

Phylogenetic and Phylodynamic Analysis of Delta Strains Circulating in Italy
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sequencing Analysis
2.3. Reference HDV Sequences
2.4. Genotyping and Phylogenetic Analysis
2.5. Phylodynamic Analysis of HDV Genotype 1
3. Results
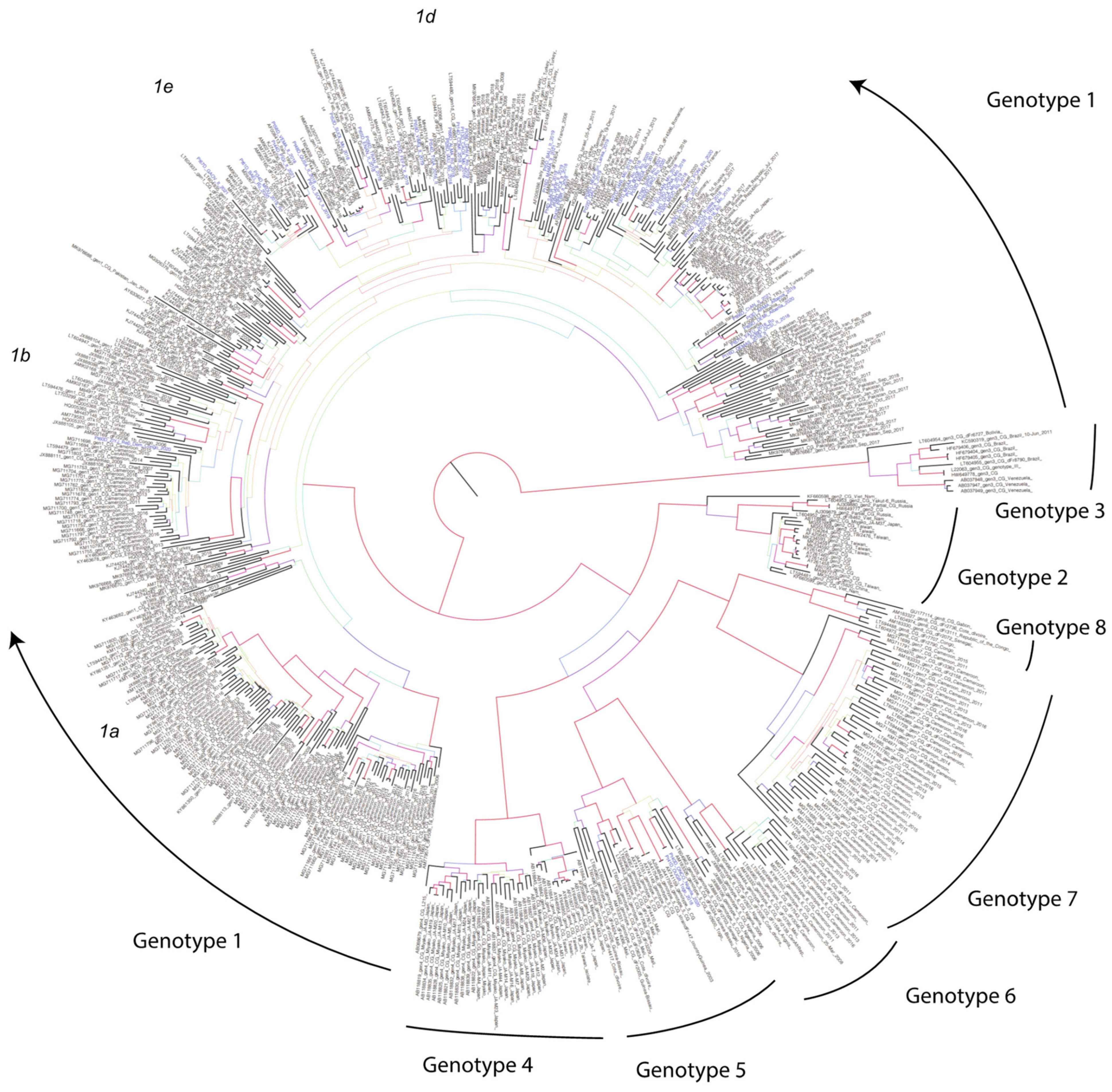
3.1. Phylogenetic Analysis
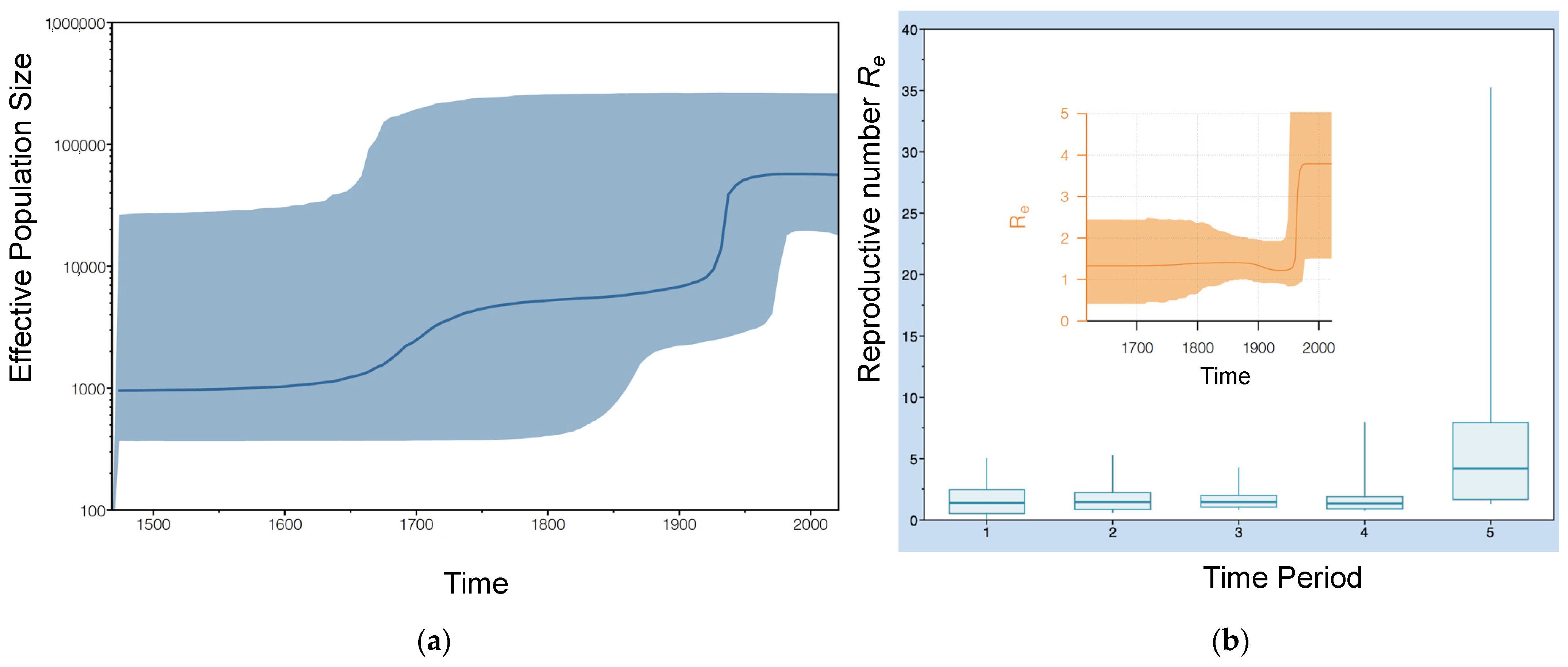
3.2. Phylodynamic Analysis
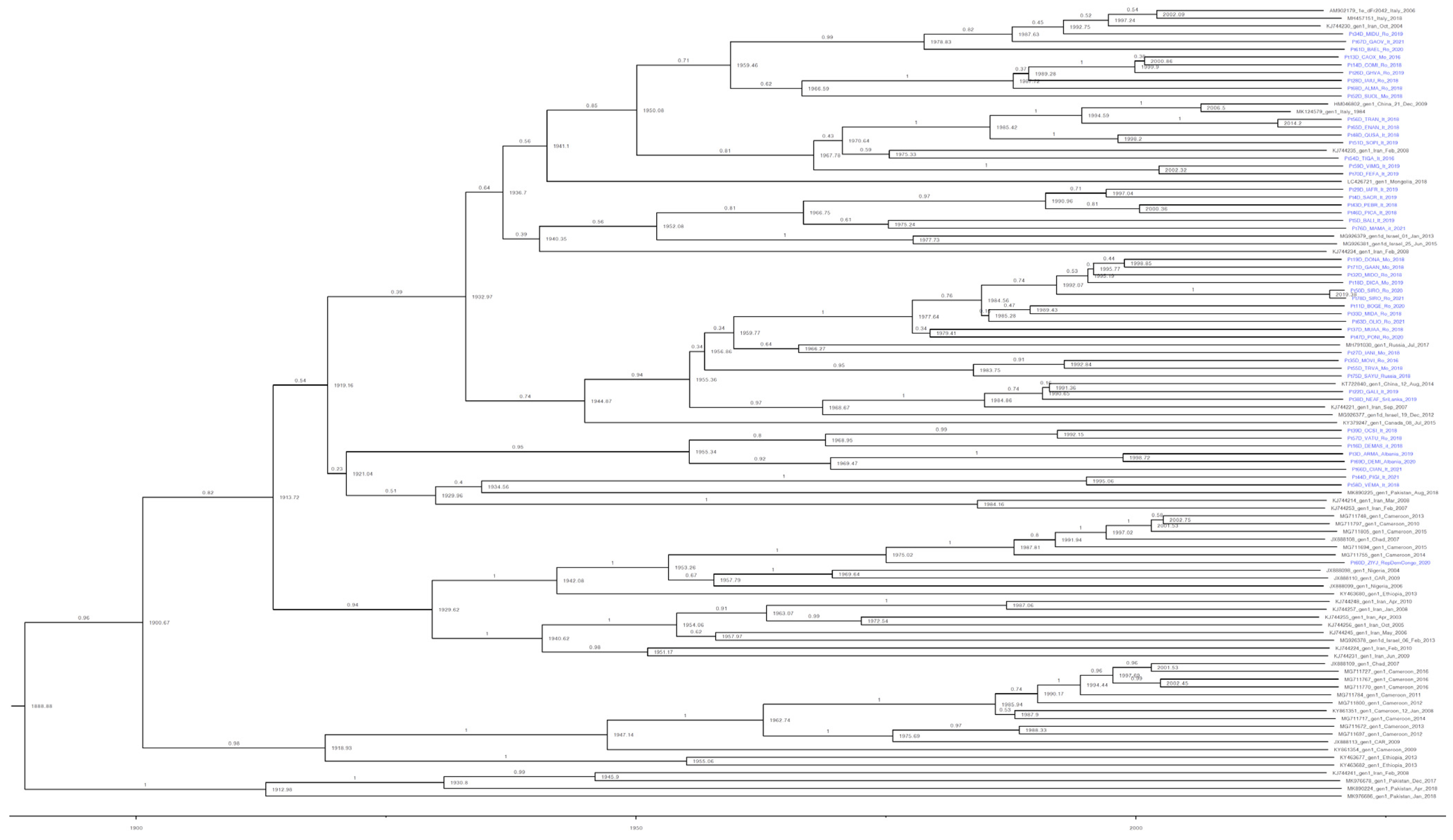
3.3. Estimating the Introduction of HDV-1 into Italy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Caviglia, G.P.; Ciancio, A.; Rizzetto, M. A Review of HDV Infection. Viruses 2022, 14, 1749. [Google Scholar] [CrossRef]
- Bender, D.; Glitscher, M.; Hildt, E. Viral hepatitis A to E: Prevalence, pathogen characteristics, and pathogenesis. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 2022, 65, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Abou-Jaoudé, G.; Sureau, C. Entry of Hepatitis Delta Virus Requires the Conserved Cysteine Residues of the Hepatitis B Virus Envelope Protein Antigenic Loop and Is Blocked by Inhibitors of Thiol-Disulfide Exchange. J. Virol. 2007, 81, 13057–13066. [Google Scholar] [CrossRef]
- Alfaiate, D.; Dény, P.; Durantel, D. Hepatitis Delta Virus: From Biological and Medical Aspects to Current and Investigational Therapeutic Options. Antiviral Res. 2015, 122, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Botelho-Souza, L.F.; Vasconcelos, M.P.A.; Dos Santos, A.d.O.; Salcedo, J.M.V.; Vieira, D.S. Hepatitis Delta: Virological and Clinical Aspects. Virol. J. 2017, 14, 177. [Google Scholar] [CrossRef]
- Lempp, F.A.; Ni, Y.; Urban, S. Hepatitis Delta Virus: Insights into a Peculiar Pathogen and Novel Treatment Options. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Dandri, M. Hepatitis B and Delta Virus: Advances on Studies about Interactions between the Two Viruses and the Infected Hepatocyte. J. Clin. Transl. Hepatol. 2015, 3, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M. Host RNA Circles and the Origin of Hepatitis Delta Virus. World J. Gastroenterol. 2014, 20, 2971–2978. [Google Scholar] [CrossRef]
- Miao, Z.; Zhang, S.; Ma, Z.; Hakim, M.S.; Wang, W.; Peppelenbosch, M.P.; Pan, Q. Recombinant Identification, Molecular Classification and Proposed Reference Genomes for Hepatitis Delta Virus. J. Viral Hepat. 2019, 26, 183–190. [Google Scholar] [CrossRef]
- Le Gal, F.; Brichler, S.; Drugan, T.; Alloui, C.; Roulot, D.; Pawlotsky, J.-M.; Dény, P.; Gordien, E. Genetic Diversity and Worldwide Distribution of the Deltavirus Genus: A Study of 2,152 Clinical Strains. Hepatology 2017, 66, 1826–1841. [Google Scholar] [CrossRef]
- Karimzadeh, H.; Usman, Z.; Frishman, D.; Roggendorf, M. Genetic Diversity of Hepatitis D Virus Genotype-1 in Europe Allows Classification into Subtypes. J. Viral Hepat. 2019, 26, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-W.; Huang, Y.-H.; Huo, T.-I.; Shih, H.H.; Sheen, I.-J.; Chen, S.-W.; Lee, P.-C.; Lee, S.-D.; Wu, J.-C. Genotypes and Viremia of Hepatitis B and D Viruses Are Associated with Outcomes of Chronic Hepatitis D Patients. Gastroenterology 2006, 130, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Ivaniushina, V.; Radjef, N.; Alexeeva, M.; Gault, E.; Semenov, S.; Salhi, M.; Kiselev, O.; Dény, P. Hepatitis Delta Virus Genotypes I and II Cocirculate in an Endemic Area of Yakutia, Russia. J. Gen. Virol. 2001, 82, 2709–2718. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Chiang, T.Y.; Sheen, I.J. Characterization and Phylogenetic Analysis of a Novel Hepatitis D Virus Strain Discovered by Restriction Fragment Length Polymorphism Analysis. J. Gen. Virol. 1998, 79 Pt 5, 1105–1113. [Google Scholar] [CrossRef]
- Hughes, S.A.; Wedemeyer, H.; Harrison, P.M. Hepatitis Delta Virus. Lancet 2011, 378, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Radjef, N.; Gordien, E.; Ivaniushina, V.; Gault, E.; Anaïs, P.; Drugan, T.; Trinchet, J.-C.; Roulot, D.; Tamby, M.; Milinkovitch, M.C.; et al. Molecular Phylogenetic Analyses Indicate a Wide and Ancient Radiation of African Hepatitis Delta Virus, Suggesting a Deltavirus Genus of at Least Seven Major Clades. J. Virol. 2004, 78, 2537–2544. [Google Scholar] [CrossRef]
- Hayashi, T.; Takeshita, Y.; Hutin, Y.J.-F.; Harmanci, H.; Easterbrook, P.; Hess, S.; van Holten, J.; Oru, E.O.; Kaneko, S.; Yurdaydin, C.; et al. The Global Hepatitis Delta Virus (HDV) Epidemic: What Gaps to Address in Order to Mount a Public Health Response? Arch. Public Health 2021, 79, 180. [Google Scholar] [CrossRef]
- World Health Organization. Global Hepatitis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Farci, P. Delta Hepatitis: An Update. J. Hepatol. 2003, 39 (Suppl. 1), S212–S219. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Shen, D.-T.; Ji, D.-Z.; Han, P.-C.; Zhang, W.-M.; Ma, J.-F.; Chen, W.-S.; Goyal, H.; Pan, S.; Xu, H.-G. Prevalence and Burden of Hepatitis D Virus Infection in the Global Population: A Systematic Review and Meta-Analysis. Gut 2019, 68, 512–521. [Google Scholar] [CrossRef]
- Miao, Z.; Zhang, S.; Ou, X.; Li, S.; Ma, Z.; Wang, W.; Peppelenbosch, M.P.; Liu, J.; Pan, Q. Estimating the Global Prevalence, Disease Progression, and Clinical Outcome of Hepatitis Delta Virus Infection. J. Infect. Dis. 2020, 221, 1677–1687. [Google Scholar] [CrossRef]
- Stockdale, A.J.; Kreuels, B.; Henrion, M.Y.R.; Giorgi, E.; Kyomuhangi, I.; de Martel, C.; Hutin, Y.; Geretti, A.M. The Global Prevalence of Hepatitis D Virus Infection: Systematic Review and Meta-Analysis. J. Hepatol. 2020, 73, 523–532. [Google Scholar] [CrossRef]
- Kamal, H.; Westman, G.; Falconer, K.; Duberg, A.-S.; Weiland, O.; Haverinen, S.; Wejstål, R.; Carlsson, T.; Kampmann, C.; Larsson, S.B.; et al. Long-Term Study of Hepatitis Delta Virus Infection at Secondary Care Centers: The Impact of Viremia on Liver-Related Outcomes. Hepatology 2020, 72, 1177–1190. [Google Scholar] [CrossRef]
- Alfaiate, D.; Clément, S.; Gomes, D.; Goossens, N.; Negro, F. Chronic Hepatitis D and Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis of Observational Studies. J. Hepatol. 2020, 73, 533–539. [Google Scholar] [CrossRef]
- Gheorghe, L.; Csiki, I.E.; Iacob, S.; Gheorghe, C.; Trifan, A.; Grigorescu, M.; Motoc, A.; Suceveanu, A.; Curescu, M.; Caruntu, F.; et al. Hepatitis Delta Virus Infection in Romania: Prevalence and Risk Factors. J. Gastrointest. Liver Dis. JGLD 2015, 24, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Smedile, A.; Lavarini, C.; Farci, P.; Aricò, S.; Marinucci, G.; Dentico, P.; Giuliani, G.; Cargnel, A.; Del Vecchio Blanco, C.; Rizzetto, M. Epidemiologic Patterns of Infection with the Hepatitis B Virus-Associated Delta Agent in Italy. Am. J. Epidemiol. 1983, 117, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Sagnelli, E.; Stroffolini, T.; Ascione, A.; Chiaramonte, M.; Craxì, A.; Giusti, G.; Piccinino, F. Decrease in HDV Endemicity in Italy. J. Hepatol. 1997, 26, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, G.B.; Stroffolini, T.; Chiaramonte, M.; Ascione, T.; Stornaiuolo, G.; Lobello, S.; Sagnelli, E.; Brunetto, M.R.; Rizzetto, M. Chronic Hepatitis D: A Vanishing Disease? An Italian Multicenter Study. Hepatology 2000, 32, 824–827. [Google Scholar] [CrossRef]
- Stroffolini, T.; Ciancio, A.; Furlan, C.; Vinci, M.; Fontana, R.; Russello, M.; Colloredo, G.; Morisco, F.; Coppola, N.; Babudieri, S.; et al. Migratory Flow and Hepatitis Delta Infection in Italy: A New Challenge at the Beginning of the Third Millennium. J. Viral Hepat. 2020, 27, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, F.; Gault, E.; Ripault, M.-P.; Serpaggi, J.; Trinchet, J.-C.; Gordien, E.; Dény, P. Eighth Major Clade for Hepatitis Delta Virus. Emerg. Infect. Dis. 2006, 12, 1447–1450. [Google Scholar] [CrossRef]
- Usman, Z.; Velkov, S.; Protzer, U.; Roggendorf, M.; Frishman, D.; Karimzadeh, H. HDVdb: A Comprehensive Hepatitis D Virus Database. Viruses 2020, 12, 538. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Drummond, A.J.; Bouckaert, R.R. Bayesian Evolutionary Analysis with BEAST; Cambridge University Press: Singapore, 2015. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biology. 2007, 7, 214. [Google Scholar] [CrossRef]
- Caviglia, G.P.; Martini, S.; Ciancio, A.; Niro, G.A.; Olivero, A.; Fontana, R.; Tandoi, F.; Rosso, C.; Romagnoli, R.; Saracco, G.M.; et al. The Hepatitis D Virus in Italy. A Vanishing Infection, Not yet a Vanished Disease. J. Adv. Res. 2021, 33, 183–187. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Romano’, L.; Zanetti, A.R. Hepatitis B Vaccination: A Historical Overview with a Focus on the Italian Achievements. Viruses 2022, 14, 1515. [Google Scholar] [CrossRef]
- Lampertico, P.; Agarwal, K.; Berg, T.; Buti, M.; Janssen, H.L.A.; Papatheodoridis, G.; Zoulim, F.; Tacke, F. EASL 2017 Clinical Practice Guidelines on the Management of Hepatitis B Virus Infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef]
- SIMIT. AISF Indicazioni Operative AISF-SIMIT per La Diagnosi e La Gestione Clinica Del Paziente Affetto Da Epatite Delta; SIMIT: Firenze, Italy, 2023. [Google Scholar]
- Karlsen, A.A.; Kyuregyan, K.K.; Isaeva, O.V.; Kichatova, V.S.; Asadi Mobarkhan, F.A.; Bezuglova, L.V.; Netesova, I.G.; Manuylov, V.A.; Pochtovyi, A.A.; Gushchin, V.A.; et al. Different Evolutionary Dynamics of Hepatitis B Virus Genotypes A and D, and Hepatitis D Virus Genotypes 1 and 2 in an Endemic Area of Yakutia, Russia. BMC Infect. Dis. 2022, 22, 452. [Google Scholar] [CrossRef]
- Celik, I.; Karatayli, E.; Cevik, E.; Kabakçi, S.G.; Karatayli, S.C.; Dinç, B.; Cinar, K.; Yalçin, K.; Idilman, R.; Yurdaydin, C.; et al. Complete Genome Sequences and Phylogenetic Analysis of Hepatitis Delta Viruses Isolated from Nine Turkish Patients. Arch. Virol. 2011, 156, 2215–2220. [Google Scholar] [CrossRef]
- Yates, R. A brief history of British drug policy: 1850–1950. Ther. Communities 2020, 41, 57–66. Available online: https://www.stir.ac.uk/research/hub/publication/1649029 (accessed on 10 August 2023). [CrossRef]
- Highlights of Transfusion Medicine History. Available online: https://www.aabb.org/news-resources/resources/transfusion-medicine/highlights-of-transfusion-medicine-history (accessed on 12 August 2023).
- Romeo, R.; Perbellini, R. Hepatitis Delta Virus: Making the Point from Virus Isolation up to 2014. World J. Hepatol. 2015, 7, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, D.; Crispim, M.A.; Santos, C.; Ferreira, F.d.L.; Fraiji, N.A.; Komninakis, S.V.; Diaz, R.S. Influence of Hepatitis B Virus (HBV) Genotype on the Clinical Course of Disease in Patients Coinfected with HBV and Hepatitis Delta Virus. J. Infect. Dis. 2009, 199, 1608–1611. [Google Scholar] [CrossRef] [PubMed]
- Bahoussi, A.N.; Wang, P.-H.; Guo, Y.-Y.; Rabbani, N.; Wu, C.; Xing, L. Global Distribution and Natural Recombination of Hepatitis D Virus: Implication of Kyrgyzstan Emerging HDVs in the Clinical Outcomes. Viruses 2022, 14, 1467. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salichos, L.; Minosse, C.; Visco-Comandini, U.; Taibi, C.; Zulian, V.; D’Offizi, G.; Pallothu, N.; McPhee, F.; Garbuglia, A.R. Phylogenetic and Phylodynamic Analysis of Delta Strains Circulating in Italy. Viruses 2023, 15, 1791. https://doi.org/10.3390/v15091791
Salichos L, Minosse C, Visco-Comandini U, Taibi C, Zulian V, D’Offizi G, Pallothu N, McPhee F, Garbuglia AR. Phylogenetic and Phylodynamic Analysis of Delta Strains Circulating in Italy. Viruses. 2023; 15(9):1791. https://doi.org/10.3390/v15091791
Chicago/Turabian StyleSalichos, Leonidas, Claudia Minosse, Ubaldo Visco-Comandini, Chiara Taibi, Verdiana Zulian, Gianpiero D’Offizi, Nayan Pallothu, Fiona McPhee, and Anna Rosa Garbuglia. 2023. "Phylogenetic and Phylodynamic Analysis of Delta Strains Circulating in Italy" Viruses 15, no. 9: 1791. https://doi.org/10.3390/v15091791