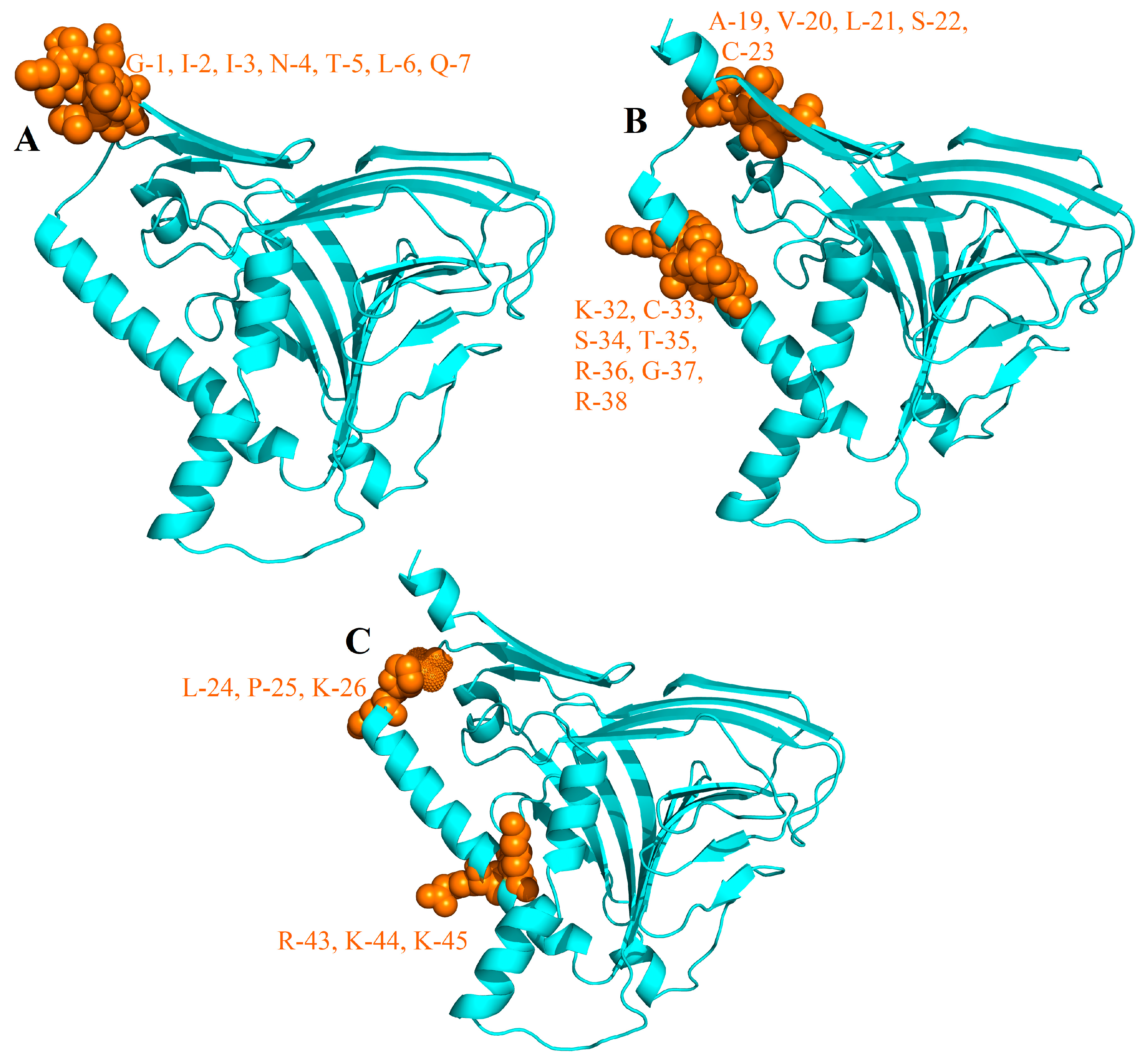
3.2.5. B-Cells Epitopes in MEV
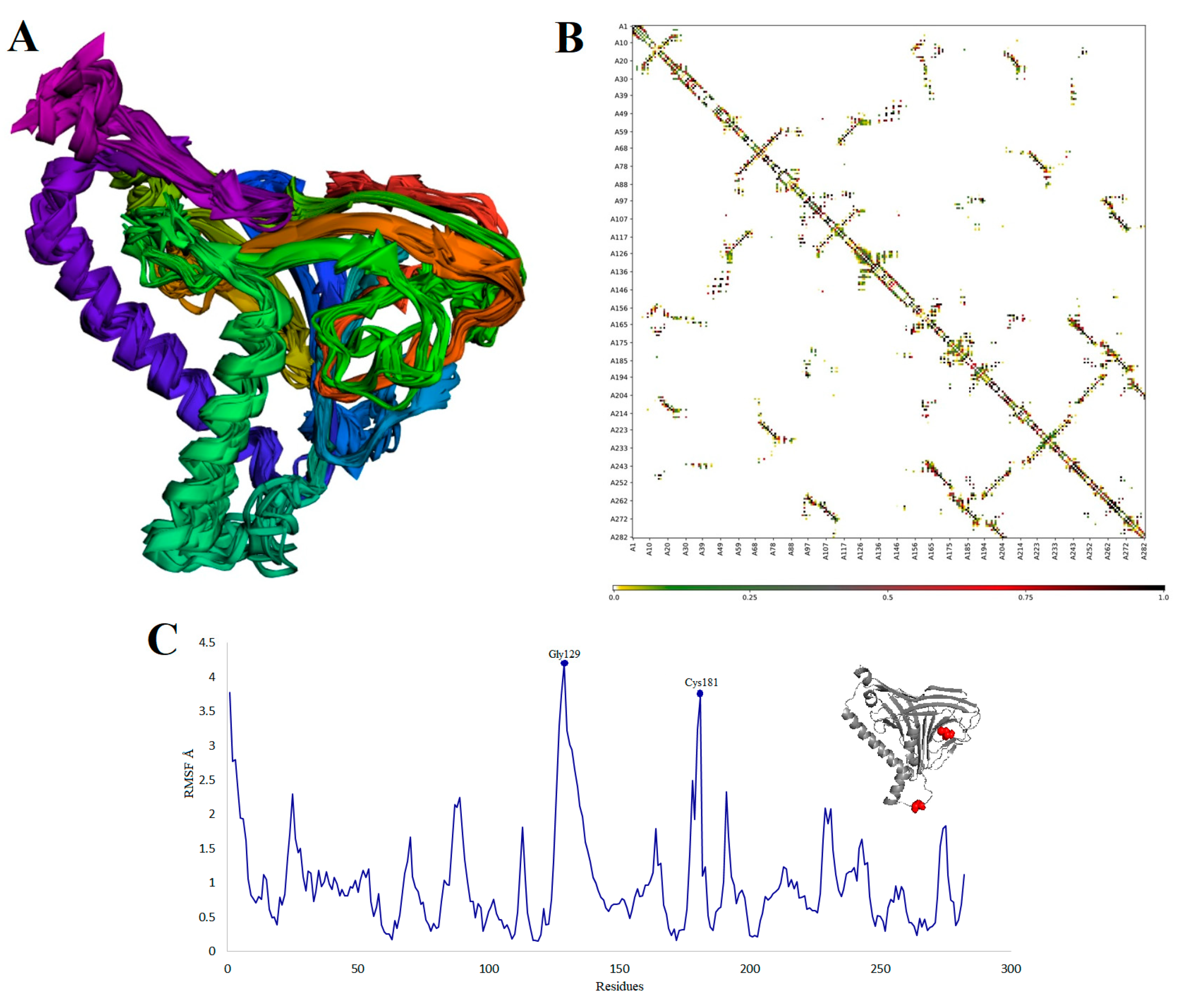
3.2.6. Interaction Analysis between Vaccine and TLR-3
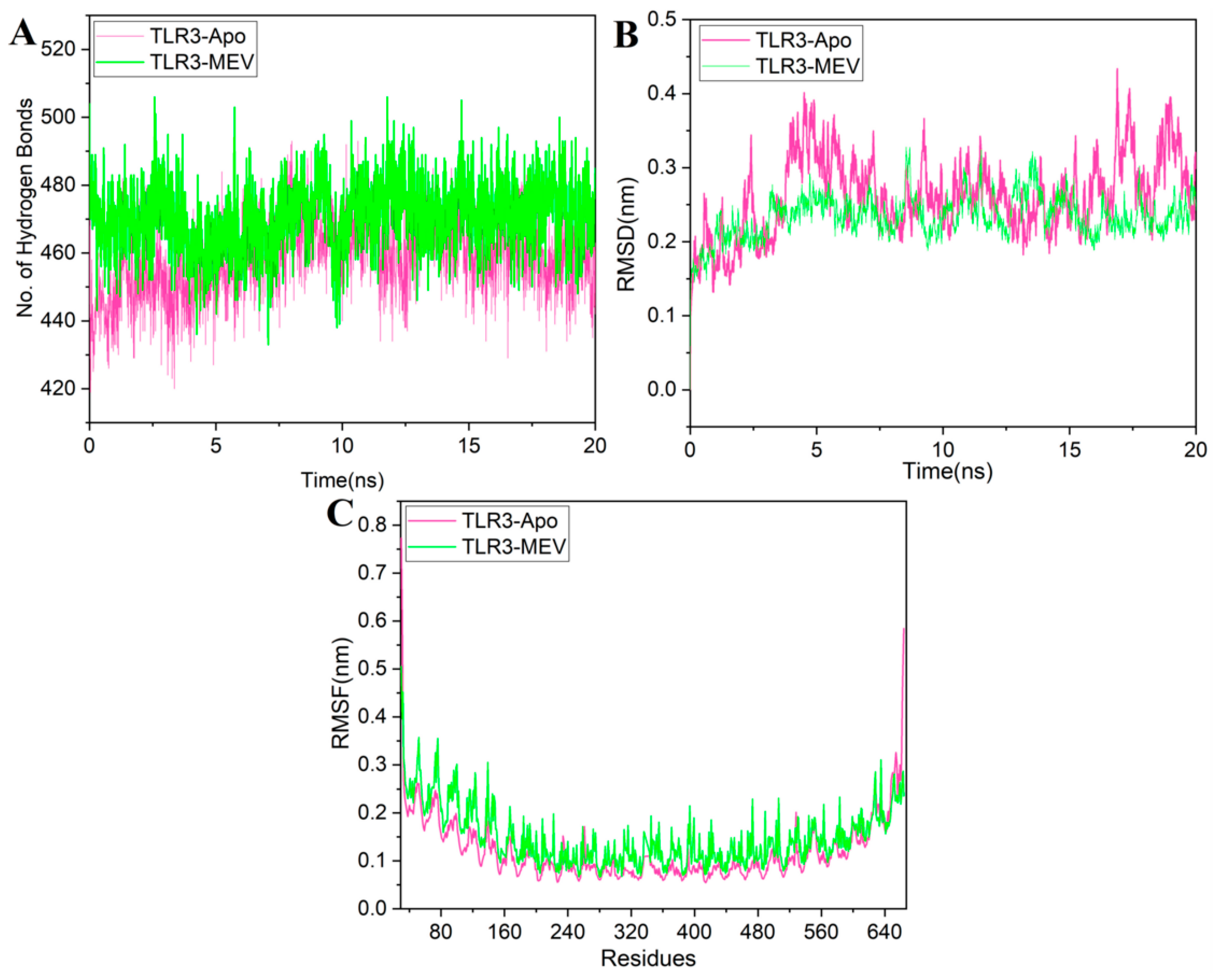
3.2.7. The Structural Integrity of the Vaccine-TLR3 Complex
3.2.8. MM/PBSA Binding Free Energy Calculation of the Simulated Vaccine-TLR3 Complex
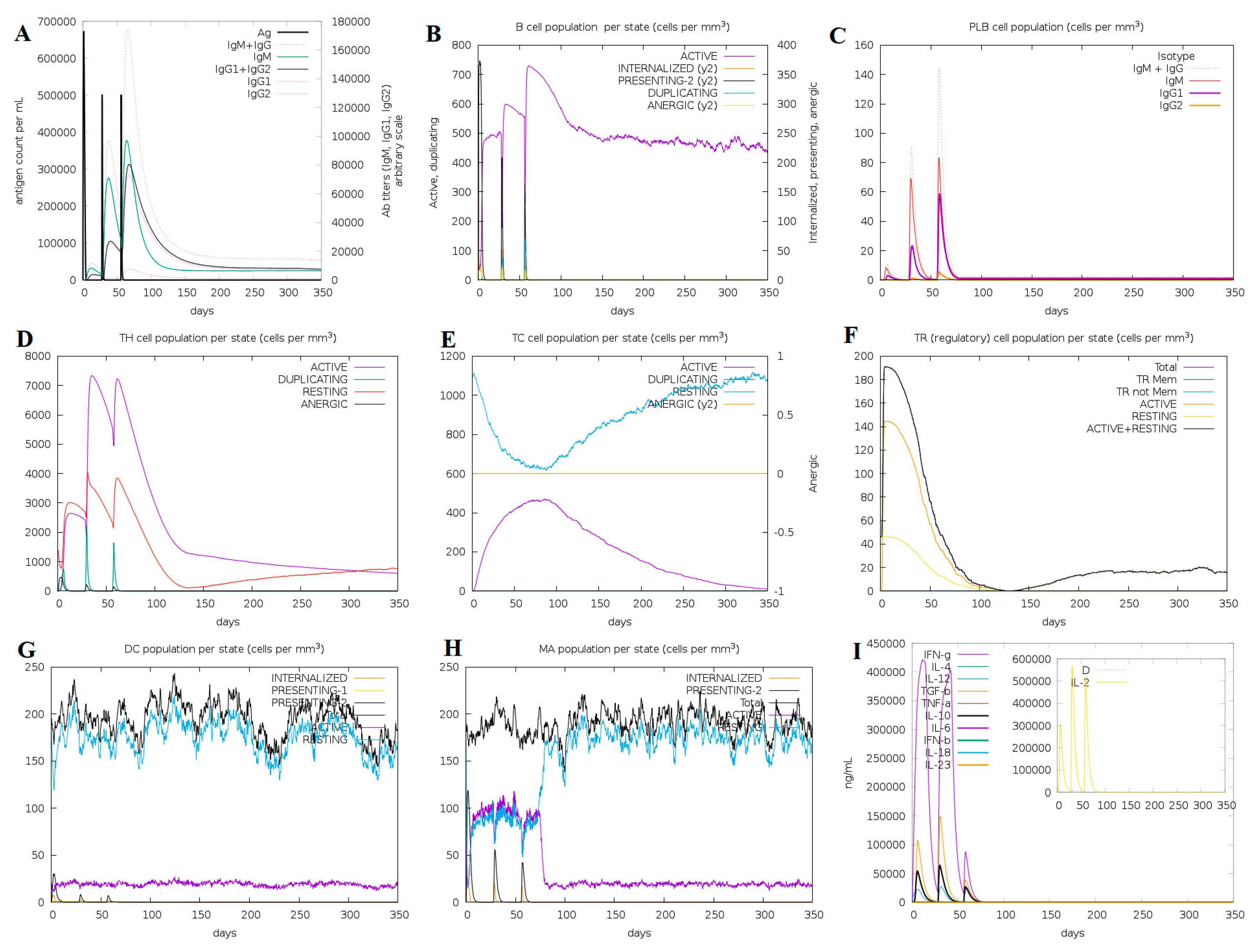
3.3. Immune Simulation for Vaccine Efficacy
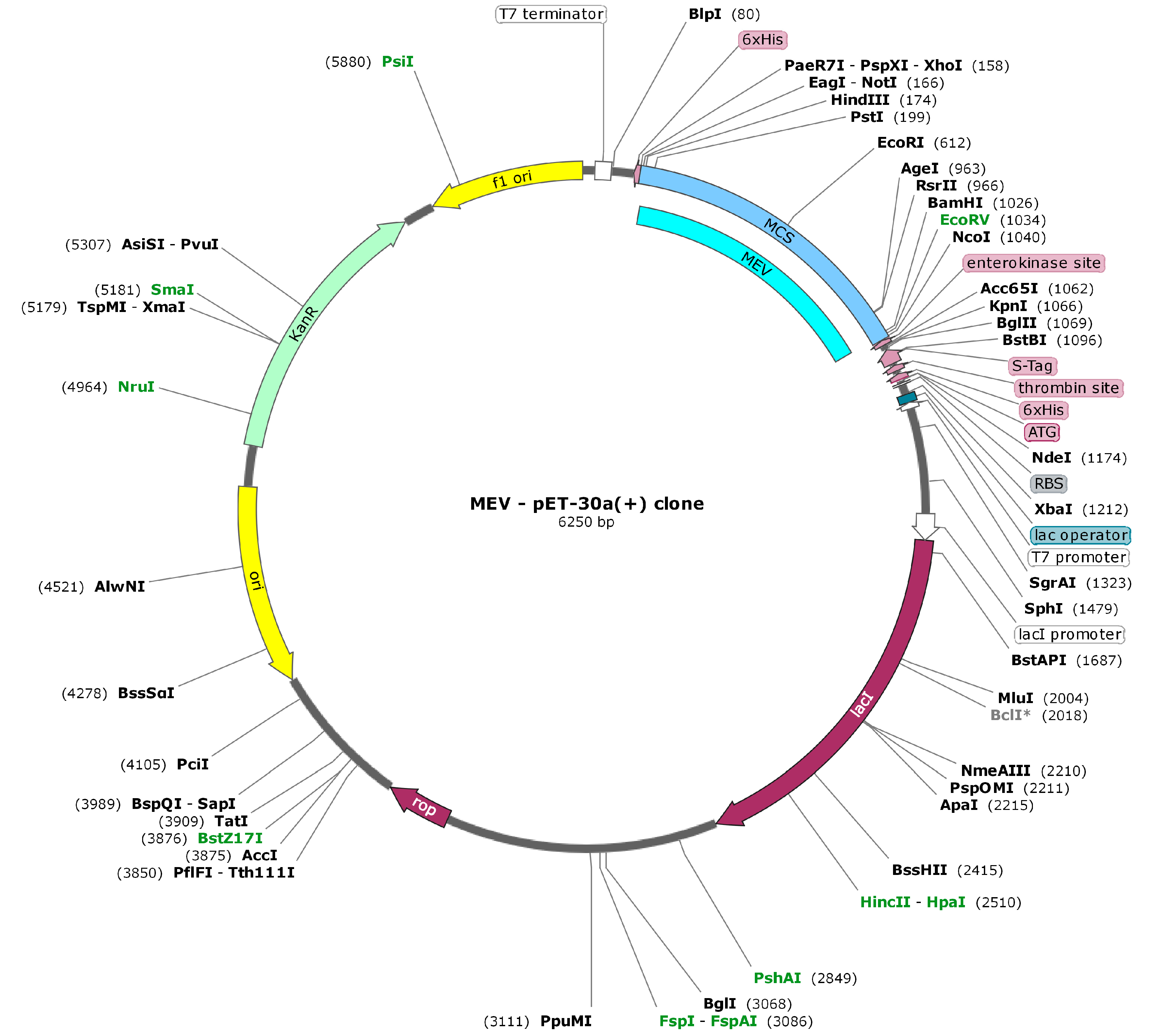
3.4. In Silico Cloning within E. coli System
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Afonso, C.L.; Amarasinghe, G.K.; Bányai, K.; Bào, Y.; Basler, C.F.; Bavari, S.; Bejerman, N.; Blasdell, K.R.; Briand, F.-X.; Bries, T. Taxonomy of the order Mononegavirales: Update 2016. Arch. Virol. 2016, 161, 2351–2360. [Google Scholar] [CrossRef]
- Mufson, M.A.; Örvell, C.; Rafnar, B.; Norrby, E. Two distinct subtypes of human respiratory syncytial virus. J. Gen. Virol. 1985, 66, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Thongpan, I.; Mauleekoonphairoj, J.; Vichiwattana, P.; Korkong, S.; Wasitthankasem, R.; Vongpunsawad, S.; Poovorawan, Y. Respiratory syncytial virus genotypes NA1, ON1, and BA9 are prevalent in Thailand, 2012–2015. PeerJ 2017, 5, e3970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, P.L.; Fearns, R.; Graham, B.S. Respiratory syncytial virus: Virology, reverse genetics, and pathogenesis of disease. In Challenges and Opportunities for Respiratory Syncytial Virus Vaccines; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–38. [Google Scholar]
- Carvajal, J.J.; Avellaneda, A.M.; Salazar-Ardiles, C.N.; Maya, J.E.; Kalergis, A.M.; Lay, M.K.-L. Host components contributing to respiratory syncytial virus pathogenesis. Front. Immunol. 2019, 10, 2152. [Google Scholar] [PubMed]
- Mastrangelo, P.; Hegele, R.G. RSV fusion: Time for a new model. Viruses 2013, 5, 873–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigwood, N.L.; Walker, C.M. Commissioned paper: Comparison of immunity to pathogens in humans, chimpanzees, and macaques. In Chimpanzees in Biomedical and Behavioral Research: Assessing the Necessity; National Academies Press (US): Washington, DC, USA, 2011. [Google Scholar]
- Schweitzer, J.W.; Justice, N.A. Respiratory Syncytial Virus Infection (RSV). Available online: https://www.ncbi.nlm.nih.gov/books/NBK459215/#__NBK459215_ai__ (accessed on 16 December 2019).
- Griffiths, C.; Drews, S.J.; Marchant, D.J. Respiratory syncytial virus: Infection, detection, and new options for prevention and treatment. Clin. Microbiol. Rev. 2017, 30, 277–319. [Google Scholar]
- Glenn, G.M.; Fries, L.F.; Thomas, D.N.; Smith, G.; Kpamegan, E.; Lu, H.; Flyer, D.; Jani, D.; Hickman, S.P.; Piedra, P.A. A randomized, blinded, controlled, dose-ranging study of a respiratory syncytial virus recombinant fusion (F) nanoparticle vaccine in healthy women of childbearing age. J. Infect. Dis. 2016, 213, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.S. Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol. Rev. 2011, 239, 149–166. [Google Scholar] [CrossRef] [Green Version]
- Crowe, J.E., Jr. Respiratory syncytial virus vaccine development. Vaccine 2001, 20, S32–S37. [Google Scholar] [CrossRef]
- Shafique, M.; Zahoor, M.A.; Arshad, M.I.; Aslam, B.; Siddique, A.B.; Rasool, M.H.; Qamar, M.U.; Usman, M. Hurdles in Vaccine Development against Respiratory Syncytial Virus. In The Burden of Respiratory Syncytial Virus Infection in the Youn; IntechOpen: London, UK, 2019. [Google Scholar]
- María, R.; Arturo, C.; Alicia, J.A.; Paulina, M.; Gerardo, A.O. The Impact of Bioinformatics on Vaccine Design and Development; InTech: Rijeka, Croatia, 2017. [Google Scholar]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Kumar Pandey, R.; Ojha, R.; Mishra, A.; Kumar Prajapati, V. Designing B-and T-cell multi-epitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. J. Cell. Biochem. 2018, 119, 7631–7642. [Google Scholar] [CrossRef] [Green Version]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef]
- Stasyk, T.; Huber, L. Spatio-temporal parameters of endosomal signaling in cancer: Implications for new treatment options. J. Cell. Biochem. 2016, 117, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.C.; Ren, E.C. Immunoinformatics: Current trends and future directions. Drug Discov. Today 2009, 14, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Bari, A.; Adeel, M.M.; Maryam, A.; Ashfaq, U.A.; Du, X.; Muneer, I.; Ahmad, H.I.; Wang, J. Peptide vaccine against chikungunya virus: Immuno-informatics combined with molecular docking approach. J. Transl. Med. 2018, 16, 298. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Saleem, S.; Ashfaq, U.A.; Bari, A.; Anwar, F.; Alqahtani, S. Epitope-based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: An immune-informatics study. J. Transl. Med. 2019, 17, 362. [Google Scholar] [CrossRef] [PubMed]
- Dar, H.A.; Zaheer, T.; Shehroz, M.; Ullah, N.; Naz, K.; Muhammad, S.A.; Zhang, T.; Ali, A. Immunoinformatics-Aided Design and Evaluation of a Potential Multi-Epitope Vaccine against Klebsiella Pneumoniae. Vaccines 2019, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Falsey, A.R.; Walsh, E.E. Safety and immunogenicity of a respiratory syncytial virus subunit vaccine (PFP-2) in ambulatory adults over age 60. Vaccine 1996, 14, 1214–1218. [Google Scholar] [CrossRef]
- Falsey, A.R.; Walsh, E.E. Safety and immunogenicity of a respiratory syncytial virus subunit vaccine (PFP-2) in the institutionalized elderly. Vaccine 1997, 15, 1130–1132. [Google Scholar] [CrossRef]
- Belshe, R.B.; Anderson, E.L.; Walsh, E.E. Immunogenicity of purified F glycoprotein of respiratory syncytial virus: Clinical and immune responses to subsequent natural infection in children. J. Infect. Dis. 1993, 168, 1024–1029. [Google Scholar] [CrossRef]
- Paradiso, P.R.; Hildreth, S.W.; Hogerman, D.A.; Speelman, D.J.; Lewin, E.B.; Oren, J.; Smith, D.H. Safety and immunogenicity of a subunit respiratory syncytial virus vaccine in children 24 to 48 months old. Pediatr. Infect. Dis. J. 1994, 13, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Tristram, D.; Welliver, R.; Mohar, C.; Hogerman, D.; Hildreth, S.; Paradiso, P. Immunogenicity and safety of respiratory syncytial virus subunit vaccine in seropositive children 18-36 months old. J. Infect. Dis. 1993, 167, 191–195. [Google Scholar] [CrossRef]
- Jones, H.G.; Ritschel, T.; Pascual, G.; Brakenhoff, J.P.; Keogh, E.; Furmanova-Hollenstein, P.; Lanckacker, E.; Wadia, J.S.; Gilman, M.S.; Williamson, R.A. Structural basis for recognition of the central conserved region of RSV G by neutralizing human antibodies. PLoS Pathog. 2018, 14, e1006935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossey, I.; Gilman, M.S.; Kabeche, S.C.; Sedeyn, K.; Wrapp, D.; Kanekiyo, M.; Chen, M.; Mas, V.; Spitaels, J.; Melero, J.A. Potent single-domain antibodies that arrest respiratory syncytial virus fusion protein in its prefusion state. Nat. Commun. 2017, 8, 14158. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2011, 39, D32. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Springer: Cham, Switzerland, 2005; pp. 571–607. [Google Scholar]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, P.; Kim, Y.; Haste-Andersen, P.; Beaver, J.; Bourne, P.E.; Bui, H.-H.; Buus, S.; Frankild, S.; Greenbaum, J. Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 2008, 36, W513–W518. [Google Scholar] [CrossRef] [Green Version]
- Reynisson, B.; Barra, C.; Peters, B.; Nielsen, M. Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data. BioRxiv 2019, 799882. [Google Scholar] [CrossRef]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.; Consortium, O.S.D.D. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhao, X.; Sun, P.; Gao, B.; Ma, Z. Conformational B-cell epitopes prediction from sequences using cost-sensitive ensemble classifiers and spatial clustering. BioMed Res. Int. 2014, 2014, 689219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [Green Version]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 153. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.N.; Sen, G.C. Novel functions of proteins encoded by viral stress-inducible genes. Pharmacol. Ther. 2004, 103, 245–259. [Google Scholar] [CrossRef]
- Fuse, S.; Molloy, M.J.; Usherwood, E.J. Immune responses against persistent viral infections: Possible avenues for immunotherapeutic interventions. Crit. Rev. Immunol. 2008, 28, 159–183. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Shahid, F.; Ali, U.; Fareed, A.Z.; Chen, L.-L. Structural modeling and conserved epitopes prediction against SARS-COV-2 structural proteins for vaccine development. Res. Sq. 2020. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [Green Version]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide protein data bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Adhikari, U.K.; Tayebi, M.; Rahman, M.M. Immunoinformatics approach for epitope-based peptide vaccine design and active site prediction against polyprotein of emerging oropouche virus. J. Immunol. Res. 2018, 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nain, Z.; Karim, M.M.; Sen, M.K.; Adhikari, U.K. Structural basis and designing of peptide vaccine using PE-PGRS family protein of Mycobacterium ulcerans—An integrated vaccinomics approach. Mol. Immunol. 2020, 120, 146–163. [Google Scholar] [CrossRef]
- Nain, Z.; Abdulla, F.; Rahman, M.M.; Karim, M.M.; Khan, M.S.A.; Sayed, S.B.; Mahmud, S.; Rahman, S.R.; Sheam, M.M.; Haque, Z. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. J. Biomol. Struct. Dyn. 2019, 1–18. [Google Scholar] [CrossRef]
- Hoover, D.M.; Wu, Z.; Tucker, K.; Lu, W.; Lubkowski, J. Antimicrobial characterization of human β-defensin 3 derivatives. Antimicrob. Agents Chemother. 2003, 47, 2804–2809. [Google Scholar] [CrossRef] [Green Version]
- Mahram, A.; Herbordt, M.C. Fast and accurate NCBI BLASTP: Acceleration with multiphase FPGA-based prefiltering. In Proceedings of the 24th ACM International Conference on Supercomputing, Tsukuba Ibaraki, Japan, June 2010; pp. 73–82. [Google Scholar]
- Walker, J.M. The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Bjellqvist, B.; Hughes, G.J.; Pasquali, C.; Paquet, N.; Ravier, F.; Sanchez, J.C.; Frutiger, S.; Hochstrasser, D. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis 1993, 14, 1023–1031. [Google Scholar] [CrossRef]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP-a server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Blaszczyk, M.; Jamroz, M.; Kmiecik, S.; Kolinski, A. CABS-fold: Server for the de novo and consensus-based prediction of protein structure. Nucleic Acids Res. 2013, 41, W406–W411. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., III; De Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengths, M.; Angles, M. Limitations of structure evaluation tools errat. Quick Guidel. Comput. Drug Des. 2018, 16, 75. [Google Scholar]
- Kuriata, A.; Gierut, A.M.; Oleniecki, T.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. CABS-flex 2.0: A web server for fast simulations of flexibility of protein structures. Nucleic Acids Res. 2018, 46, W338–W343. [Google Scholar] [CrossRef] [Green Version]
- Kurcinski, M.; Oleniecki, T.; Ciemny, M.P.; Kuriata, A.; Kolinski, A.; Kmiecik, S. CABS-flex standalone: A simulation environment for fast modeling of protein flexibility. Bioinformatics 2019, 35, 694–695. [Google Scholar] [CrossRef]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A new ensemble learning framework for improved linear B-cell epitope prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A. PDBsum new things. Nucleic Acids Res. 2009, 37, D355–D359. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Oostenbrink, C.; van Gunsteren, W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tahir ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural Basis of SARS-CoV-2 3CLpro and Anti-COVID-19 Drug Discovery from Medicinal Plants. J. Pharm. Anal. 2020. [Google Scholar] [CrossRef]
- Muneer, I.; Ul Qamar, M.T.; Tusleem, K.; Abdul Rauf, S.; Hussain, H.M.J.; Siddiqi, A.R. Discovery of selective inhibitors for cyclic AMP response element-binding protein: A combined ligand and structure-based resources pipeline. Anticancer Drugs 2019, 30, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Maryam, A.; Muneer, I.; Xing, F.; Ashfaq, U.A.; Khan, F.A.; Anwar, F.; Geesi, M.H.; Khalid, R.R.; Rauf, S.A.; et al. Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Sci. Rep. 2019, 9, 1433. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; Tahir ul Qamar, M.; Alqahtani, S.M. Pharmacoinformatics and Molecular Dynamic Simulation Studies Reveal Potential Inhibitors of SARS-CoV-2 Main Protease 3CLpro. Preprints 2020, 2020020308. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Wu, J.C.; Yan, C.; Wang, Y.; Luo, R.; Gonzales, M.B.; Dalby, K.N.; Ren, P. Virtual screening using molecular simulations. Proteins Struct. Funct. Bioinform. 2011, 79, 1940–1951. [Google Scholar] [CrossRef] [Green Version]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]
- Kroger, A.T. General Recommendations on Immunization; US Department of Health and Human Services, Public Health Servic, Centers for Disease Control: Atlanta, GA, USA, 2013.
- Chauhan, V.; Singh, M.P. Immuno-informatics approach to design a multi-epitope vaccine to combat cytomegalovirus infection. Eur. J. Pharm. Sci. 2020, 147, 105279. [Google Scholar] [CrossRef]
- Castiglione, F.; Mantile, F.; De Berardinis, P.; Prisco, A. How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput. Math. Methods Med. 2012, 2012, 842329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, S.; Ahmad, S.; Azam, S.S. Immuno-informatics Characterization SARS-CoV-2 Spike Glycoprotein for Prioritization of Epitope based Multivalent Peptide Vaccine. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Tahir ul Qamar, M.; Rehman, A.; Ashfaq, U.A.; Awan, M.Q.; Fatima, I.; Shahid, F.; Chen, L.-L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Munch, R.; Nortemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Econome, J.G.; Schutt, A.; Klco, S.; Cantor, C.R. A physical map of the Escherichia coli K12 genome. Science 1987, 236, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Xu, J. RaptorX: Exploiting structure information for protein alignment by statistical inference. Proteins Struct. Funct. Bioinform. 2011, 79, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Pandey, R.K.; Narula, A.; Naskar, M.; Srivastava, S.; Verma, P.; Malik, R.; Shah, P.; Prajapati, V.K. Exploring dual inhibitory role of febrifugine analogues against Plasmodium utilizing structure-based virtual screening and molecular dynamic simulation. J. Biomol. Struct. Dyn. 2017, 35, 791–804. [Google Scholar] [CrossRef]
- Nezafat, N.; Karimi, Z.; Eslami, M.; Mohkam, M.; Zandian, S.; Ghasemi, Y. Designing an efficient multi-epitope peptide vaccine against Vibrio cholerae via combined immunoinformatics and protein interaction based approaches. Comput. Biol. Chem. 2016, 62, 82–95. [Google Scholar] [CrossRef]
- Livingston, B.; Crimi, C.; Newman, M.; Higashimoto, Y.; Appella, E.; Sidney, J.; Sette, A. A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J. Immunol. 2002, 168, 5499–5506. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef] [PubMed]
- Lund, F.E. Cytokine-producing B lymphocytes—Key regulators of immunity. Curr. Opin. Immunol. 2008, 20, 332–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehan Khalid, R.; Tahir Ul Qamar, M.; Maryam, A.; Ashique, A.; Anwar, F.; H Geesi, M.; Siddiqi, A.R. Comparative Studies of the Dynamics Effects of BAY60-2770 and BAY58-2667 Binding with Human and Bacterial H-NOX Domains. Molecules 2018, 23, 2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- McLellan, J.S.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 2011, 85, 7788–7796. [Google Scholar] [CrossRef] [Green Version]
- Jaberolansar, N.; Toth, I.; Young, P.R.; Skwarczynski, M. Recent advances in the development of subunit-based RSV vaccines. Expert Rev. Vaccines 2016, 15, 53–68. [Google Scholar] [CrossRef]
- McLellan, J.S.; Ray, W.C.; Peeples, M.E. Structure and function of respiratory syncytial virus surface glycoproteins. In Challenges and Opportunities for Respiratory Syncytial Virus Vaccines; Springer: Berlin/Heidelberg, Germany, 2013; pp. 83–104. [Google Scholar]
- Takimoto, T.; Hurwitz, J.L.; Coleclough, C.; Prouser, C.; Krishnamurthy, S.; Zhan, X.; Boyd, K.; Scroggs, R.A.; Brown, B.; Nagai, Y. Recombinant Sendai virus expressing the G glycoprotein of respiratory syncytial virus (RSV) elicits immune protection against RSV. J. Virol. 2004, 78, 6043–6047. [Google Scholar] [CrossRef] [Green Version]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 9902–9907. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Allan, M.F.; Maranas, C.D. OptMAVEn-2.0: De novo design of variable antibody regions against targeted antigen epitopes. Antibodies 2018, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Nimrod, G.; Fischman, S.; Austin, M.; Herman, A.; Keyes, F.; Leiderman, O.; Hargreaves, D.; Strajbl, M.; Breed, J.; Klompus, S. Computational design of epitope-specific functional antibodies. Cell Rep. 2018, 25, 2121–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolf-Bryfogle, J.; Kalyuzhniy, O.; Kubitz, M.; Weitzner, B.D.; Hu, X.; Adachi, Y.; Schief, W.R.; Dunbrack, R.L., Jr. RosettaAntibodyDesign (RAbD): A general framework for computational antibody design. PLoS Comput. Biol. 2018, 14, e1006112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisitkun, T.; Hoffert, J.D.; Saeed, F.; Knepper, M.A. NHLBI-AbDesigner: An online tool for design of peptide-directed antibodies. Am. J. Physiol. Cell Physiol. 2012, 302, C154–C164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Pandey, K.R.; Khatoon, N.; Narula, A.; Mishra, A.; Prajapati, V.K. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 2017, 7, 9232. [Google Scholar] [CrossRef] [PubMed]
- Broberg, E.K.; Waris, M.; Johansen, K.; Snacken, R.; Penttinen, P.; Network, E.I.S. Seasonality and geographical spread of respiratory syncytial virus epidemics in 15 European countries, 2010 to 2016. Eurosurveillance 2018, 23, 17-00284. [Google Scholar] [CrossRef] [Green Version]
- Ikram, A.; Zaheer, T.; Awan, F.M.; Obaid, A.; Naz, A.; Hanif, R.; Paracha, R.Z.; Ali, A.; Naveed, A.K.; Janjua, H.A. Exploring NS3/4A, NS5A and NS5B proteins to design conserved subunit multi-epitope vaccine against HCV utilizing immunoinformatics approaches. Sci. Rep. 2018, 8, 16107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Cell. Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.K.; Ojha, R.; Aathmanathan, V.S.; Krishnan, M.; Prajapati, V.K. Immunoinformatics approaches to design a novel multi-epitope subunit vaccine against HIV infection. Vaccine 2018, 36, 2262–2272. [Google Scholar] [CrossRef]
- Kazi, A.; Chuah, C.; Majeed, A.B.A.; Leow, C.H.; Lim, B.H.; Leow, C.Y. Current progress of immunoinformatics approach harnessed for cellular-and antibody-dependent vaccine design. Pathog. Glob. Health 2018, 112, 123–131. [Google Scholar] [CrossRef]
- He, R.; Yang, X.; Liu, C.; Chen, X.; Wang, L.; Xiao, M.; Ye, J.; Wu, Y.; Ye, L. Efficient control of chronic LCMV infection by a CD4 T cell epitope-based heterologous prime-boost vaccination in a murine model. Cell. Mol. Immunol. 2018, 15, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Cai, Y.; Chen, J.; Ye, X.; Mao, S.; Zhu, S.; Xue, X.; Chen, S.; Zhang, L. Evaluation of tandem Chlamydia trachomatis MOMP multi-epitopes vaccine in BALB/c mice model. Vaccine 2017, 35, 3096–3103. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Jang, Y.-S. The development of mucosal vaccines for both mucosal and systemic immune induction and the roles played by adjuvants. Clin. Exp. Vaccine Res. 2017, 6, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, J.; Czerkinsky, C. Mucosal immunity and vaccines. Nat. Med. 2005, 11, S45–S53. [Google Scholar] [CrossRef] [PubMed]
- Ogra, P.L.; Faden, H.; Welliver, R.C. Vaccination strategies for mucosal immune responses. Clin. Microbiol. Rev. 2001, 14, 430–445. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, B.; Ashfaq, U.A.; Rahman, M.U.; Masoud, M.S.; Yousaf, M.Z. Conserved B and T cell epitopes prediction of ebola virus glycoprotein for vaccine development: An immuno-informatics approach. Microb. Pathog. 2019, 132, 243–253. [Google Scholar] [CrossRef]
- Shahid, F.; Ashfaq, U.A.; Javaid, A.; Khalid, H. Immunoinformatics guided rational design of a next generation multi epitope based peptide (MEBP) vaccine by exploring Zika virus proteome. Infect. Genet. Evol. 2020, 80, 104199. [Google Scholar] [CrossRef]
- Pandey, R.K.; Dahiya, S.; Mahita, J.; Sowdhamini, R.; Prajapati, V.K. Vaccination and immunization strategies to design Aedes aegypti salivary protein based subunit vaccine tackling Flavivirus infection. Int. J. Biol. Macromol. 2019, 122, 1203–1211. [Google Scholar] [CrossRef]
- Kesherwani, V.; Tarang, S. An immunoinformatic approach to universal therapeutic vaccine design against BK virus. Vaccine 2019, 37, 3457–3463. [Google Scholar] [CrossRef]
- Azim, K.F.; Hasan, M.; Hossain, M.N.; Somana, S.R.; Hoque, S.F.; Bappy, M.N.I.; Chowdhury, A.T.; Lasker, T. Immunoinformatics approaches for designing a novel multi epitope peptide vaccine against human norovirus (Norwalk virus). Infect. Genet. Evol. 2019, 74, 103936. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Tool Utilized | Template | Ramachandran Plot | ||
|---|---|---|---|---|---|
| Favored Region | Allowed Region | Disallowed Region | |||
| G | RaptorX | 6blhG | 89.2% | 8.1% | 2.7% |
| F | RaptorX | 4mmrB | 96.0% | 2.9% | 1.2% |
| Sr. No | Epitope | Protein | Position | HLA Alleles | Antigenicity Score | Binding Score (kcal/mol) | # H-bonds |
|---|---|---|---|---|---|---|---|
| MHC I | |||||||
| 1 | KSIAQITLSILA | G | 36–47 | A*3201 | 0.6 | −13.36 | 6 |
| 2 | YLTQNPQLGISF | G | 90–101 | B*1502 | 1.3 | −14.24 | 2 |
| 3 | TTKQRQNKPPNK | G | 147–158 | A*3001 | 0.7 | −21.38 | 12 |
| 4 | CSICSNNPTCWA | G | 173–184 | B*5801 | 0.6 | −14.44 | 4 |
| 5 | CSNNPTCWAICK | G | 176–186 | A*1101 | 0.6 | −19.11 | 12 |
| 6 | SSEGNISPSQVY | G | 269–280 | B*4403 | 0.7 | −14.98 | 6 |
| 7 | ETVIEFQQKNNR | F | 218–229 | A*3303 | 1.3 | −15.95 | 17 |
| 8 | DTPCWKLHTSPL | F | 310–321 | B*0702 | 0.6 | −12.49 | 4 |
| 9 | SVSFFPLAETCK | F | 348–359 | A*0301 | 0.7 | −14.85 | 10 |
| 10 | FFPLAETCKVQS | F | 351–362 | B*3503 | 0.6 | −12.93 | 1 |
| 11 | SLYVKGEPIINF | F | 466–477 | B*1502 | 0.7 | −13.82 | 3 |
| 12 | CKARSTPVTLSK | F | 550–561 | A*3001 | 1.2 | −18.88 | 17 |
| 13 | RSTPVTLSKDQL | F | 553–564 | B*5801 | 0.7 | −11.62 | 5 |
| MHC II | |||||||
| 14 | AIIFIASANNKVTLT | G | 58–72 | DRB1_0410 | 0.76 | −12.04 | 5 |
| 15 | IIFIASANNKVTLTT | G | 59–73 | DRB1_0410 | 0.67 | −13.39 | 4 |
| B-Cell Epitope | Position | Antigenicity |
|---|---|---|
| LTQNPQLGISFAAY | 67 | 1.15 |
| SSEGNISPSQVYAA | 126 | 0.67 |
| IGKCSTRGRKCCRR | 30 | 1.27 |
| LPKEEQIGKCSTRG | 24 | 0.75 |
| RQNKPPNKAAYCSI | 85 | 0.76 |
| STPVTLSKDQLGPG | 232 | 0.70 |
| LAETCKAAYFFPLA | 177 | 0.57 |
| SPLAAYSVSFFPLA | 165 | 0.82 |
| NNPTCWAICKAAYS | 113 | 0.59 |
| YAAYETVIEFQQKN | 137 | 1.19 |
| LSKAAYRSTPVTLS | 225 | 1.10 |
| AAYCSICSNNPTCW | 93 | 0.57 |
| YSLYVKGEPIINFA | 200 | 0.73 |
| SFAAYTTKQRQNKP | 76 | 0.85 |
| FAAYCKARSTPVTL | 212 | 1.22 |
| AAYCSNNPTCWAIC | 108 | 0.82 |
| QKNNRAAYDTPCWK | 148 | 0.77 |
| VSFFPLAETCKAAY | 172 | 0.60 |
| GEPIINFAAYCKAR | 206 | 0.56 |
| AYDTPCWKLHTSPL | 154 | 0.64 |
| CKAAYSSEGNISPS | 121 | 0.77 |
| Parameters | Value |
|---|---|
| HADDOCK v.2.2 score | −151.2 ± 2.3 |
| Cluster size | 17 |
| RMSD from the overall lowest-energy structure | 1.3 ± 0.1 |
| Van der Waals energy | −107.5 ± 1.9 |
| Electrostatic energy | −411.5 ± 19.7 |
| Desolvation energy | 7.0 ± 1.6 |
| Restraints violation energy | 0.0 ± 0.0 |
| Buried Surface Area | 3680.6 ± 26.9 |
| Z-Score | 0 |
| Energy Terms | TLR3-MEV Complex |
|---|---|
| ΔEvdW | −394.240 ± 48.875 kJ/moL |
| ΔEelec | −3451.087 ± 187.883 kJ/moL |
| ΔESA | −59.371 ± 7.180 kJ/moL |
| ΔGpolar | 1169.754 ± 248.542 kJ/moL |
| ΔGbinding | −2734.944 ± 175.446 kJ/moL |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tahir ul Qamar, M.; Shokat, Z.; Muneer, I.; Ashfaq, U.A.; Javed, H.; Anwar, F.; Bari, A.; Zahid, B.; Saari, N. Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach. Vaccines 2020, 8, 288. https://doi.org/10.3390/vaccines8020288
Tahir ul Qamar M, Shokat Z, Muneer I, Ashfaq UA, Javed H, Anwar F, Bari A, Zahid B, Saari N. Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach. Vaccines. 2020; 8(2):288. https://doi.org/10.3390/vaccines8020288
Chicago/Turabian StyleTahir ul Qamar, Muhammad, Zeeshan Shokat, Iqra Muneer, Usman Ali Ashfaq, Hamna Javed, Farooq Anwar, Amna Bari, Barira Zahid, and Nazamid Saari. 2020. "Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach" Vaccines 8, no. 2: 288. https://doi.org/10.3390/vaccines8020288