
Modeling the Immune Response for Pathogenic and Nonpathogenic Orthohantavirus Infections in Human Lung Microvasculature Endothelial Cells

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Immunofluorescence Microscopy
2.3. Gene Expression Assay
2.4. Multiplex Immunoassay
2.5. Caspase-3/7 Glo Activity Assay
2.6. Least Squares Fit
3. Results
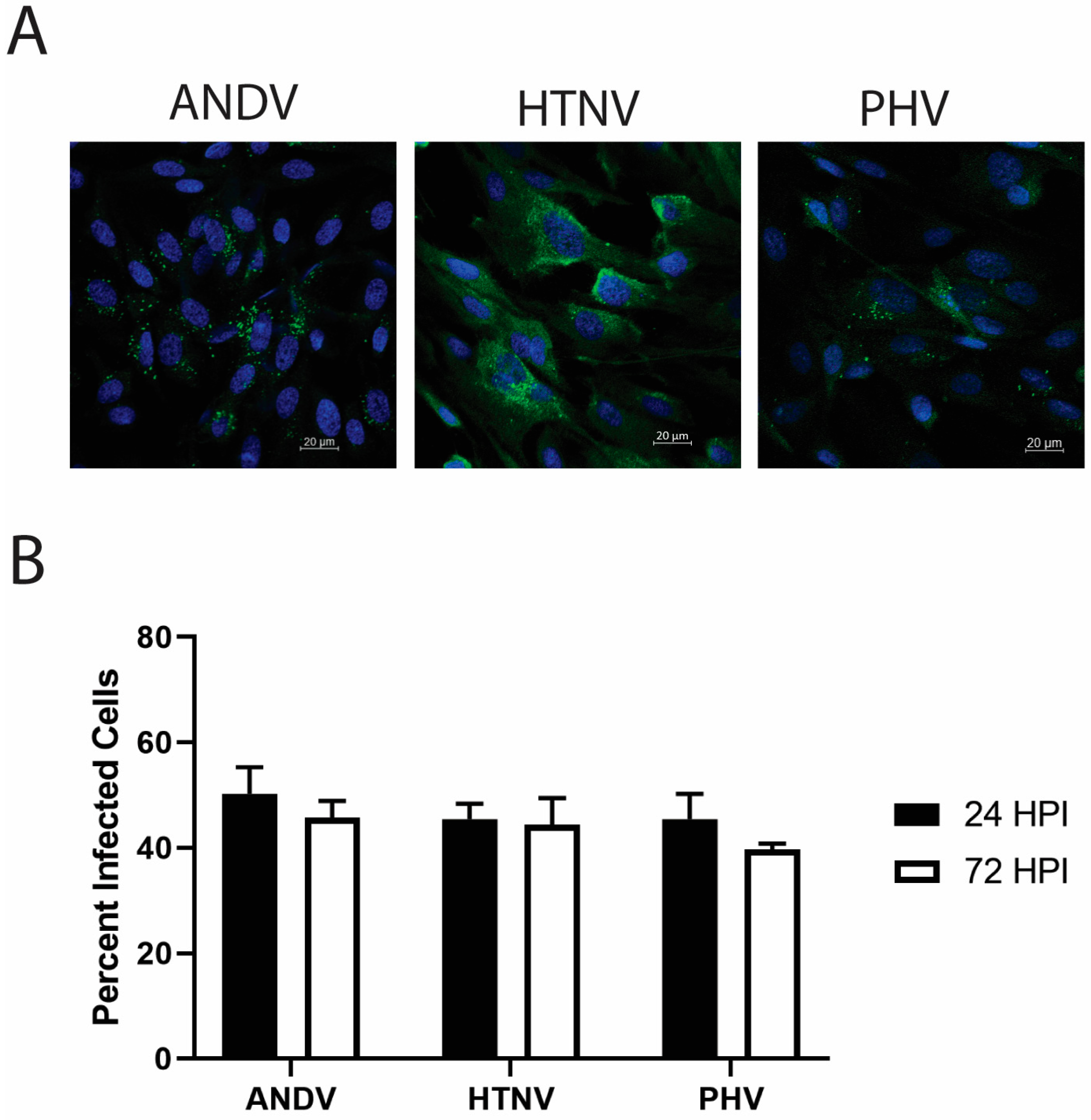
3.1. Infection of a Male HLMVEC Donor by ANDV, HTNV, or PHV
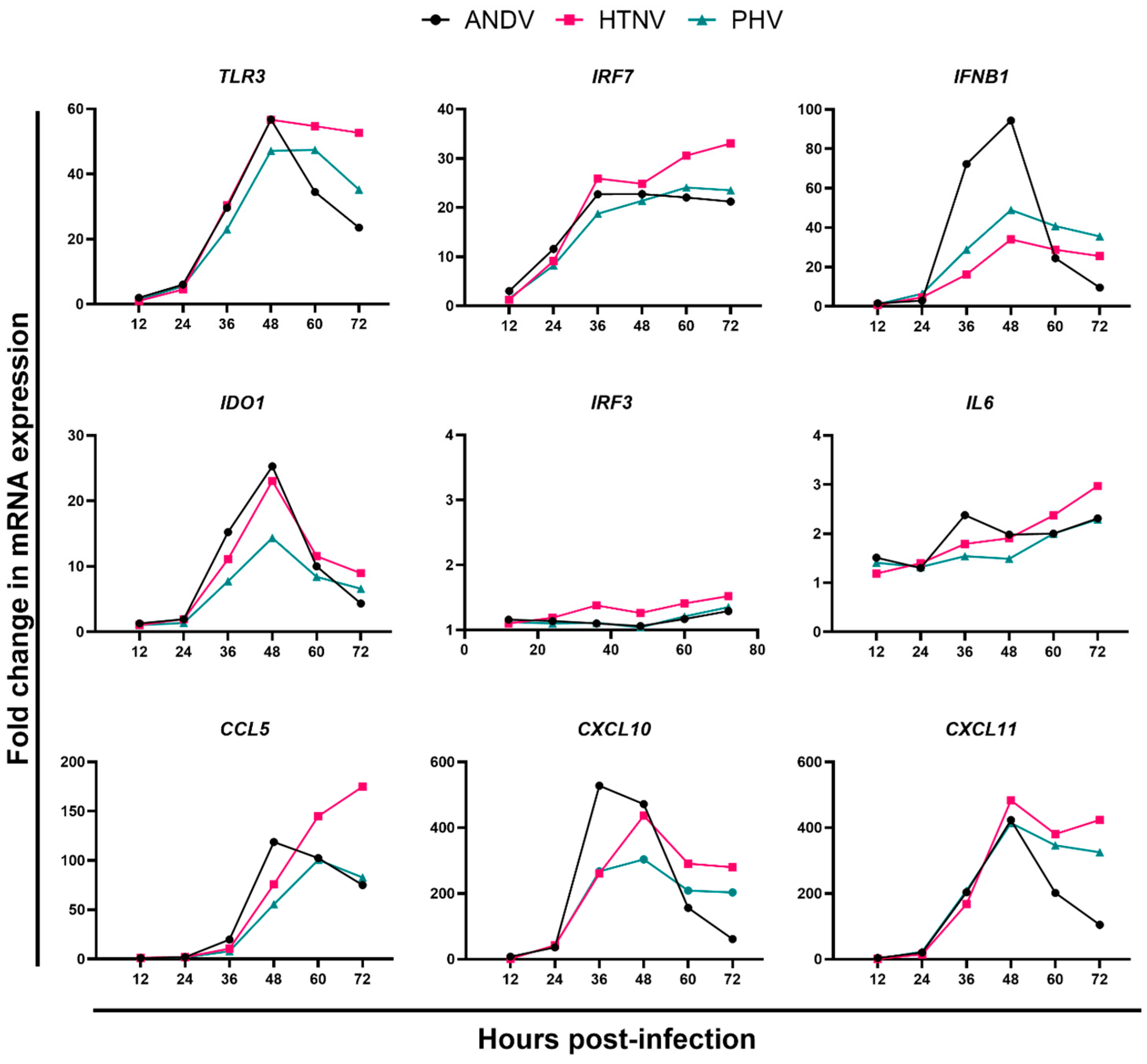
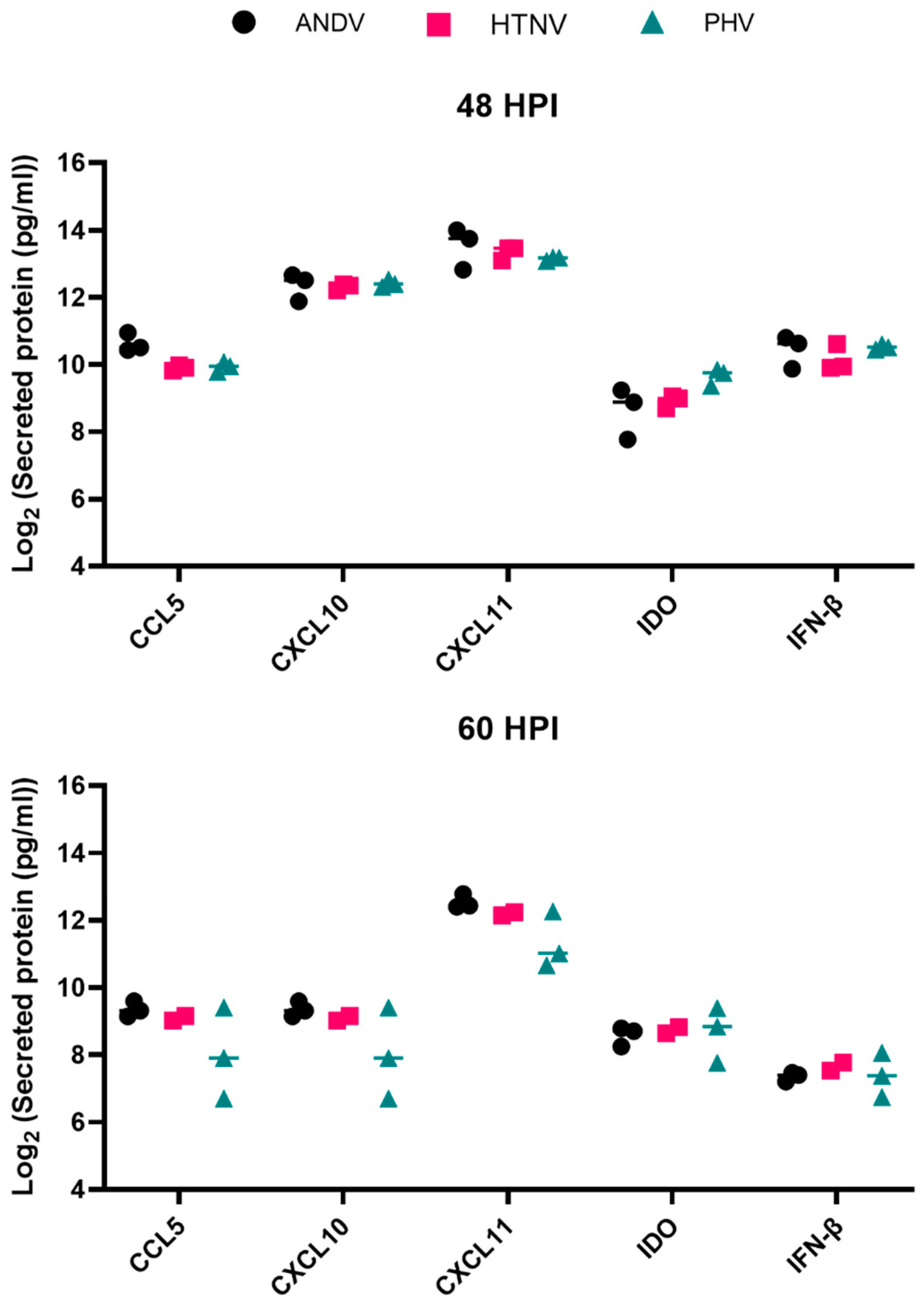
3.2. Gene and Protein Expression Profiles from Male HLMVEC Donor following ANDV, HTNV, or PHV
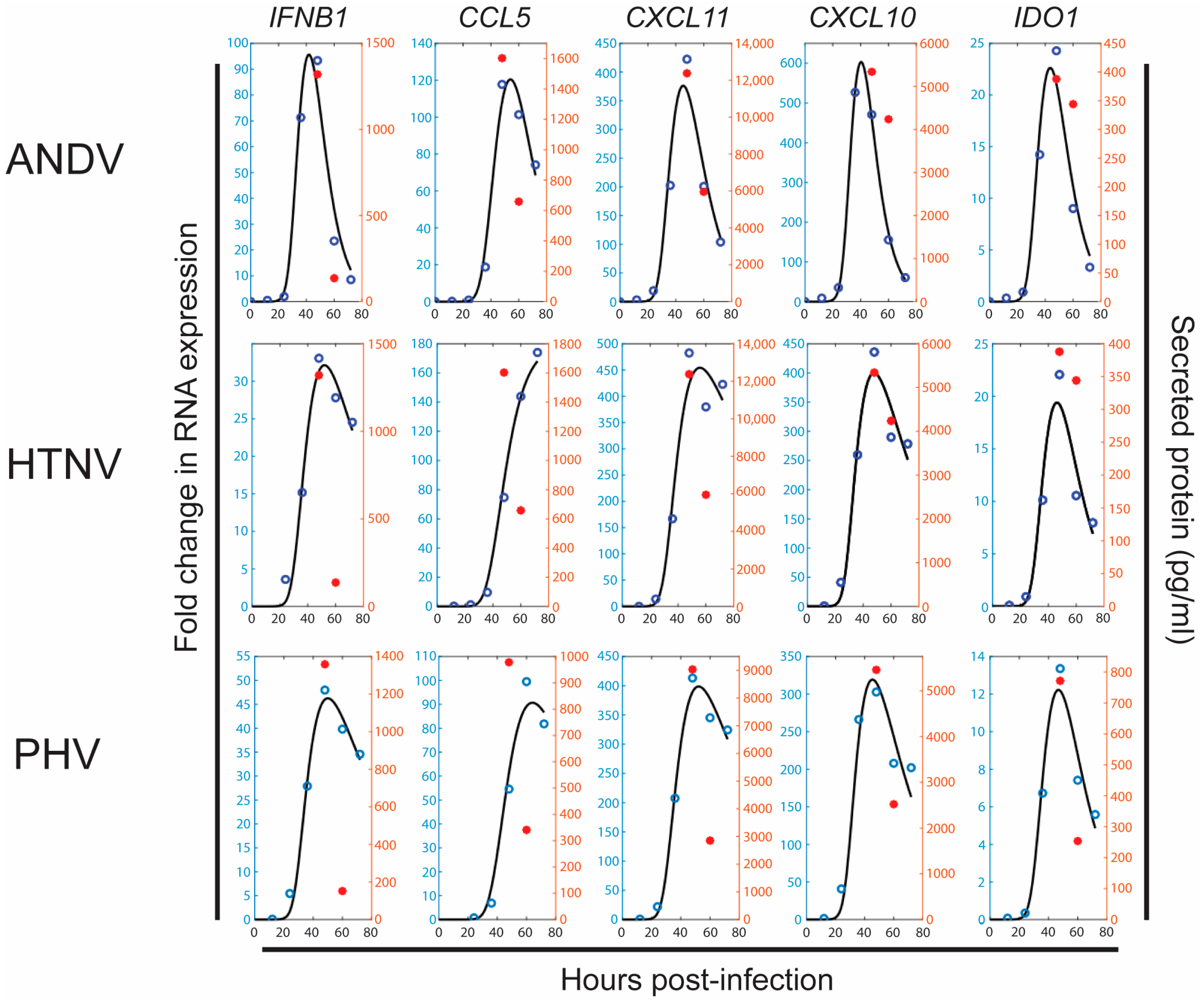
3.3. Mathematical Model of the Gene Expression Profiles and Secreted Protein Levels
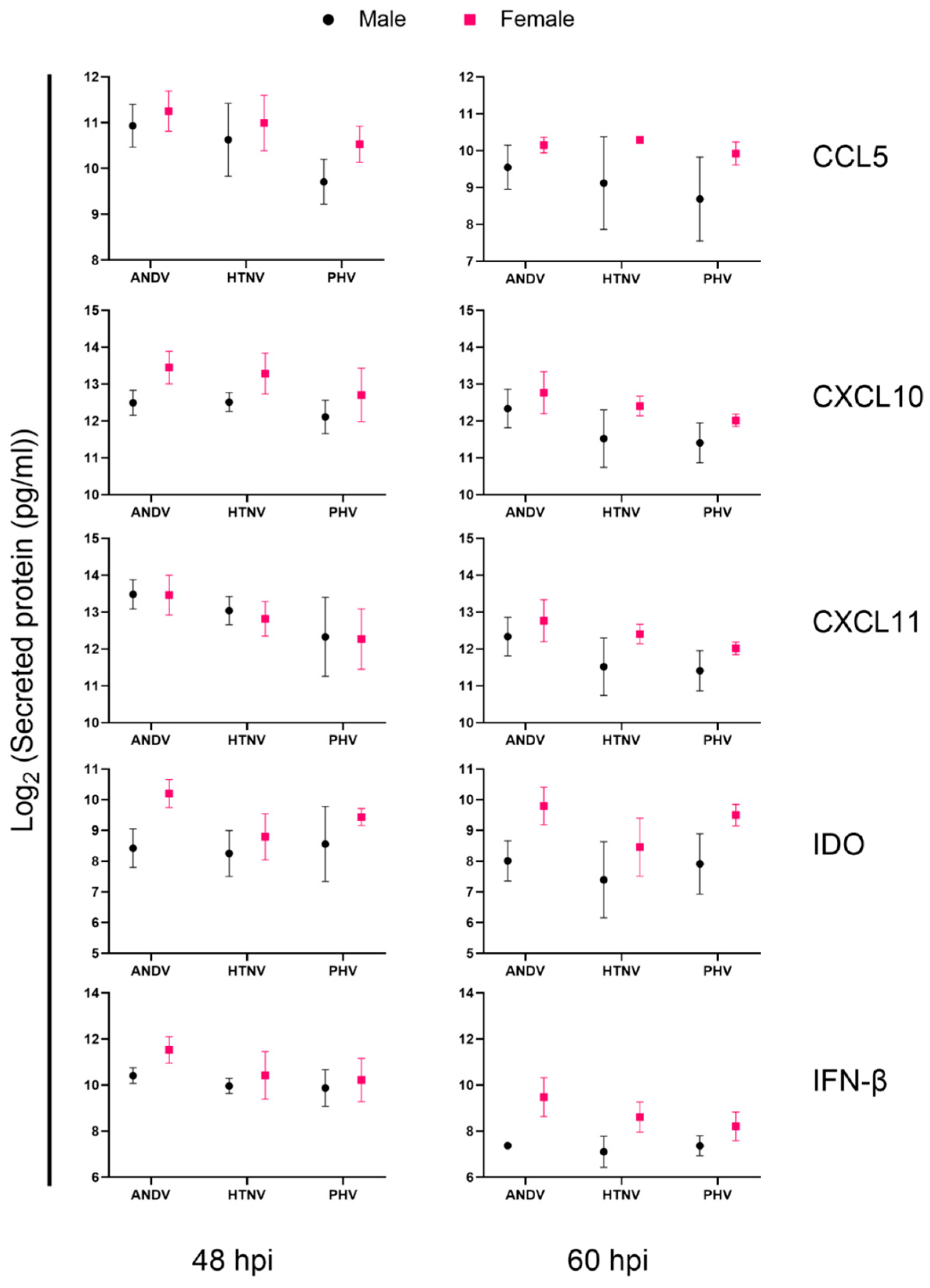
3.4. Immune Responses of Male and Female Donors following Infection
3.5. ANDV Infection Inhibits Cell Death
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schmaljohn, C.S.; Dalrymple, J.M. Analysis of Hantaan virus RNA: Evidence for a new genus of bunyaviridae. Virology 1983, 131, 482–491. [Google Scholar] [PubMed]
- Maes, P.; Alkhovsky, S.V.; Bào, Y.; Beer, M.; Birkhead, M.; Briese, T.; Buchmeier, M.J.; Calisher, C.H.; Charrel, R.N.; Choi, I.R.; et al. Taxonomy of the family Arenaviridae and the order Bunyavirales: Update 2018. Arch. Virol. 2018, 163, 2295–2310. [Google Scholar] [PubMed]
- Laenen, L.; Vergote, V.; Calisher, C.H.; Klempa, B.; Klingström, J.; Kuhn, J.H.; Maes, P. Hantaviridae: Current Classification and Future Perspectives. Viruses 2019, 11, 788. [Google Scholar] [PubMed]
- Jonsson, C.B.; Figueiredo, L.T.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar]
- Plyusnin, A.; Sironen, T. Evolution of hantaviruses: Co-speciation with reservoir hosts for more than 100 MYR. Virus Res. 2014, 187, 22–26. [Google Scholar]
- Lee, H.W.; Lee, P.W.; Johnson, K.M. Isolation of the etiologic agent of Korean Hemorrhagic fever. J. Infect. Dis. 1978, 137, 298–308. [Google Scholar]
- Nichol, S.T.; Spiropoulou, C.F.; Morzunov, S.; Rollin, P.E.; Ksiazek, T.G.; Feldmann, H.; Sanchez, A.; Childs, J.; Zaki, S.; Peters, C.J. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science 1993, 262, 914–917. [Google Scholar]
- Avšič-Županc, T.; Saksida, A.; Korva, M. Hantavirus infections. Clin. Microbiol. Infect. 2019, 21, e6–e16. [Google Scholar]
- Outinen, T.K.; Laine, O.K.; Mäkelä, S.; Pörsti, I.; Huhtala, H.; Vaheri, A.; Mustonen, J. Thrombocytopenia associates with the severity of inflammation and variables reflecting capillary leakage in Puumala Hantavirus infection, an analysis of 546 Finnish patients. Infect. Dis. 2016, 48, 682–687. [Google Scholar]
- Duchin, J.S.; Koster, F.T.; Peters, C.; Simpson, G.L.; Tempest, B.; Zaki, S.R.; Ksiazek, T.G.; Rollin, P.E.; Nichol, S.; Umland, E.T.; et al. Hantavirus Pulmonary Syndrome: A Clinical Description of 17 Patients with a Newly Recognized Disease. N. Engl. J. Med. 1994, 330, 949–955. [Google Scholar]
- MacNeil, A.; Nichol, S.T.; Spiropoulou, C.F. Hantavirus pulmonary syndrome. Virus Res. 2011, 162, 138–147. [Google Scholar]
- Borges, A.A.; Campos, G.M.; Moreli, M.L.; Morosouza, R.; Saggioro, F.P.; Figueiredo, G.G.; Livonesi, M.C.; Moraesfigueiredo, L. Role of mixed Th1 and Th2 serum cytokines on pathogenesis and prognosis of hantavirus pulmonary syndrome. Microbes Infect. 2008, 10, 1150–1157. [Google Scholar]
- Linderholm, M.; Ahlm, C.; Settergren, B.; Waage, A.; Tärnvik, A. Elevated plasma levels of tumor necrosis factor (TNF)-alpha, soluble TNF receptors, interleukin (IL)-6, and IL-10 in patients with hemorrhagic fever with renal syndrome. J. Infect. Dis. 1996, 173, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Saksida, A.; Wraber, B.; Avšič-Županc, T. Serum levels of inflammatory and regulatory cytokines in patients with hemorrhagic fever with renal syndrome. BMC Infect. Dis. 2011, 11, 142. [Google Scholar]
- Kim, Y.K.; Lee, S.C.; Kim, C.; Heo, S.T.; Choi, C.; Kim, J.M. Clinical and laboratory predictors of oliguric renal failure in haemorrhagic fever with renal syndrome caused by Hantaan virus. J. Infect. 2007, 54, 381–386. [Google Scholar] [PubMed]
- Turčinov, D.; Puljiz, I.; Markotić, A.; Kuzman, I.; Begovac, J. Clinical and laboratory findings in patients with oliguric and non-oliguric Hantavirus haemorrhagic fever with renal syndrome: An analysis of 128 patients. Clin. Microbiol. Infect. 2013, 19, 674–679. [Google Scholar]
- Peters, C.J.; Simpson, G.L.; Levy, H. Spectrum of hantavirus infection: Hemorrhagic fever with renal syndrome and hantavirus pulmonary syndrome. Annu. Rev. Med. 1999, 50, 531–545. [Google Scholar]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Mäkelä, S.; Mustonen, J. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar]
- Jonsson, C.B.; Hooper, J.; Mertz, G. Treatment of hantavirus pulmonary syndrome. Antivir. Res. 2008, 78, 162–169. [Google Scholar]
- Zaki, S.R.; Greer, P.W.; Coffield, L.M.; Goldsmith, C.S.; Nolte, K.B.; Foucar, K.; Feddersen, R.M.; E Zumwalt, R.; Miller, G.L.; Khan, A.S. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am. J. Pathol. 1995, 146, 552–579. [Google Scholar]
- Settergren, B.; Ahlm, C.; Alexeyev, O.; Billheden, J.; Stegmayr, B. Pathogenetic and clinical aspects of the renal involvement in hemorrhagic fever with renal syndrome. Renal Fail. 1997, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Poljak, M.; Županc, T.A. Immunohistochemical Detection of Hantaan Virus Antigen in Renal Tissue from Patient with Hemorrhagic Fever with Renal Syndrome. Nephron 1994, 67, 252. [Google Scholar] [CrossRef] [PubMed]
- Angulo, J.; Martínez-Valdebenito, C.; Marco, C.; Galeno, H.; Villagra, E.; Vera, L.; Lagos, N.; Becerra, N.; Mora, J.; Bermúdez, A.; et al. Serum levels of interleukin-6 are linked to the severity of the disease caused by Andes Virus. PLoS Negl. Trop. Dis. 2017, 11, e0005757. [Google Scholar] [CrossRef]
- Wang, P.Z.; Li, Z.-D.; Yu, H.-T.; Zhang, Y.; Wang, W.; Jiang, W.; Bai, X.-F. Elevated serum concentrations of inflammatory cytokines and chemokines in patients with haemorrhagic fever with renal syndrome. J. Int. Med. Res. 2012, 40, 648–656. [Google Scholar] [CrossRef]
- Morzunov, S.P.; Khaiboullina, S.F.; Jeor, S.S.; Rizvanov, A.A.; Lombardi, V.C. Multiplex Analysis of Serum Cytokines in Humans with Hantavirus Pulmonary Syndrome. Front. Immunol. 2015, 6, 432. [Google Scholar] [PubMed]
- Gavrilovskaya, I.; Gorbunova, E.; Koster, F.; Mackow, E. Elevated VEGF Levels in Pulmonary Edema Fluid and PBMCs from Patients with Acute Hantavirus Pulmonary Syndrome. Adv. Virol. 2012, 2012, 674360. [Google Scholar] [CrossRef] [PubMed]
- Maleki, K.T.; García, M.; Iglesias, A.; Alonso, D.; Ciancaglini, M.; Hammar, U.; Ljunggren, H.-G.; Schierloh, P.; Martínez, V.P.; Klingström, J. Serum Markers Associated with Severity and Outcome of Hantavirus Pulmonary Syndrome. J. Infect. Dis. 2019, 219, 1832–1840. [Google Scholar] [CrossRef]
- Kilpatrick, E.D.; Terajima, M.; Koster, F.T.; Catalina, M.D.; Cruz, J.; Ennis, F.A. Role of specific CD8+ T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J. Immunol. 2004, 172, 3297–3304. [Google Scholar] [CrossRef]
- Lewis, R.M.; Lee, H.W.; See, A.F.; Parrish, D.B.; Moon, J.S.; Kim, D.J.; Cosgriff, T.M. Changes in populations of immune effector cells during the course of haemorrhagic fever with renal syndrome. Trans. R. Soc. Trop. Med. Hyg. 1991, 85, 282–286. [Google Scholar] [CrossRef]
- Björkström, N.K.; Lindgren, T.; Stoltz, M.; Fauriat, C.; Braun, M.; Evander, M.; Michaëlsson, J.; Malmberg, K.-J.; Klingström, J.; Ahlm, C.; et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J. Exp. Med. 2010, 208, 13–21. [Google Scholar]
- Braun, M.; Björkström, N.K.; Gupta, S.; Sundström, K.; Ahlm, C.; Klingström, J.; Ljunggren, H.-G. Cell Activation in Human Hantavirus Infection Explained by Virus-Induced IL-15/IL15Rα Expression. PLoS Pathog. 2014, 10, e1004521. [Google Scholar] [CrossRef] [PubMed]
- Lundkvist, Å.; Hörling, J.; Niklasson, B. The humoral response to Puumala virus infection (nephropathia epidemica) investigated by viral protein specific immunoassays. Arch. Virol. 1993, 130, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Groen, J.; Gerding, M.; Jordans, J.G.M.; Clement, J.P.; Osterhaus, A.D.M.E. Class and subclass distribution of hantavirus-specific serum antibodies at different times after the onset of nephropathia epidemica. J. Med. Virol. 1994, 43, 39–43. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, A.; Comer, J.A.; Ksiazek, T.G.; Rollin, P.E. Sin Nombre Virus–Specific Immunoglobulin M and G Kinetics in Hantavirus Pulmonary Syndrome and the Role Played by Serologic Responses in Predicting Disease Outcome. J. Infect. Dis. 2010, 202, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Bostik, P.; Winter, J.; Ksiazek, T.G.; Rollin, P.E.; Villinger, F.; Zaki, S.R.; Peters, C.J.; Ansari, A.A. Sin nombre virus (SNV) Ig isotype antibody response during acute and convalescent phases of hantavirus pulmonary syndrome. Emerg. Infect. Dis. 2000, 6, 184–187. [Google Scholar] [CrossRef]
- Witkowski, P.T.; Bourquain, D.; Bankov, K.; Auste, B.; Dabrowski, P.W.; Nitsche, A.; Krüger, D.H.; Schaade, L. Infection of human airway epithelial cells by different subtypes of Dobrava-Belgrade virus reveals gene expression patterns corresponding to their virulence potential. Virology 2016, 493, 189–201. [Google Scholar] [CrossRef]
- Handke, W.; Oelschlegel, R.; Franke, R.; Krüger, D.H.; Rang, A. Hantaan Virus Triggers TLR3-Dependent Innate Immune Responses. J. Immunol. 2009, 182, 2849–2858. [Google Scholar] [CrossRef]
- Alff, P.J.; Sen, N.; Gorbunova, E.; Gavrilovskaya, I.N.; Mackow, E.R. The NY-1 Hantavirus Gn Cytoplasmic Tail Coprecipitates TRAF3 and Inhibits Cellular Interferon Responses by Disrupting TBK1-TRAF3 Complex Formation. J. Virol. 2008, 82, 9115–9122. [Google Scholar] [CrossRef]
- Alff, P.J.; Gavrilovskaya, I.N.; Gorbunova, E.; Endriss, K.; Chong, Y.; Geimonen, E.; Sen, N.; Reich, N.C.; Mackow, E.R. The Pathogenic NY-1 Hantavirus G1 Cytoplasmic Tail Inhibits RIG-I- and TBK-1-Directed Interferon Responses. J. Virol. 2006, 80, 9676–9686. [Google Scholar] [CrossRef]
- Taylor, S.L.; Frias-Staheli, N.; García-Sastre, A.; Schmaljohn, C.S. Hantaan virus nucleocapsid protein binds to importin alpha proteins and inhibits tumor necrosis factor alpha-induced activation of nuclear factor kappa B. J. Virol. 2009, 83, 1271–1279. [Google Scholar] [CrossRef]
- Geimonen, E.; Neff, S.; Raymond, T.; Kocer, S.S.; Gavrilovskaya, I.N.; Mackow, E.R. Pathogenic and nonpathogenic hantaviruses differentially regulate endothelial cell responses. Proc. Natl. Acad. Sci. USA 2002, 99, 13837–13842. [Google Scholar] [CrossRef] [PubMed]
- Khaiboullina, S.F.; Morzunov, S.P.; Jeor, S.C.S.; Rizvanov, A.A.; Lombardi, V.C. Hantavirus Infection Suppresses Thrombospondin-1 Expression in Cultured Endothelial Cells in a Strain-Specific Manner. Front. Microbiol. 2016, 7, 1077. [Google Scholar] [CrossRef]
- Prescott, J.; Ye, C.; Sen, G.; Hjelle, B. Induction of innate immune response genes by Sin Nombre hantavirus does not require viral replication. J. Virol. 2005, 79, 15007–15015. [Google Scholar] [CrossRef] [PubMed]
- Spiropoulou, C.F.; Albariño, C.G.; Ksiazek, T.G.; Rollin, P.E. Andes and Prospect Hill hantaviruses differ in early induction of interferon although both can downregulate interferon signaling. J. Virol. 2007, 81, 2769–2776. [Google Scholar] [CrossRef]
- Sundstrom, J.B.; Mcmullan, L.; Spiropoulou, C.F.; Hooper, W.C.; Ansari, A.A.; Peters, C.J.; Rolling, P.E. Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J. Virol. 2001, 75, 6070–6085. [Google Scholar] [CrossRef] [PubMed]
- Matthys, V.S.; Cimica, V.; Dalrymple, N.A.; Glennon, N.B.; Bianco, C.; Mackow, E.R. Hantavirus GnT Elements Mediate TRAF3 Binding and Inhibit RIG-I/TBK1-Directed Beta Interferon Transcription by Blocking IRF3 Phosphorylation. J. Virol. 2014, 88, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Ontiveros, S.J.; Li, Q.; Jonsson, C.B. Modulation of apoptosis and immune signaling pathways by the Hantaan virus nucleocapsid protein. Virology 2010, 401, 165–178. [Google Scholar] [CrossRef]
- Jonsson, C.B.; Yong, S.J.; Ontiveros, K.C. Hantaan Virus. In Molecular Detection of Human Viral Pathogens; Liu, L., Ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 649–658. [Google Scholar]
- Holmgren, A.M.; McConkey, C.A.; Shin, S. Outrunning the Red Queen: Bystander activation as a means of outpacing innate immune subversion by intracellular pathogens. Cell. Mol. Immunol. 2017, 14, 14–21. [Google Scholar] [CrossRef]
- Dahlquist, K.D.; Fitzpatrick, B.G.; Camacho, E.T.; Entzminger, S.D.; Wanner, N.C. Parameter Estimation for Gene Regulatory Networks from Microarray Data: Cold Shock Response in Saccharomyces cerevisiae. Bull. Math. Biol. 2015, 77, 1457–1492. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- De Boer, R.J.; Perelson, A.S. Target cell limited and immune control models of HIV infection: A comparison. J. Theor. Biol. 1998, 190, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Perelson, A.S.; Nelson, P.W. Mathematical Analysis of HIV-I: Dynamics in Vivo. SIAM Rev. 1999, 41, 3–44. [Google Scholar] [CrossRef]
- Fosse, J.H.; Haraldsen, G.; Falk, K.; Edelmann, R. Endothelial Cells in Emerging Viral Infections. Front. Cardiovasc. Med. 2021, 8, 619690. [Google Scholar] [CrossRef] [PubMed]
- Seber, G.A.F.; Wild, C.J. Nonlinear Regression; John Wiley & Sons: New York, NY, USA, 1989. [Google Scholar]
- Brocato, R.L.; Altamura, L.A.; Carey, B.D.; Perley, C.C.; Blancett, C.D.; Minogue, T.D.; Hooper, J.W. Comparison of transcriptional responses between pathogenic and nonpathogenic hantavirus infections in Syrian hamsters using NanoString. PLoS Neglect. Trop. Dis. 2021, 15, e0009592. [Google Scholar] [CrossRef]
- Outinen, T.K.; Mäkelä, S.M.; Ala-Houhala, I.O.; Huhtala, H.S.; Hurme, M.; Libraty, D.H.; Oja, S.S.; Pörsti, I.H.; Syrjänen, J.T.; Vaheri, A.; et al. High activity of indoleamine 2,3-dioxygenase is associated with renal insufficiency in Puumala hantavirus induced nephropathia epidemica. J. Med. Virol. 2011, 83, 731–737. [Google Scholar] [CrossRef]
- Koivula, T.T.; Tuulasvaara, A.; Hetemäki, I.; Hurme, M.; Mäkelä, S.; Mustonen, J.; Vaheri, A.; Arstila, T.P. Indoleamine 2,3-dioxygenase activity is associated with regulatory T cell response in acute Puumala hantavirus infection. Pathog. Dis. 2017, 75, ftw114. [Google Scholar] [CrossRef]
- Becerra, A.; Warke, R.V.; Xhaja, K.; Evans, B.; Evans, J.; Martin, K.; de Bosch, N.; Rothman, A.L.; Bosch, I. Increased activity of indoleamine 2,3-dioxygenase in serum from acutely infected dengue patients linked to gamma interferon antiviral function. J. Gen. Virol. 2009, 90 Pt 4, 810–817. [Google Scholar] [CrossRef]
- Larrea, E.; Riezu-Boj, J.I.; Gil-Guerrero, L.; Casares, N.; Aldabe, R.; Sarobe, P.; Civeira, M.P.; Heeney, J.L.; Rollier, C.; Verstrepen, B.; et al. Upregulation of Indoleamine 2,3-Dioxygenase in Hepatitis C Virus Infection. J. Virol. 2007, 81, 3662–3666. [Google Scholar] [CrossRef]
- Lepiller, Q.; Soulier, E.; Li, Q.; Lambotin, M.; Barths, J.; Fuchs, D.; Stoll-Keller, F.; Liang, T.J.; Barth, H. Antiviral and Immunoregulatory Effects of Indoleamine-2,3-Dioxygenase in Hepatitis C Virus Infection. J. Innate Immun. 2015, 7, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Lin, C.F.; Yeh, T.H. Influenza A virus infection induces indoleamine 2,3-dioxygenase (IDO) expression and modulates subsequent inflammatory mediators in nasal epithelial cells. Acta Otolaryngol. 2020, 140, 149–156. [Google Scholar] [CrossRef]
- Solà-Riera, C.; García, M.; Ljunggren, H.-G.; Klingström, J. Hantavirus inhibits apoptosis by preventing mitochondrial membrane potential loss through up-regulation of the pro-survival factor BCL-2. PLoS Pathog. 2020, 16, e1008297. [Google Scholar] [CrossRef] [PubMed]
- Solà-Riera, C.; Gupta, S.; Maleki, K.T.; González-Rodriguez, P.; Saidi, D.; Zimmer, C.L.; Vangeti, S.; Rivino, L.; Leo, Y.-S.; Lye, D.C.; et al. Hantavirus Inhibits TRAIL-Mediated Killing of Infected Cells by Downregulating Death Receptor 5. Cell Rep. 2019, 28, 2124–2139.e6. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.P.; Bellomo, C.M.; Cacace, M.L.; Suárez, P.; Bogni, L.; Padula, P.J. Hantavirus pulmonary syndrome in Argentina, 1995–2008. Emerg. Infect. Dis. 2010, 16, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Marks, M.A.; Li, W.; Glass, G.E.; Fang, L.-Q.; Ma, J.-Q.; Cao, W.-C. Sex differences in the incidence and case fatality rates from hemorrhagic fever with renal syndrome in China, 2004–2008. Clin. Infect. Dis. 2011, 52, 1414–1421. [Google Scholar] [CrossRef]
- Brauer, F.; Nohel, J.A. The Qualitative Theory of Ordinary Differential Equations: An Introduction; University Mathematics Series; W. A. Benjamin: New York, NY, USA, 1969; 314p. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2559 | 2572 | 2551 | 438Z013.1 | |
|---|---|---|---|---|
| Sex | Male | Male | Female | Female |
| Age | 42 | 19 | 34 | 53 |
| Race | Caucasian | Hispanic | Hispanic | Caucasian |
| Vendor | Cell App Inc. | Cell App Inc. | Cell App Inc. | Promo Cell |
| Variable | Description | Parameter | Value |
|---|---|---|---|
| E | Uninfected endothelial cell | ρ | 10−5 |
| I | Infected endothelial cell | γ | 1/6 |
| IA | Actively infected endothelial cell | δ1 | 1/12 |
| Bi | Bystander endothelial cell stage i | δ2 | 1/4 |
| Gi | Activated gene i | aij, ri | Estimated |
| Parameter | Description | Units |
|---|---|---|
| f(t) | Time-dependent activation rate of infected cells I | hpi−1 |
| γ | Maximum transition rate to actively infected cell | hpi−1 |
| ρ | External signaling rate per actively infected cell IA | (hpi × infected cell) −1 |
| δ1 | Rate external signaling decreases in actively infected cell IA | hpi−1 |
| δ2 | Transition rate between bystander cell stages | hpi−1 |
| aij, j = 1, 3, 4, 5 i = 1, 2 | Fold change rate in gene Gj within a bystander cell Bi | (fold change)/(hpi × bystander cell) |
| ai2, i = 1, 2 | Fold change rate in gene G2 within a bystander cell Bi+2 | (fold change)/(hpi × bystander cell) |
| ri, i = 1, 2, 3, 4, 5 | Rate gene expression level Gi decreased within bystander cells | hpi−1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, E.P.; Nandi, A.; Nam, V.; Allen, L.J.S.; Trindade, A.A.; Kosiewicz, M.M.; Jonsson, C.B. Modeling the Immune Response for Pathogenic and Nonpathogenic Orthohantavirus Infections in Human Lung Microvasculature Endothelial Cells. Viruses 2023, 15, 1806. https://doi.org/10.3390/v15091806
Williams EP, Nandi A, Nam V, Allen LJS, Trindade AA, Kosiewicz MM, Jonsson CB. Modeling the Immune Response for Pathogenic and Nonpathogenic Orthohantavirus Infections in Human Lung Microvasculature Endothelial Cells. Viruses. 2023; 15(9):1806. https://doi.org/10.3390/v15091806
Chicago/Turabian StyleWilliams, Evan P., Aadrita Nandi, Victoria Nam, Linda J. S. Allen, A. Alexandre Trindade, Michele M. Kosiewicz, and Colleen B. Jonsson. 2023. "Modeling the Immune Response for Pathogenic and Nonpathogenic Orthohantavirus Infections in Human Lung Microvasculature Endothelial Cells" Viruses 15, no. 9: 1806. https://doi.org/10.3390/v15091806