HIV-1 Virological Synapse is not Simply a Copycat of the Immunological Synapse

Abstract
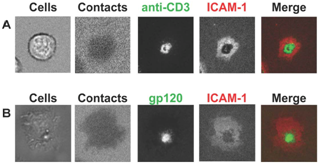
:
1. Introduction
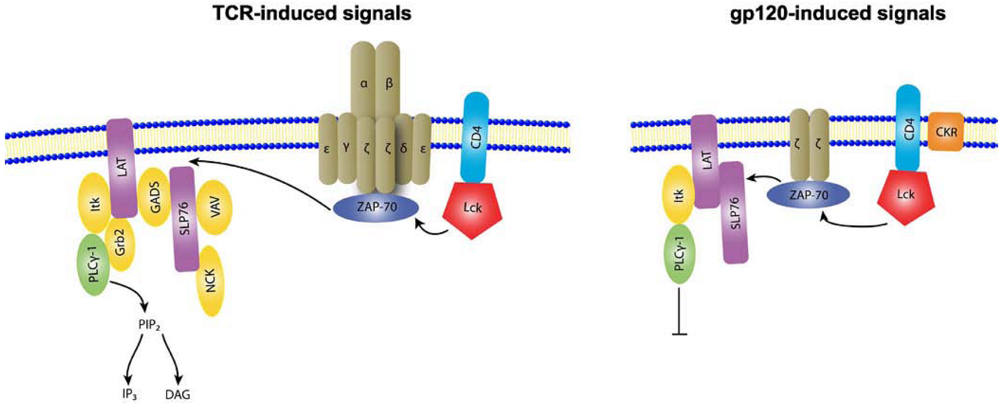
2. Supramolecular structures of IS versus VS

3. Structural stability of IS versus VS
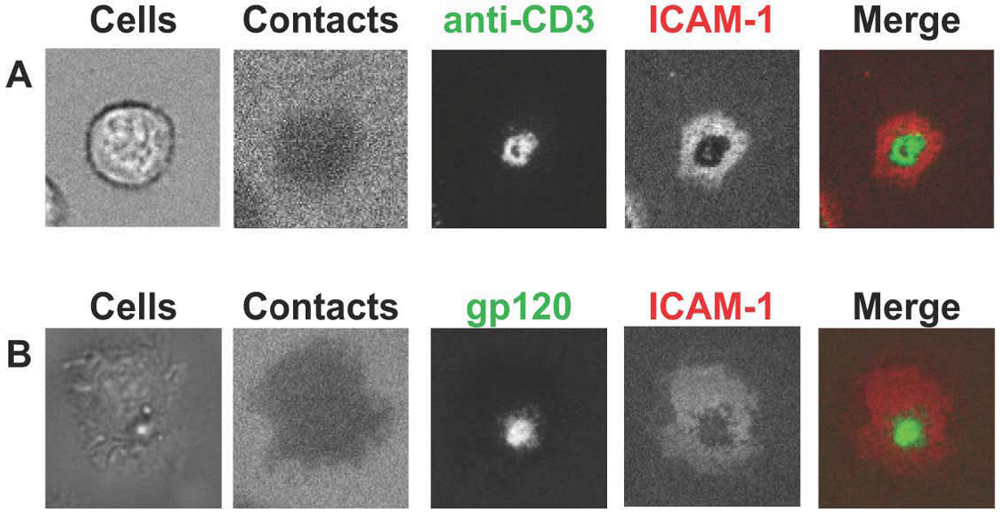
4. T cell activation induced by IS versus VS

5. Cytoskeleton dynamics in IS versus VS


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunological Synapse | Virological Synapse | |
|---|---|---|
| Initial trigger | TCR-pMHC | gp120-CD4 |
| Adhesion molecules involved | LFA-1-ICAM-1 | LFA-1-ICAM-1,2 & 3 |
| Synaptic structureNascent | MCs: TCR and LFA-1(LFA-1 essential) | MCs: GP120 and LFA-1(LFA-1 optional) |
| Mature | cSMAC: TCR-pMHCpSMAC: LFA-1-ICAM-1dSMAC: CD45MCs: TCR and LFA-1 | cSMAC: gp120-CD4pSMAC LFA-1-ICAM-1dSMAC: NDMCs: ND |
| Synapse duration | Hours | 20-30 minutes (planar bilayer system)10 min to several hours (cell-cell system) |
| Signaling Initiation | TCR MCs(sustained in MCs) | gp120 MCs(sustained in cSMAC) |
| Termination | cSMAC | ND |
| Cytoskeleton: Actin | Required for MCs formation but not for stabilitycSMAC depleted of actin | Required for MCs formation and stabilitycSMAC depleted of actin |
| MTOC polarization | Yes | No: within the target T cellYes: within the infected cell |
| Requirement for signaling | Intact actin required for signaling | ND |
6. Conclusions
Acknowledgments
References
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Hubner, W.; McNerney, G.P.; Chen, P.; Dale, B.M.; Gordon, R.E.; Chuang, F.Y.; Li, X.D.; Asmuth, D.M.; Huser, T.; Chen, B.K. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 2009, 323, 1743–1747. [Google Scholar] [CrossRef] [PubMed]
- Aubert, M.; Yoon, M.; Sloan, D.D.; Spear, P.G.; Jerome, K.R. The virological synapse facilitates herpes simplex virus entry into T cells. J. Virol. 2009, 83, 6171–6183. [Google Scholar] [CrossRef] [PubMed]
- York, I.A.; Johnson, D.C. Direct contact with herpes simplex virus-infected cells results in inhibition of lymphokine-activated killer cells because of cell-to-cell spread of virus. J. Infect. Dis. 1993, 168, 1127–1132. [Google Scholar] [PubMed]
- Cameron, P.U.; Freudenthal, P.S.; Barker, J.M.; Gezelter, S.; Inaba, K.; Steinman, R.M. Dendritic cells exposed to human immunodeficiency virus type-1 transmit a vigorous cytopathic infection to CD4+ T cells. Science 1992, 257, 383–387. [Google Scholar] [PubMed]
- Rudnicka, D.; Feldmann, J.; Porrot, F.; Wietgrefe, S.; Guadagnini, S.; Prevost, M.C.; Estaquier, J.; Haase, A.; Sol-Foulon, N.; Schwartz, O. Simultaneous HIV Cell-to-Cell Transmission To Multiple Targets Through Polysynapses. J. Virol. 2009, 83, 6234–6246. [Google Scholar] [CrossRef] [PubMed]
- Arrighi, J.F.; Pion, M.; Garcia, E.; Escola, J.M.; van Kooyk, Y.; Geijtenbeek, T.B.; Piguet, V. DC-SIGN-mediated infectious synapse formation enhances X4 HIV-1 transmission from dendritic cells to T cells. J. Exp. Med. 2004, 200, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.; Pion, M.; Pelchen-Matthews, A.; Collinson, L.; Arrighi, J.F.; Blot, G.; Leuba, F.; Escola, J.M.; Demaurex, N.; Marsh, M.; Piguet, V. HIV-1 trafficking to the dendritic cell-T-cell infectious synapse uses a pathway of tetraspanin sorting to the immunological synapse. Traffic 2005, 6, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.J.; Reuter, M.A.; McDonald, D. HIV traffics through a specialized, surface-accessible intracellular compartment during trans-infection of T cells by mature dendritic cells. PLoS Pathog. 2008, 4, e1000134. [Google Scholar] [CrossRef] [PubMed]
- Groot, F.; Welsch, S.; Sattentau, Q.J. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 2008, 111, 4660–4663. [Google Scholar] [CrossRef] [PubMed]
- Alfsen, A.; Yu, H.; Magerus-Chatinet, A.; Schmitt, A.; Bomsel, M. HIV-1-infected blood mononuclear cells form an integrin- and agrin-dependent viral synapse to induce efficient HIV-1 transcytosis across epithelial cell monolayer. Mol. Biol. Cell 2005, 16, 4267–4279. [Google Scholar] [CrossRef] [PubMed]
- Carreno, M.P.; Krieff, C.; Irinopoulou, T.; Kazatchkine, M.D.; Belec, L. Enhanced transcytosis of R5-tropic human immunodeficiency virus across tight monolayer of polarized human endometrial cells under pro-inflammatory conditions. Cytokine 2002, 20, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Kohler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C.; Onfelt, B.; Sattentau, Q.; Davis, D.M. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Piguet, V.; Sattentau, Q. Dangerous liaisons at the virological synapse. J. Clin. Invest. 2004, 114, 605–610. [Google Scholar] [PubMed]
- Dustin, M.L.; Bromley, S.K.; Kan, Z.; Peterson, D.A.; Unanue, E.R. Antigen receptor engagement delivers a stop signal to migrating T lymphocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 3909–3913. [Google Scholar] [CrossRef]
- Vasiliver-Shamis, G.; Tuen, M.; Wu, T.W.; Starr, T.; Cameron, T.O.; Thomson, R.; Kaur, G.; Liu, J.; Visciano, M.L.; Li, H.; Kumar, R.; Ansari, R.; Han, D.P.; Cho, M.W.; Dustin, M.L.; Hioe, C.E. Human immunodeficiency virus type 1 envelope gp120 induces a stop signal and virological synapse formation in noninfected CD4+ T cells. J. Virol. 2008, 82, 9445–9457. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hubner, W.; Spinelli, M.A.; Chen, B.K. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 2007, 81, 12582–12595. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Hope, T.J. Live cell imaging of the HIV-1 life cycle. Trends Microbiol. 2008, 16, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D. The cell biology of HIV-1 virion genesis. Cell Host Microbe 2009, 5, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Wang, J.J.; Spearman, P. Human immunodeficiency virus type-1 gag and host vesicular trafficking pathways. Curr. Top. Microbiol. Immunol. 2009, 339, 67–84. [Google Scholar] [PubMed]
- Hioe, C.E.; Bastiani, L.; Hildreth, J.E.; Zolla-Pazner, S. Role of cellular adhesion molecules in HIV type 1 infection and their impact on virus neutralization. AIDS Res. Hum. Retroviruses 1998, 14 Suppl 3, S247–S254. [Google Scholar] [PubMed]
- Hioe, C.E.; Chien, P.C.; Lu, C.; Springer, T.A.; Wang, X.H.; Bandres, J.; Tuen, M. LFA-1 expression on target cells promotes human immunodeficiency virus type 1 infection and transmission. J. Virol. 2001, 75, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Bastiani, L.; Laal, S.; Kim, M.; Zolla-Pazner, S. Host cell-dependent alterations in envelope components of human immunodeficiency virus type 1 virions. J. Virol. 1997, 71, 3444–3450. [Google Scholar] [PubMed]
- Fortin, J.F.; Cantin, R.; Lamontagne, G.; Tremblay, M. Host-derived ICAM-1 glycoproteins incorporated on human immunodeficiency virus type 1 are biologically active and enhance viral infectivity. J. Virol. 1997, 71, 3588–3596. [Google Scholar] [PubMed]
- Rizzuto, C.D.; Sodroski, J.G. Contribution of virion ICAM-1 to human immunodeficiency virus infectivity and sensitivity to neutralization. J. Virol. 1997, 71, 4847–4851. [Google Scholar] [PubMed]
- Frank, I.; Stoiber, H.; Godar, S.; Stockinger, H.; Steindl, F.; Katinger, H.W.; Dierich, M.P. Acquisition of host cell-surface-derived molecules by HIV-1. Aids 1996, 10, 1611–1620. [Google Scholar] [PubMed]
- Garrido, M.; Mozos, A.; Martinez, A.; Garcia, F.; Serafin, A.; Morente, V.; Caballero, M.; Gil, C.; Fumero, E.; Miro, J.M.; Climent, N.; Gatell, J.M.; Alos, L. HIV-1 upregulates intercellular adhesion molecule-1 gene expression in lymphoid tissue of patients with chronic HIV-1 infection. J. Acquir. Immune Defic. Syndr. 2007, 46, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Paquette, J.S.; Fortin, J.F.; Blanchard, L.; Tremblay, M.J. Level of ICAM-1 surface expression on virus producer cells influences both the amount of virion-bound host ICAM-1 and human immunodeficiency virus type 1 infectivity. J. Virol. 1998, 72, 9329–9336. [Google Scholar] [PubMed]
- Tardif, M.R.; Tremblay, M.J. Presence of host ICAM-1 in human immunodeficiency virus type 1 virions increases productive infection of CD4+ T lymphocytes by favoring cytosolic delivery of viral material. J. Virol. 2003, 77, 12299–12309. [Google Scholar] [CrossRef] [PubMed]
- Groot, F.; Kuijpers, T.W.; Berkhout, B.; de Jong, E.C. Dendritic cell-mediated HIV-1 transmission to T cells of LAD-1 patients is impaired due to the defect in LFA-1. Retrovirology 2006, 3, 75. [Google Scholar] [CrossRef] [PubMed]
- Tardif, M.R.; Tremblay, M.J. LFA-1 is a key determinant for preferential infection of memory CD4+ T cells by human immunodeficiency virus type 1. J. Virol. 2005, 79, 13714–13724. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Mitar, I.; Sattentau, Q.J. Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J. Virol. 2007, 81, 13916–13921. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. Multiscale analysis of T cell activation: correlating in vitro and in vivo analysis of the immunological synapse. Curr. Top. Microbiol. Immunol. 2009, 334, 47–70. [Google Scholar] [PubMed]
- Poo, W.J.; Conrad, L.; Janeway, C.A. Receptor-directed focusing of lymphokine release by helper T cells. Nature 1988, 332, 378–380. [Google Scholar] [CrossRef] [PubMed]
- Campi, G.; Varma, R.; Dustin, M.L. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 2005, 202, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Varma, R.; Campi, G.; Yokosuka, T.; Saito, T.; Dustin, M.L. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 2006, 25, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Sakata-Sogawa, K.; Kobayashi, W.; Hiroshima, M.; Hashimoto-Tane, A.; Tokunaga, M.; Dustin, M.L.; Saito, T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 2005, 6, 1253–1262. [Google Scholar] [CrossRef]
- Delon, J.; Kaibuchi, K.; Germain, R. N. Exclusion of CD43 from the immunological synapse is mediated by phosphorylation-regulated relocation of the cytoskeletal adaptor moesin. Immunity 2001, 15, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Allenspach, E.J.; Cullinan, P.; Tong, J.; Tang, Q.; Tesciuba, A.G.; Cannon, J.L.; Takahashi, S.M.; Morgan, R.; Burkhardt, J.K.; Sperling, A.I. ERM-dependent movement of CD43 defines a novel protein complex distal to the immunological synapse. Immunity 2001, 15, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.S.; Dustin, M.L. Making the T cell receptor go the distance: a topological view of T cell activation. Immunity 1997, 6, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lo, P.F.; Zal, T.; Gascoigne, N.R.; Smith, B.A.; Levin, S.D.; Grey, H.M. CD28 plays a critical role in the segregation of PKC theta within the immunologic synapse. Proc. Natl. Acad. Sci. USA 2002, 99, 9369–9373. [Google Scholar] [CrossRef]
- Monks, C.R.; Freiberg, B.A.; Kupfer, H.; Sciaky, N.; Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 1998, 395, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, B.A.; Kupfer, H.; Maslanik, W.; Delli, J.; Kappler, J.; Zaller, D.M.; Kupfer, A. Staging and resetting T cell activation in SMACs. Nat. Immunol. 2002, 3, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.; Lin, J.; Littman, D.R.; Shaw, A.S.; Allen, P.M. Regulated movement of CD4 in and out of the immunological synapse. J. Immunol. 2008, 181, 8248–8257. [Google Scholar] [PubMed]
- Molon, B.; Gri, G.; Bettella, M.; Gomez-Mouton, C.; Lanzavecchia, A.; Martinez, A.C.; Manes, S.; Viola, A. T cell costimulation by chemokine receptors. Nat. Immunol. 2005, 6, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A. Adhesion receptors of the immune system. Nature 1990, 346, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Fooksman, D.R.; Vardhana, S.; Vasiliver-Shamis, G.; Liese, J.; Blair, D.; Waite, J.; Sacristan, C.; Victora, G.; Zanin-Zhorov, A.; Dustin, M.L. Functional Anatomy of T Cell Activation and Synapse Formation. Annu. Rev. Immunol. 2009, 28, 79–105. [Google Scholar] [CrossRef]
- Liu, J.; Bartesaghi, A.; Borgnia, M.J.; Sapiro, G.; Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature 2008, 455, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Kwong, P.D.; Hendrickson, W.A. Dimeric association and segmental variability in the structure of human CD4. Nature 1997, 387, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Vasiliver-Shamis, G.; Cho, M.W.; Hioe, C.E.; Dustin, M.L. Human immunodeficiency virus type 1 envelope gp120-induced partial T-cell receptor signaling creates an F-actin-depleted zone in the virological synapse. J. Virol. 2009, 83, 11341–11355. [Google Scholar] [CrossRef] [PubMed]
- Vardhana, S.; Choudhuri, K.; Varma, R.; Dustin, M.L. Essential role of ubiquitin and TSG101 protein in formation and function of the central supramolecular activation cluster. Immunity 2010, 32, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; Myszka, D.G.; Sundquist, W.I. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Perez, O.D.; Nolan, G.P. Resistance is futile: assimilation of cellular machinery by HIV-1. Immunity 2001, 15, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Donnadieu, E.; Bismuth, G.; Trautmann, A. Antigen recognition by helper T cells elicits a sequence of distinct changes of their shape and intracellular calcium. Curr. Biol. 1994, 4, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Negulescu, P.A.; Krasieva, T.B.; Khan, A.; Kerschbaum, H.H.; Cahalan, M.D. Polarity of T cell shape, motility, and sensitivity to antigen. Immunity 1996, 4, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Sims, T.N.; Soos, T.J.; Xenias, H.S.; Dubin-Thaler, B.; Hofman, J.M.; Waite, J.C.; Cameron, T.O.; Thomas, V.K.; Varma, R.; Wiggins, C.H.; Sheetz, M.P.; Littman, D.R.; Dustin, M.L. Opposing effects of PKCtheta and WASp on symmetry breaking and relocation of the immunological synapse. Cell 2007, 129, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; McKall-Faienza, K.; Schmits, R.; Bouchard, D.; Beach, J.; Speiser, D.E.; Mak, T.W.; Ohashi, P.S. Distinct roles for LFA-1 and CD28 during activation of naive T cells: adhesion versus costimulation. Immunity 1997, 7, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse: a molecular machine controlling T cell activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Colman, D.R. Neural and immunological synaptic relations. Science 2002, 298, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, J.C.; Majorovits, E.; Bossi, G.; Fuller, S.; Griffiths, G.M. Centrosome polarization delivers secretory granules to the immunological synapse. Nature 2006, 443, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.; Welsch, S.; Jolly, C.; Briggs, J.A.; Vaux, D.; Sattentau, Q.J. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J. Virol. 2010, 84, 3516–3527. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Sattentau, Q.J. Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J. Virol. 2007, 81, 7873–7884. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Sattentau, Q.J. Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J. Virol. 2005, 79, 12088–12094. [Google Scholar] [CrossRef] [PubMed]
- Arthos, J.; Cicala, C.; Martinelli, E.; Macleod, K.; Van Ryk, D.; Wei, D.; Xiao, Z.; Veenstra, T.D.; Conrad, T.P.; Lempicki, R.A.; McLaughlin, S.; Pascuccio, M.; Gopaul, R.; McNally, J.; Cruz, C.C.; Censoplano, N.; Chung, E.; Reitano, K.N.; Kottilil, S.; Goode, D.J.; Fauci, A.S. HIV-1 envelope protein binds to and signals through integrin alpha4beta7, the gut mucosal homing receptor for peripheral T cells. Nat. Immunol. 2008, 9, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Costantino, C.M.; Ploegh, H.L.; Hafler, D.A. Cathepsin S regulates class II MHC processing in human CD4+ HLA-DR+ T cells. J. Immunol. 2009, 183, 945–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Tuen, M.; Virland, D.; Cohen, S.; Mehra, N.K.; Munz, C.; Abdelwahab, S.; Garzino-Demo, A.; Hioe, C.E. Antigen stimulation induces HIV envelope gp120-specific CD4(+) T cells to secrete CCR5 ligands and suppress HIV infection. Virology 2007, 369, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N.; Lehmann, M.; Clauss, K.; Nienhaus, G.U.; Piguet, V.; Kirchhoff, F. The inability to disrupt the immunological synapse between infected human T cells and APCs distinguishes HIV-1 from most other primate lentiviruses. J. Clin. Invest. 2009, 119, 2965–2975. [Google Scholar] [PubMed]
- Thoulouze, M.I.; Sol-Foulon, N.; Blanchet, F.; Dautry-Varsat, A.; Schwartz, O.; Alcover, A. Human immunodeficiency virus type-1 infection impairs the formation of the immunological synapse. Immunity 2006, 24, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Rauch, S.; Fackler, O.T. HIV-1 Nef employs two distinct mechanisms to modulate Lck subcellular localization and TCR induced actin remodeling. PLoS One 2007, 2, e1212. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.M.; Eickel, N.; Haller, C.; Schindler, M.; Fackler, O.T. Inhibition of T-cell receptor-induced actin remodeling and relocalization of Lck are evolutionarily conserved activities of lentiviral Nef proteins. J. Virol. 2009, 83, 11528–11539. [Google Scholar] [CrossRef] [PubMed]
- Douek, D.C.; Brenchley, J.M.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Okamoto, Y.; Casazza, J.P.; Kuruppu, J.; Kunstman, K.; Wolinsky, S.; Grossman, Z.; Dybul, M.; Oxenius, A.; Price, D.A.; Connors, M.; Koup, R.A. HIV preferentially infects HIV-specific CD4+ T cells. Nature 2002, 417, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Hill, B.J.; Ambrozak, D.R.; Price, D.A.; Guenaga, F.J.; Casazza, J.P.; Kuruppu, J.; Yazdani, J.; Migueles, S.A.; Connors, M.; Roederer, M.; Douek, D.C.; Koup, R.A. T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: implications for HIV pathogenesis. J. Virol. 2004, 78, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; Schacker, T.W.; Hill, B.J.; Douek, D.C.; Routy, J.P.; Haddad, E.K.; Sekaly, R.P. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Hermankova, M.; Siliciano, J.D.; Zhou, Y.; Monie, D.; Chadwick, K.; Margolick, J.B.; Quinn, T.C.; Siliciano, R.F. Analysis of human immunodeficiency virus type 1 gene expression in latently infected resting CD4+ T lymphocytes in vivo. J. Virol. 2003, 77, 7383–7392. [Google Scholar] [CrossRef] [PubMed]
- Bousso, P. T-cell activation by dendritic cells in the lymph node: lessons from the movies. Nat. Rev. Immunol. 2008, 8, 675–684. [Google Scholar] [CrossRef]
- Dustin, M.L.; Miller, J.M.; Ranganath, S.; Vignali, D.A.; Viner, N.J.; Nelson, C.A.; Unanue, E.R. TCR-mediated adhesion of T cell hybridomas to planar bilayers containing purified MHC class II/peptide complexes and receptor shedding during detachment. J. Immunol. 1996, 157, 2014–2021. [Google Scholar] [PubMed]
- Scholer, A.; Hugues, S.; Boissonnas, A.; Fetler, L.; Amigorena, S. Intercellular adhesion molecule-1-dependent stable interactions between T cells and dendritic cells determine CD8+ T cell memory. Immunity 2008, 28, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.H.; Sidhu, S.S.; Chan, A.C. Regulation of a late phase of T cell polarity and effector functions by Crtam. Cell 2008, 132, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Beal, A.M.; Anikeeva, N.; Varma, R.; Cameron, T.O.; Vasiliver-Shamis, G.; Norris, P.J.; Dustin, M.L.; Sykulev, Y. Kinetics of early T cell receptor signaling regulate the pathway of lytic granule delivery to the secretory domain. Immunity 2009, 31, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.A.; Reeves, J.D.; Garg, H.; Foley, B.; Doms, R.W.; Blumenthal, R. Kinetic studies of HIV-1 and HIV-2 envelope glycoprotein-mediated fusion. Retrovirology 2006, 3, 90. [Google Scholar] [CrossRef] [PubMed]
- Brainard, D.M.; Tharp, W.G.; Granado, E.; Miller, N.; Trocha, A.K.; Ren, X.H.; Conrad, B.; Terwilliger, E.F.; Wyatt, R.; Walker, B.D.; Poznansky, M.C. Migration of antigen-specific T cells away from CXCR4-binding human immunodeficiency virus type 1 gp120. J. Virol. 2004, 78, 5184–5193. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Schwartz, D.H.; Clements, J.E.; Hildreth, J.E. CD4-independent, CCR5-dependent simian immunodeficiency virus infection and chemotaxis of human cells. J. Virol. 2000, 74, 6720–6724. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Schwartz, D.H.; Hildreth, J.E. T cell-tropic HIV gp120 mediates CD4 and CD8 cell chemotaxis through CXCR4 independent of CD4: implications for HIV pathogenesis. J. Immunol. 1999, 162, 6263–6267. [Google Scholar] [PubMed]
- Krementsov, D.N.; Weng, J.; Lambele, M.; Roy, N.H.; Thali, M. Tetraspanins regulate cell-to-cell transmission of HIV-1. Retrovirology 2009, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Krementsov, D.N.; Khurana, S.; Roy, N.H.; Thali, M. Formation of syncytia is repressed by tetraspanins in human immunodeficiency virus type 1-producing cells. J. Virol. 2009, 83, 7467–7474. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Dustin, M.L.; Philips, M.R. Small GTPases and LFA-1 reciprocally modulate adhesion and signaling. Immunol. Rev. 2007, 218, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Kobayashi, W.; Sakata-Sogawa, K.; Takamatsu, M.; Hashimoto-Tane, A.; Dustin, M.L.; Tokunaga, M.; Saito, T. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C theta translocation. Immunity 2008, 29, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, H.; Siliciano, J.D.; Siliciano, R.F. Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J. Virol. 2005, 79, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Korin, Y.D.; Zack, J.A. Progression to the G1b phase of the cell cycle is required for completion of human immunodeficiency virus type 1 reverse transcription in T cells. J. Virol. 1998, 72, 3161–3168. [Google Scholar] [PubMed]
- Cicala, C.; Arthos, J.; Censoplano, N.; Cruz, C.; Chung, E.; Martinelli, E.; Lempicki, R.A.; Natarajan, V.; VanRyk, D.; Daucher, M.; Fauci, A.S. HIV-1 gp120 induces NFAT nuclear translocation in resting CD4+ T-cells. Virology 2006, 345, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Cicala, C.; Arthos, J.; Selig, S.M.; Dennis, G.; Hosack, D.A.; Van Ryk, D.; Spangler, M.L.; Steenbeke, T.D.; Khazanie, P.; Gupta, N.; Yang, J.; Daucher, M.; Lempicki, R.A.; Fauci, A.S. HIV envelope induces a cascade of cell signals in non-proliferating target cells that favor virus replication. Proc. Natl. Acad. Sci. USA 2002, 99, 9380–9385. [Google Scholar] [CrossRef]
- Misse, D.; Gajardo, J.; Oblet, C.; Religa, A.; Riquet, N.; Mathieu, D.; Yssel, H.; Veas, F. Soluble HIV-1 gp120 enhances HIV-1 replication in non-dividing CD4+ T cells, mediated via cell signaling and Tat cofactor overexpression. Aids 2005, 19, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Kinter, A.L.; Umscheid, C.A.; Arthos, J.; Cicala, C.; Lin, Y.; Jackson, R.; Donoghue, E.; Ehler, L.; Adelsberger, J.; Rabin, R.L.; Fauci, A.S. HIV envelope induces virus expression from resting CD4+ T cells isolated from HIV-infected individuals in the absence of markers of cellular activation or apoptosis. J. Immunol. 2003, 170, 2449–2455. [Google Scholar] [PubMed]
- Cicala, C.; Arthos, J.; Ruiz, M.; Vaccarezza, M.; Rubbert, A.; Riva, A.; Wildt, K.; Cohen, O.; Fauci, A.S. Induction of phosphorylation and intracellular association of CC chemokine receptor 5 and focal adhesion kinase in primary human CD4+ T cells by macrophage-tropic HIV envelope. J. Immunol. 1999, 163, 420–426. [Google Scholar] [PubMed]
- Su, S.B.; Gong, W.; Grimm, M.; Utsunomiya, I.; Sargeant, R.; Oppenheim, J.J.; Ming Wang, J. Inhibition of tyrosine kinase activation blocks the down-regulation of CXC chemokine receptor 4 by HIV-1 gp120 in CD4+ T cells. J. Immunol. 1999, 162, 7128–7132. [Google Scholar] [PubMed]
- Davis, C.B.; Dikic, I.; Unutmaz, D.; Hill, C.M.; Arthos, J.; Siani, M.A.; Thompson, D.A.; Schlessinger, J.; Littman, D.R. Signal transduction due to HIV-1 envelope interactions with chemokine receptors CXCR4 or CCR5. J. Exp. Med. 1997, 186, 1793–1798. [Google Scholar] [CrossRef] [PubMed]
- Melar, M.; Ott, D.E.; Hope, T.J. Physiological levels of virion-associated human immunodeficiency virus type 1 envelope induce coreceptor-dependent calcium flux. J. Virol. 2007, 81, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Cicala, C.; Arthos, J.; Rubbert, A.; Selig, S.; Wildt, K.; Cohen, O.J.; Fauci, A.S. HIV-1 envelope induces activation of caspase-3 and cleavage of focal adhesion kinase in primary human CD4(+) T cells. Proc. Natl. Acad. Sci. USA 2000, 97, 1178–1183. [Google Scholar] [CrossRef]
- Arthos, J.; Cicala, C.; Selig, S.M.; White, A.A.; Ravindranath, H.M.; Van Ryk, D.; Steenbeke, T.D.; Machado, E.; Khazanie, P.; Hanback, M.S.; Hanback, D.B.; Rabin, R.L.; Fauci, A.S. The role of the CD4 receptor versus HIV coreceptors in envelope-mediated apoptosis in peripheral blood mononuclear cells. Virology 2002, 292, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Biard-Piechaczyk, M.; Robert-Hebmann, V.; Richard, V.; Roland, J.; Hipskind, R.A.; Devaux, C. Caspase-dependent apoptosis of cells expressing the chemokine receptor CXCR4 is induced by cell membrane-associated human immunodeficiency virus type 1 envelope glycoprotein (gp120). Virology 2000, 268, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Roggero, R.; Robert-Hebmann, V.; Harrington, S.; Roland, J.; Vergne, L.; Jaleco, S.; Devaux, C.; Biard-Piechaczyk, M. Binding of human immunodeficiency virus type 1 gp120 to CXCR4 induces mitochondrial transmembrane depolarization and cytochrome c-mediated apoptosis independently of Fas signaling. J. Virol. 2001, 75, 7637–7650. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.P.; Grivel, J.C.; Boasso, A.; Hardy, A.W.; Chougnet, C.; Dolan, M.J.; Yagita, H.; Lifson, J.D.; Shearer, G.M. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood 2005, 106, 3524–3531. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; Schnepple, D.J.; Badley, A.D. Acquired T-cell sensitivity to TRAIL mediated killing during HIV infection is regulated by CXCR4-gp120 interactions. Aids 2005, 19, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Trushin, S.A.; Algeciras-Schimnich, A.; Vlahakis, S.R.; Bren, G.D.; Warren, S.; Schnepple, D. J.; Badley, A.D. Glycoprotein 120 binding to CXCR4 causes p38-dependent primary T cell death that is facilitated by, but does not require cell-associated CD4. J. Immunol. 2007, 178, 4846–4853. [Google Scholar] [PubMed]
- Vasiliver-Shamis, G. New York University School of Medicine. 2008. [Google Scholar]
- Edinger, A.L.; Clements, J.E.; Doms, R.W. Chemokine and orphan receptors in HIV-2 and SIV tropism and pathogenesis. Virology 1999, 260, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Billadeau, D.D.; Nolz, J.C.; Gomez, T.S. Regulation of T-cell activation by the cytoskeleton. Nat. Rev. Immunol. 2007, 7, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Rosen, D.; Berke, G. Spatial relationships of microtubule-organizing centers and the contact area of cytotoxic T lymphocytes and target cells. J. Cell. Biol. 1982, 95, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Kupfer, A.; Dennert, G.; Singer, S.J. Polarization of the Golgi apparatus and the microtubule-organizing center within cloned natural killer cells bound to their targets. Proc. Natl. Acad. Sci. USA 1983, 80, 7224–7228. [Google Scholar] [CrossRef]
- Kupfer, A.; Mosmann, T.R.; Kupfer, H. Polarized expression of cytokines in cell conjugates of helper T cells and splenic B cells. Proc. Natl. Acad. Sci. USA 1991, 88, 775–779. [Google Scholar] [CrossRef]
- Nejmeddine, M.; Negi, V.S.; Mukherjee, S.; Tanaka, Y.; Orth, K.; Taylor, G.P.; Bangham, C.R. HTLV-1-Tax and ICAM-1 act on T-cell signal pathways to polarize the microtubule-organizing center at the virological synapse. Blood 2009, 114, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Nejmeddine, M.; Barnard, A.L.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R. Human T-lymphotropic virus, type 1, tax protein triggers microtubule reorientation in the virological synapse. J. Biol. Chem. 2005, 280, 29653–29660. [Google Scholar] [CrossRef] [PubMed]
- Quann, E.J.; Merino, E.; Furuta, T.; Huse, M. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nat. Immunol. 2009, 10, 627–635. [Google Scholar] [CrossRef] [PubMed]
- DeMond, A.L.; Mossman, K.D.; Starr, T.; Dustin, M.L.; Groves, J.T. T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation. Biophys. J. 2008, 94, 3286–3292. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, Y.; Douglass, A.D.; Varma, R.; Dustin, M.L.; Vale, R.D. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc. Natl. Acad. Sci. USA 2007, 104, 20296–20301. [Google Scholar] [CrossRef]
- Gomez, T.S.; Billadeau, D.D. T cell activation and the cytoskeleton: you can't have one without the other. Adv. Immunol. 2008, 97, 1–64. [Google Scholar] [PubMed]
- Barrero-Villar, M.; Cabrero, J.R.; Gordon-Alonso, M.; Barroso-Gonzalez, J.; Alvarez-Losada, S.; Munoz-Fernandez, M.A.; Sanchez-Madrid, F.; Valenzuela-Fernandez, A. Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J. Cell Sci. 2009, 122, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Baranda, S.; Gomez-Mouton, C.; Rojas, A.; Martinez-Prats, L.; Mira, E.; Ana Lacalle, R.; Valencia, A.; Dimitrov, D.S.; Viola, A.; Delgado, R.; Martinez, A.C.; Manes, S. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat. Cell Biol. 2007, 9, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Hildreth, J.E.; Schwartz, D.H. Actin-dependent receptor colocalization required for human immunodeficiency virus entry into host cells. J. Virol. 1998, 72, 5251–5255. [Google Scholar] [PubMed]
- Campbell, E.M.; Nunez, R.; Hope, T.J. Disruption of the actin cytoskeleton can complement the ability of Nef to enhance human immunodeficiency virus type 1 infectivity. J. Virol. 2004, 78, 5745–5755. [Google Scholar] [CrossRef] [PubMed]
- Yoder, A.; Yu, D.; Dong, L.; Iyer, S.R.; Xu, X.; Kelly, J.; Liu, J.; Wang, W.; Vorster, P.J.; Agulto, L.; Stephany, D.A.; Cooper, J.N.; Marsh, J.W.; Wu, Y. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 2008, 134, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Belkina, N.V.; Shaw, S. HIV infection of T cells: actin-in and actin-out. Sci. Signal. 2009, 2, pe23. [Google Scholar] [CrossRef]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.; Grigorov, B.; Senserrich, J.; Clotet, B.; Darlix, J.L.; Muriaux, D.; Este, J.A. A clathrin-dynamin-dependent endocytic pathway for the uptake of HIV-1 by direct T cell-T cell transmission. Antiviral. Res. 2008, 80, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Krausslich, H.G. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J. Virol. 2005, 79, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Bryceson, Y.T.; Meckel, T.; Vasiliver-Shamis, G.; Dustin, M.L.; Long, E.O. Integrin-dependent organization and bidirectional vesicular traffic at cytotoxic immune synapses. Immunity 2009, 31, 99–109. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Vasiliver-Shamis, G.; Dustin, M.L.; Hioe, C.E. HIV-1 Virological Synapse is not Simply a Copycat of the Immunological Synapse. Viruses 2010, 2, 1239-1260. https://doi.org/10.3390/v2051239
Vasiliver-Shamis G, Dustin ML, Hioe CE. HIV-1 Virological Synapse is not Simply a Copycat of the Immunological Synapse. Viruses. 2010; 2(5):1239-1260. https://doi.org/10.3390/v2051239
Chicago/Turabian StyleVasiliver-Shamis, Gaia, Michael L. Dustin, and Catarina E. Hioe. 2010. "HIV-1 Virological Synapse is not Simply a Copycat of the Immunological Synapse" Viruses 2, no. 5: 1239-1260. https://doi.org/10.3390/v2051239