
Pharmacokinetics of the Antiviral Lectin Griffithsin Administered by Different Routes Indicates Multiple Potential Uses

Abstract
:1. Introduction
2. Materials and Methods
2.1. Lectin Reagents
2.2. Animal Housing and Care
2.3. Pharmacokinetic Study and Sample Collection after Single Dose Administration
2.4. Active Mass Balance Rat Treatment and Sample Collection
2.5. Chronic Oral Dosing Treatment and Sample Collection
2.6. GRFT Capture Immunoassay Using Purified Influenza Hemagglutinin
2.7. Evaluation of Anti-HIV Activity of Fecal Extracts
2.8. Histopathology
2.9. Pharmacokinetic Analysis
2.10. Statistical Analyses
3. Results
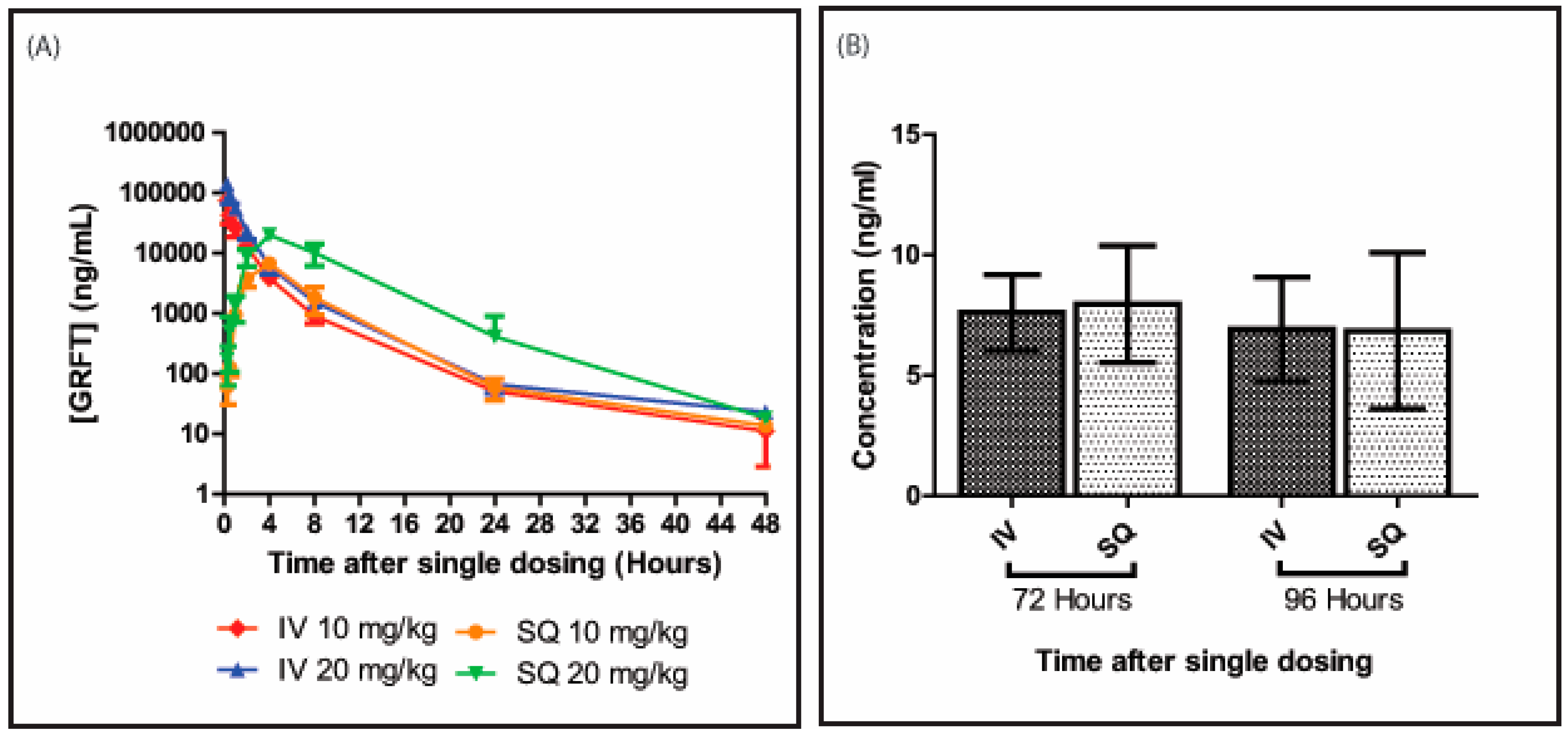
3.1. GRFT’s Bioavailability after Intravenous Administration
3.2. GRFT’s Bioavailability after Subcutaneous Administration
3.3. GRFT’s Pharmacokinetic Parameters after Parenteral Administration
3.4. Pharmacokinetic Parameters after Oral Administration
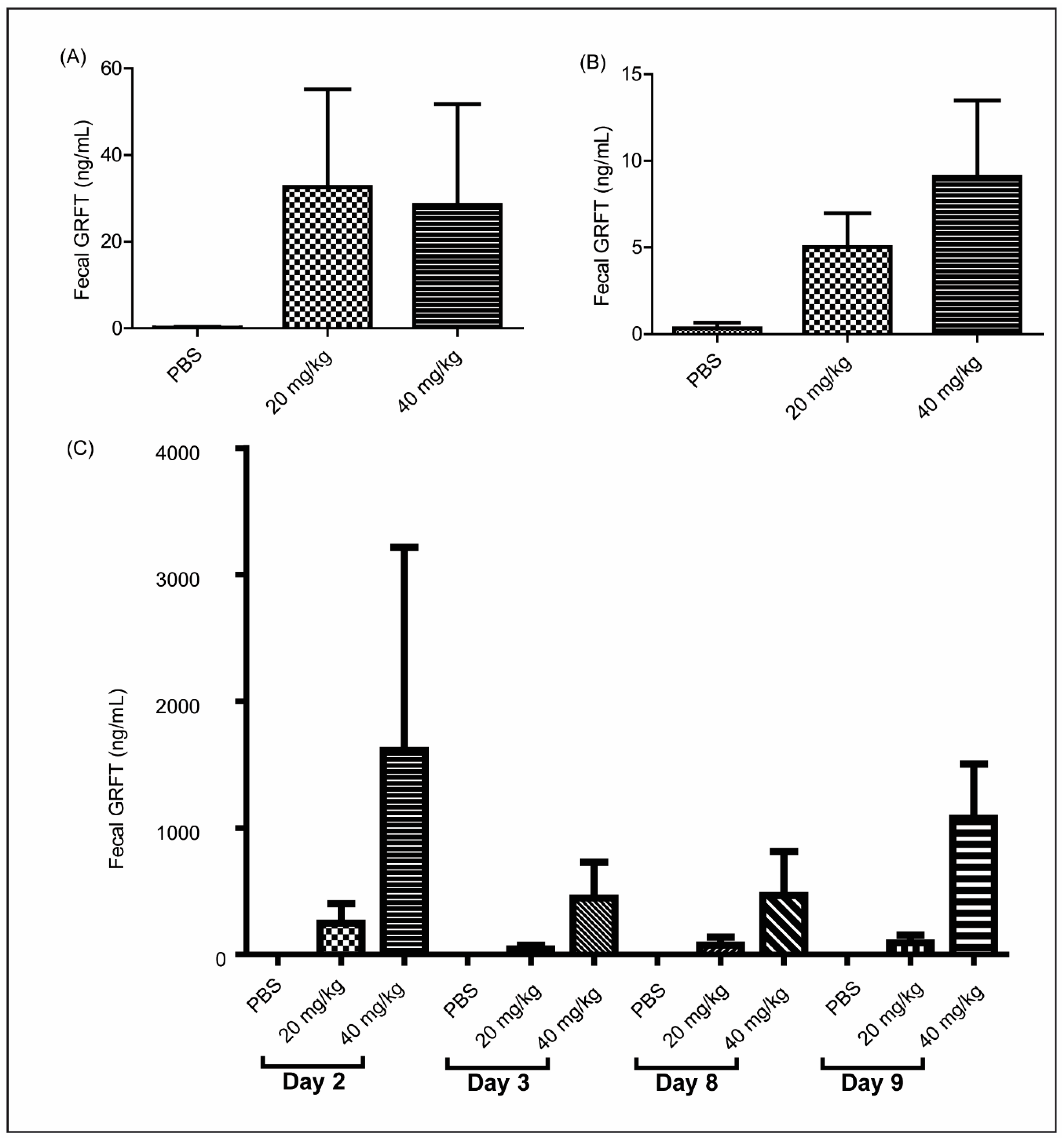
3.5. GRFT Is Found in Fecal Extracts after Chronic Oral Treatment
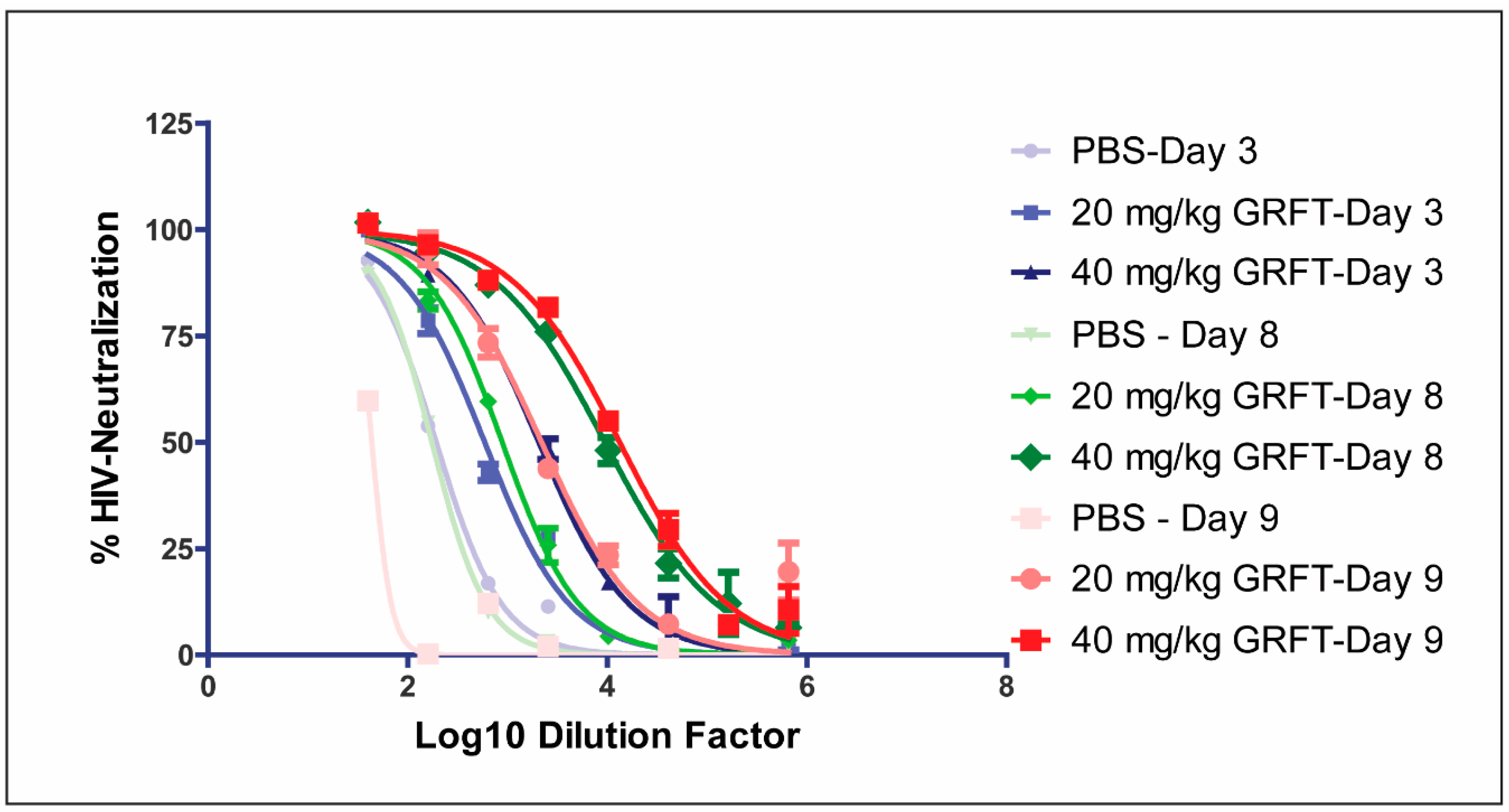
3.6. Fecal Extracts after Chronic Oral Treatment Show Neutralization Activity Against HIV
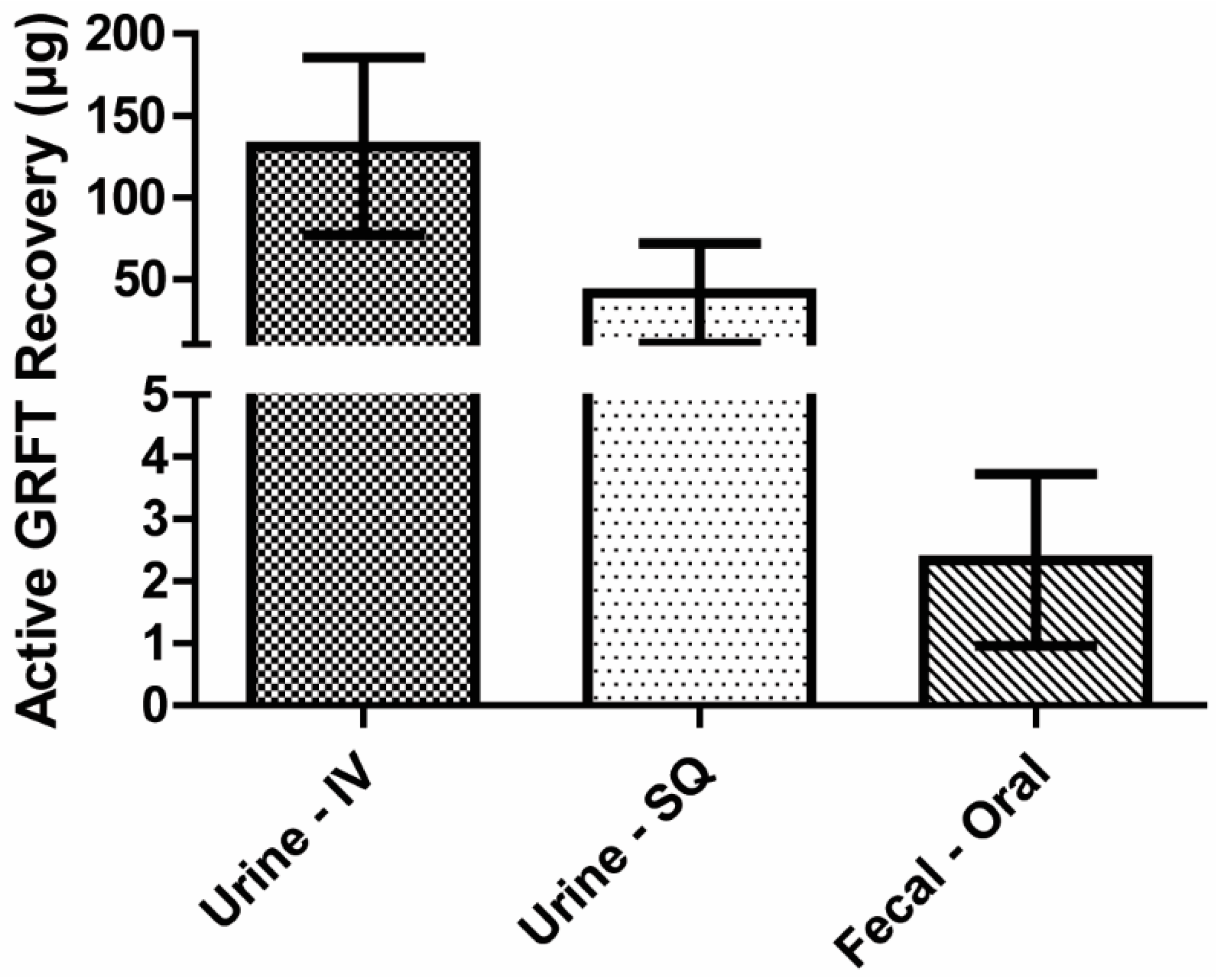
3.7. Mass Balance of Active GRFT
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Michelow, I.C.; Dong, M.; Mungall, B.A.; Yantosca, L.M.; Lear, C.; Ji, X.; Karpel, M.; Rootes, C.L.; Brudner, M.; Houen, G.; et al. A novel L-ficolin/mannose-binding lectin chimeric molecule with enhanced activity against Ebola virus. J. Biol. Chem. 2010, 285, 24729–24739. [Google Scholar] [CrossRef] [PubMed]
- Michelow, I.C.; Lear, C.; Scully, C.; Prugar, L.I.; Longley, C.B.; Yantosca, L.M.; Ji, X.; Karpel, M.; Brudner, M.; Takahashi, K.; et al. High-dose mannose-binding lectin therapy for Ebola virus infection. J. Infect. Dis. 2011, 203, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Albecka, A.; Belouzard, S.; Vercauteren, K.; Verhoye, L.; Wychowski, C.; Leroux-Roels, G.; Palmer, K.E.; Dubuisson, J. Griffithsin has antiviral activity against hepatitis C virus. Antimicrob. Agents Chemother. 2011, 55, 5159–5167. [Google Scholar] [CrossRef] [PubMed]
- Swanson, M.D.; Winter, H.C.; Goldstein, I.J.; Markovitz, D.M. A lectin isolated from bananas is a potent inhibitor of HIV replication. J. Biol. Chem. 2010, 285, 8646–8655. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, K.B.; Gray, E.S.; Mufhandu, H.; McMahon, J.B.; Chakauya, E.; O’Keefe, B.R.; Chikwamba, R.; Morris, L. The lectins griffithsin, cyanovirin-N and scytovirin inhibit HIV-1 binding to the DC-SIGN receptor and transfer to CD4+ cells. Virology 2012, 423, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Huskens, D.; Schols, D. Algal lectins as potential HIV microbicide candidates. Marine Drugs 2012, 10, 1476–1497. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; O’Keefe, B.R.; Sowder, R.C., 2nd; Bringans, S.; Gardella, R.; Berg, S.; Cochran, P.; Turpin, J.A.; Buckheit, R.W., Jr.; McMahon, J.B.; et al. Isolation and characterization of griffithsin, a novel HIV-inactivating protein, from the red alga Griffithsia sp. J. Biol. Chem. 2005, 280, 9345–9353. [Google Scholar] [CrossRef] [PubMed]
- Moulaei, T.; Shenoy, S.R.; Giomarelli, B.; Thomas, C.; McMahon, J.B.; Dauter, Z.; O’Keefe, B.R.; Wlodawer, A. Monomerization of viral entry inhibitor griffithsin elucidates the relationship between multivalent binding to carbohydrates and anti-HIV activity. Structure 2010, 18, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowska, N.E.; O’Keefe, B.R.; Mori, T.; Zhu, C.; Giomarelli, B.; Vojdani, F.; Palmer, K.E.; McMahon, J.B.; Wlodawer, A. Domain-swapped structure of the potent antiviral protein griffithsin and its mode of carbohydrate binding. Structure 2006, 14, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowska, N.E.; Shenoy, S.R.; O’Keefe, B.R.; McMahon, J.B.; Palmer, K.E.; Dwek, R.A.; Wormald, M.R.; Wlodawer, A. Crystallographic, thermodynamic, and molecular modeling studies of the mode of binding of oligosaccharides to the potent antiviral protein griffithsin. Proteins 2007, 67, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowska, N.E.; Shenoy, S.R.; O’Keefe, B.R.; Wlodawer, A. Crystallographic studies of the complexes of antiviral protein griffithsin with glucose and N-acetylglucosamine. Protein Sci. 2007, 16, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Bonomelli, C.; Doores, K.J.; Dunlop, D.C.; Thaney, V.; Dwek, R.A.; Burton, D.R.; Crispin, M.; Scanlan, C.N. The glycan shield of HIV is predominantly oligomannose independently of production system or viral clade. PLoS ONE 2011, 6, e23521. [Google Scholar] [CrossRef] [PubMed]
- Doores, K.J.; Bonomelli, C.; Harvey, D.J.; Vasiljevic, S.; Dwek, R.A.; Burton, D.R.; Crispin, M.; Scanlan, C.N. Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc. Natl. Acad. Sci. USA 2010, 107, 13800–13805. [Google Scholar] [CrossRef] [PubMed]
- Vigerust, D.J.; Shepherd, V.L. Virus glycosylation: Role in virulence and immune interactions. Trends Microbiol. 2007, 15, 211–218. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, B.R.; Vojdani, F.; Buffa, V.; Shattock, R.J.; Montefiori, D.C.; Bakke, J.; Mirsalis, J.; d’Andrea, A.L.; Hume, S.D.; Bratcher, B.; et al. Scaleable manufacture of HIV-1 entry inhibitor griffithsin and validation of its safety and efficacy as a topical microbicide component. Proc. Natl. Acad. Sci. USA 2009, 106, 6099–6104. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, K.B.; Gray, E.S.; Lambson, B.E.; Moore, P.L.; Choge, I.A.; Mlisana, K.; Karim, S.S.; McMahon, J.; O’Keefe, B.; Chikwamba, R.; et al. Mannose-rich glycosylation patterns on HIV-1 subtype C gp120 and sensitivity to the lectins, Griffithsin, Cyanovirin-N and Scytovirin. Virology 2010, 402, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Nixon, B.; Stefanidou, M.; Mesquita, P.M.; Fakioglu, E.; Segarra, T.; Rohan, L.; Halford, W.; Palmer, K.E.; Herold, B.C. Griffithsin protects mice from genital herpes by preventing cell-to-cell spread. J. Virol. 2013, 87, 6257–6269. [Google Scholar] [CrossRef] [PubMed]
- Ishag, H.Z.; Li, C.; Huang, L.; Sun, M.X.; Wang, F.; Ni, B.; Malik, T.; Chen, P.Y.; Mao, X. Griffithsin inhibits Japanese encephalitis virus infection in vitro and in vivo. Arch. Virol. 2013, 158, 349–358. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, B.R.; Giomarelli, B.; Barnard, D.L.; Shenoy, S.R.; Chan, P.K.; McMahon, J.B.; Palmer, K.E.; Barnett, B.W.; Meyerholz, D.K.; Wohlford-Lenane, C.L.; et al. Broad-spectrum in vitro activity and in vivo efficacy of the antiviral protein griffithsin against emerging viruses of the family Coronaviridae. J. Virol. 2010, 84, 2511–2521. [Google Scholar] [CrossRef] [PubMed]
- Ferir, G.; Huskens, D.; Palmer, K.E.; Boudreaux, D.M.; Swanson, M.D.; Markovitz, D.M.; Balzarini, J.; Schols, D. Combinations of griffithsin with other carbohydrate-binding agents demonstrate superior activity against HIV Type 1, HIV Type 2, and selected carbohydrate-binding agent-resistant HIV Type 1 strains. AIDS Res. Hum. Retrovir. 2012, 28, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Ferir, G.; Palmer, K.E.; Schols, D. Synergistic activity profile of griffithsin in combination with tenofovir, maraviroc and enfuvirtide against HIV-1 clade C. Virology 2011, 417, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Barton, C.; Kouokam, J.C.; Lasnik, A.B.; Foreman, O.; Cambon, A.; Brock, G.; Montefiori, D.C.; Vojdani, F.; McCormick, A.A.; O’Keefe, B.R.; et al. Activity of and effect of subcutaneous treatment with the broad-spectrum antiviral lectin griffithsin in two laboratory rodent models. Antimicrob. Agents Chemother. 2014, 58, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Kouokam, J.C.; Huskens, D.; Schols, D.; Johannemann, A.; Riedell, S.K.; Walter, W.; Walker, J.M.; Matoba, N.; O’Keefe, B.R.; Palmer, K.E. Investigation of griffithsin’s interactions with human cells confirms its outstanding safety and efficacy profile as a microbicide candidate. PLoS ONE 2011, 6, e22635. [Google Scholar] [CrossRef] [PubMed]
- Montefiori, D.C. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Current Protocols in Immunology 2005. [Google Scholar] [CrossRef]
- Macdougall, I.C.; Roberts, D.E.; Coles, G.A.; Williams, J.D. Clinical pharmacokinetics of epoetin (recombinant human erythropoietin). Clin. Pharmacokinet. 1991, 20, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Azanza, J.R.; Sadaba, B.; Gomez-Guiu, A. Monoclonal antibodies: Pharmacokinetics as a basis for new dosage regimens? J. Oncol. Pharm. Pract. 2015, 21, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Dostalek, M.; Gardner, I.; Gurbaxani, B.M.; Rose, R.H.; Chetty, M. Pharmacokinetics, pharmacodynamics and physiologically-based pharmacokinetic modelling of monoclonal antibodies. Clin. Pharmacokinet. 2013, 52, 83–124. [Google Scholar] [CrossRef] [PubMed]
- Moncla, B.J.; Pryke, K.; Rohan, L.C.; Graebing, P.W. Degradation of naturally occurring and engineered antimicrobial peptides by proteases. Adv. Biosci. Biotechnol. 2011, 2, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Prevention, Interim guidance for clinicians considering the use of preexposure prophylaxis for the prevention of HIV infection in heterosexually active adults. MMWR. Morb. Mortal. Wkly. Rep. 2012, 61, 586–589. [Google Scholar]
- Interim guidance for PrEP in MSM. CDC: Do not expand to other risk groups yet. AIDS Alert 2011, 26, 44–45.
- Flexner, C. Post-exposure prophylaxis revisited: New CDC guidelines. Centers for Disease Control and Prevention. In The Hopkins HIV Report: A Bimonthly Newsletter for Healthcare Providers; Johns Hopkins University AIDS Service: Baltimore, MD, USA, 1998; Volume 10, pp. 2–3. [Google Scholar]
- Young, T.N.; Arens, F.J.; Kennedy, G.E.; Laurie, J.W.; Rutherford, G. Antiretroviral post-exposure prophylaxis (PEP) for occupational HIV exposure. Cochrane Database Systematic Rev. 2007, 1. [Google Scholar] [CrossRef]
- Post-exposure prophylaxis for HIV. Drug Ther. Bull. 2011, 49, 30–33.
- Hardy, E.J. Testing and treatment after non-occupational exposures to STDs and HIV. Med. Health Rhode Island 2012, 95, 258–261. [Google Scholar]
- Nikolopoulos, G.; Tsiodras, S.; Bonovas, S.; Hatzakis, A. Antiretrovirals for HIV exposure prophylaxis. Curr. Med. Chem. 2012, 19, 5924–5939. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, G.; Ahlman, H.; Kewenter, J. A comparison of small intestinal transit time between the rat and the guinea-pig. Acta Chir. Scand. 1976, 142, 537–540. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intravenous | Subcutaneous | ||||
|---|---|---|---|---|---|
| Parameter | Unit | 10 mg/kg (n = 4) | 20 mg/kg (n = 3) | 10 mg/kg (n = 4) | 20 mg/kg (n = 4) |
| absorption half life | hour | 0.5 ± 0.1 | 0.5 ± 0.2 | 1.3 ± 0.3 | 1.6 ± 0.4 |
| distribution half life | hour | 1.7 ± 0.3 | 2.1 ± 0.7 | 2.1 ± 0.9 | 2.8 ± 1.2 |
| elimination half life | hour | 10.7 ± 4.6 | 17.5 ± 6.1 | 13.8 ± 6.8 | 6.6 ± 1.9 |
| AUC | mg-h/L | 105.7 ± 16.9 | 203.6 ± 27.6 | 45.6 ± 9.9 | 183.2 ± 45.3 |
| VD | L | 0.4 ± 0.1 | 0.6 ± 0.2 | 1.2 ± 0.6 | 0.2 ± 0.1 |
| Clearance | L/h | 0.03 ± 0.01 | 0.02 ± 0.01 | 0.06 ± 0.01 | 0.02 ± 0.01 |
| CMAX | µg/mL | 81.8 ± 25.7 | 176.0 ± 26.7 | 6.6 ± 0.6 | 19.7 ± 2.4 |
| Day | Treatment | Fecal GRFT (ng/mL) | Interpolated ID50 |
|---|---|---|---|
| 3 | PBS | 0 | 192 |
| 20 mg/kg GRFT | 23 | 590 | |
| 40 mg/kg GRFT | 447 | 2059 | |
| 8 | PBS | 0 | 174 |
| 20 mg/kg GRFT | 46 | 885 | |
| 40 mg/kg GRFT | 460 | 9452 | |
| 9 | PBS | 0 | 43 |
| 20 mg/kg GRFT | 140 | 2195 | |
| 40 mg/kg GRFT | 1073 | 13,307 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barton, C.; Kouokam, J.C.; Hurst, H.; Palmer, K.E. Pharmacokinetics of the Antiviral Lectin Griffithsin Administered by Different Routes Indicates Multiple Potential Uses. Viruses 2016, 8, 331. https://doi.org/10.3390/v8120331
Barton C, Kouokam JC, Hurst H, Palmer KE. Pharmacokinetics of the Antiviral Lectin Griffithsin Administered by Different Routes Indicates Multiple Potential Uses. Viruses. 2016; 8(12):331. https://doi.org/10.3390/v8120331
Chicago/Turabian StyleBarton, Christopher, J. Calvin Kouokam, Harrell Hurst, and Kenneth E. Palmer. 2016. "Pharmacokinetics of the Antiviral Lectin Griffithsin Administered by Different Routes Indicates Multiple Potential Uses" Viruses 8, no. 12: 331. https://doi.org/10.3390/v8120331