
HSV-1 Infection of Epithelial Dendritic Cells Is a Critical Strategy for Interfering with Antiviral Immunity

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cell Lines
2.3. Virus
2.4. Isolation of Dendritic Cells from Mouse Skin and Bone Marrow
2.5. Analysis of Virus Growth in Cells
2.6. Mouse Experiment Design
2.7. Immunofluorescence and Confocal Microscopy
2.8. Quantification of Viral Load by qRT-PCR
2.9. Cytokine Analysis
2.10. Histopathology
2.11. Neutralizing Antibody Detection
2.12. Flow Cytometry Analysis
2.13. ELISPOT Assay
2.14. Identification of the Effect of HVEM Deficiency
2.15. Statistical Analysis
3. Results
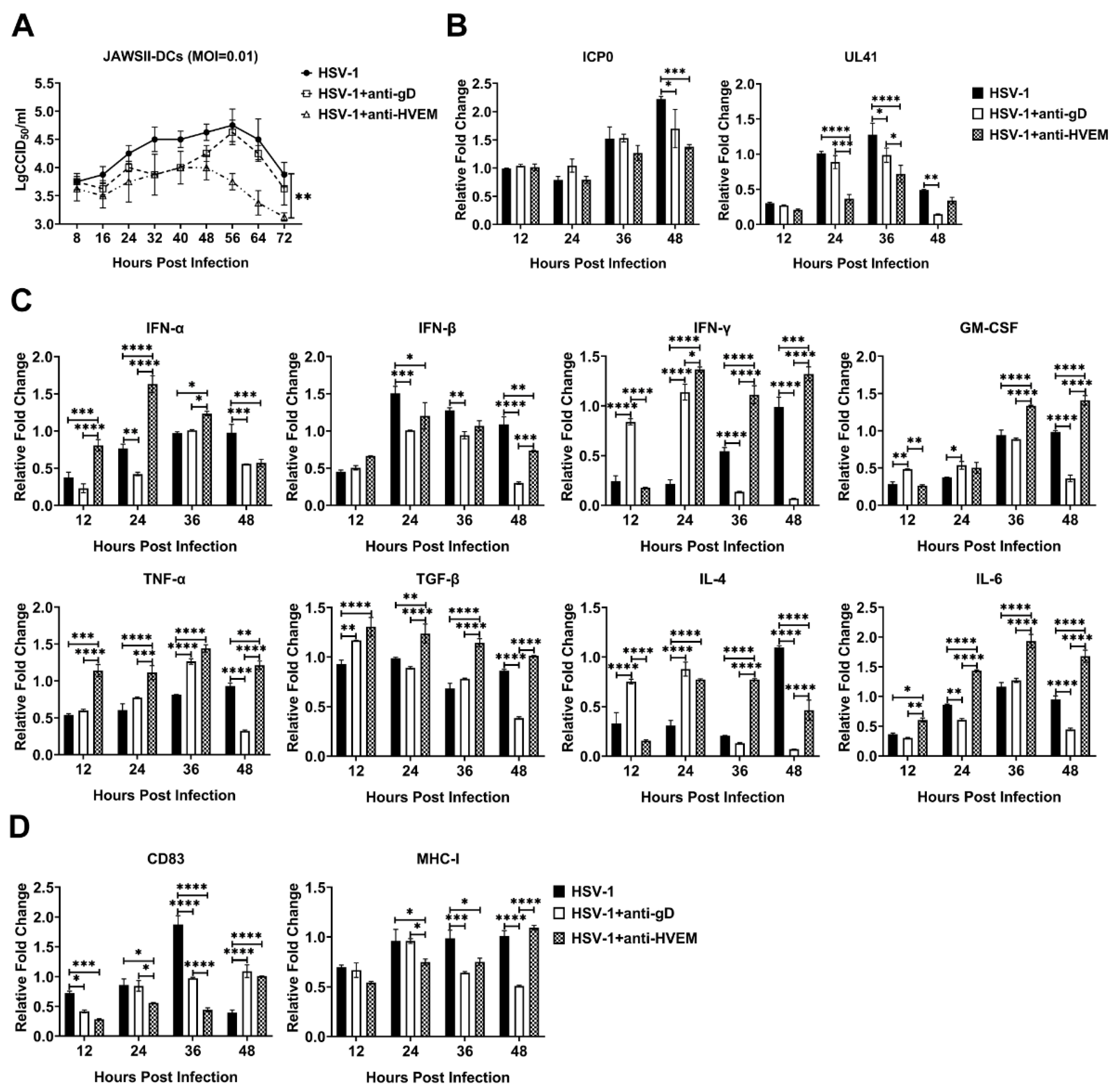
3.1. HSV-1 Enters Dendritic Cells via gD Binding to HVEM and Replicates in the Cells
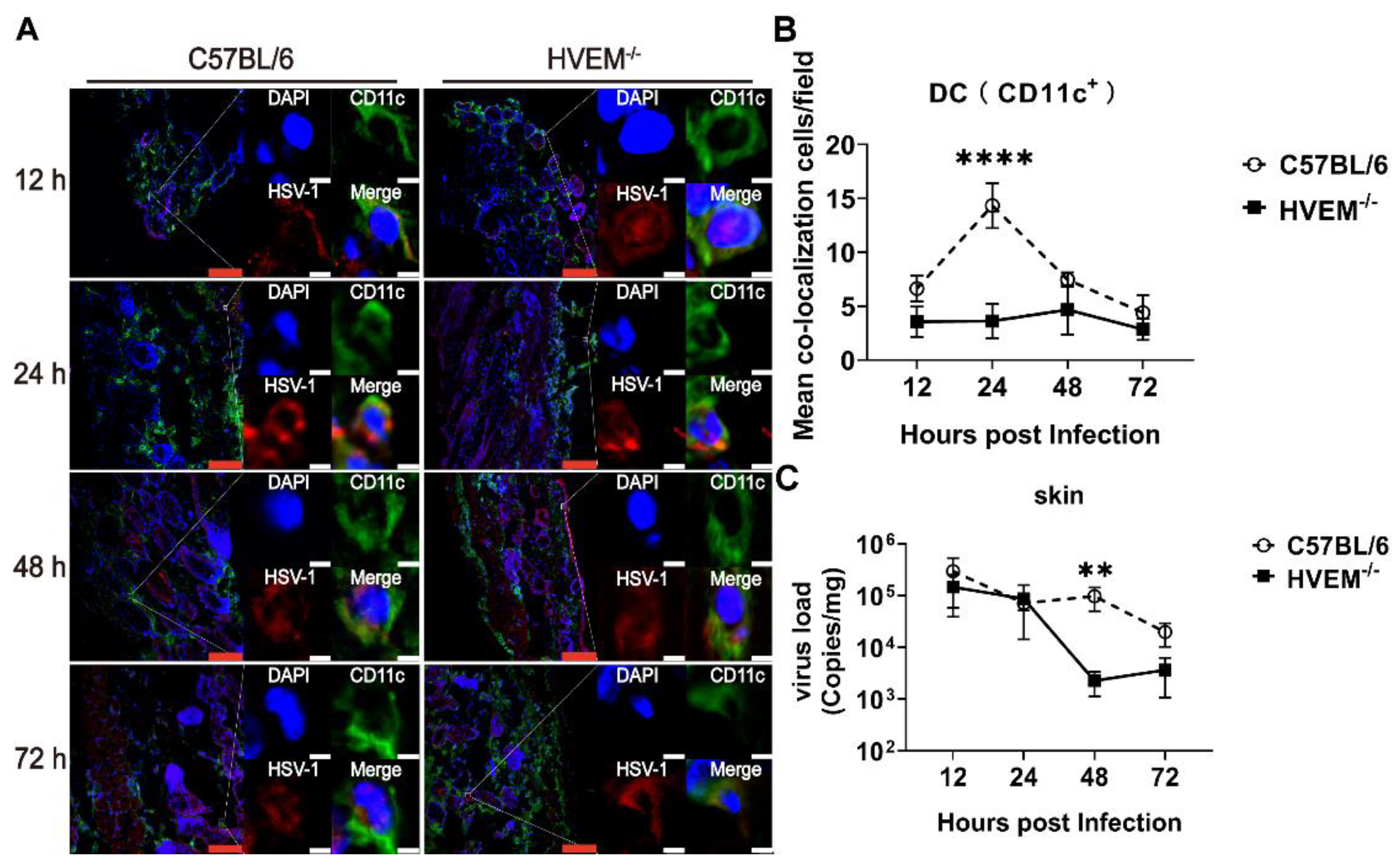
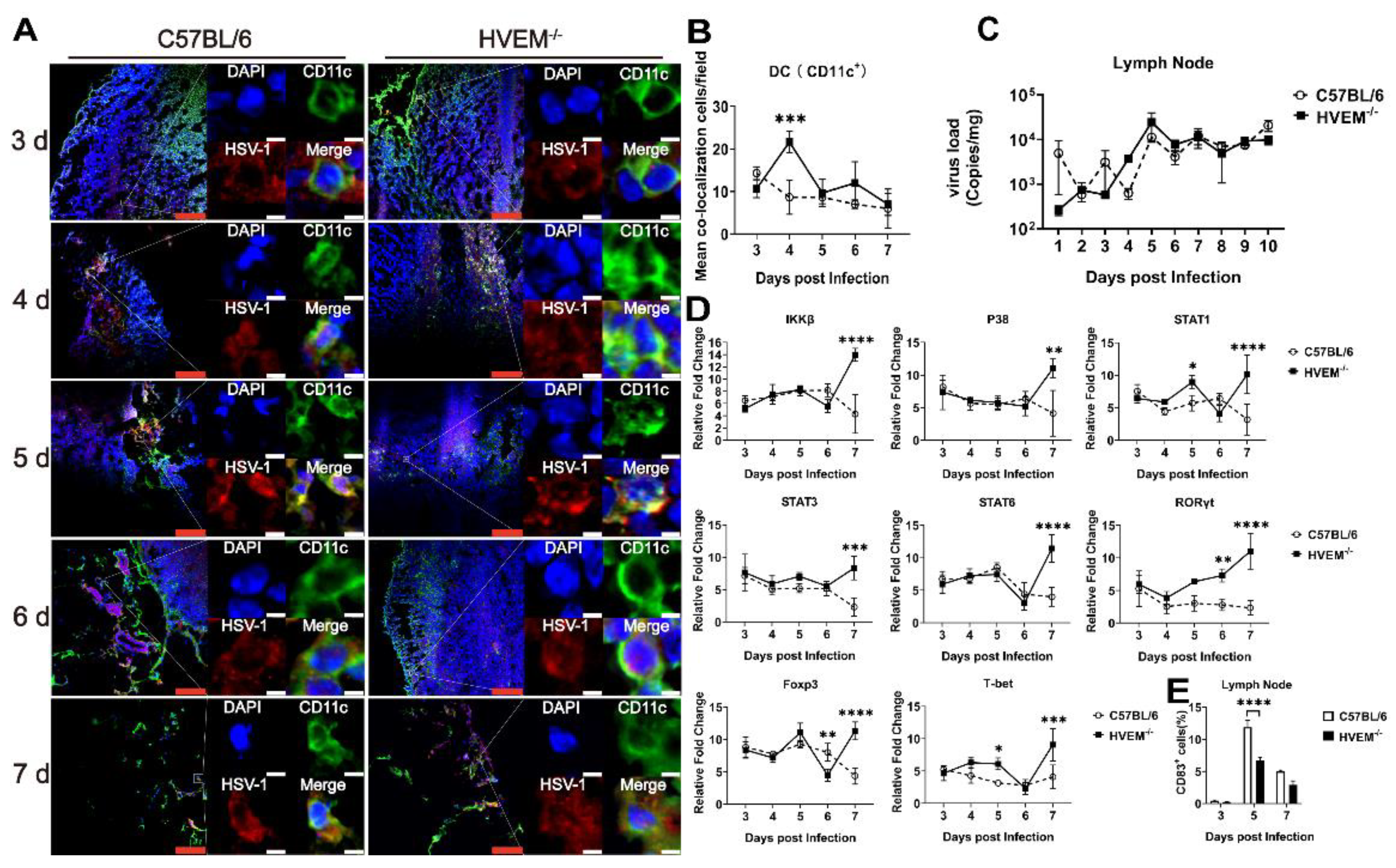
3.2. HSV-1 Enters Dendritic Cells in Epithelial Tissue via the Interaction between gD and HVEM and Replicates in the Cells
3.3. HSV-1 Infection in Dendritic Cells Restrains the Activation of Innate Immunity and Inflammatory Reactions
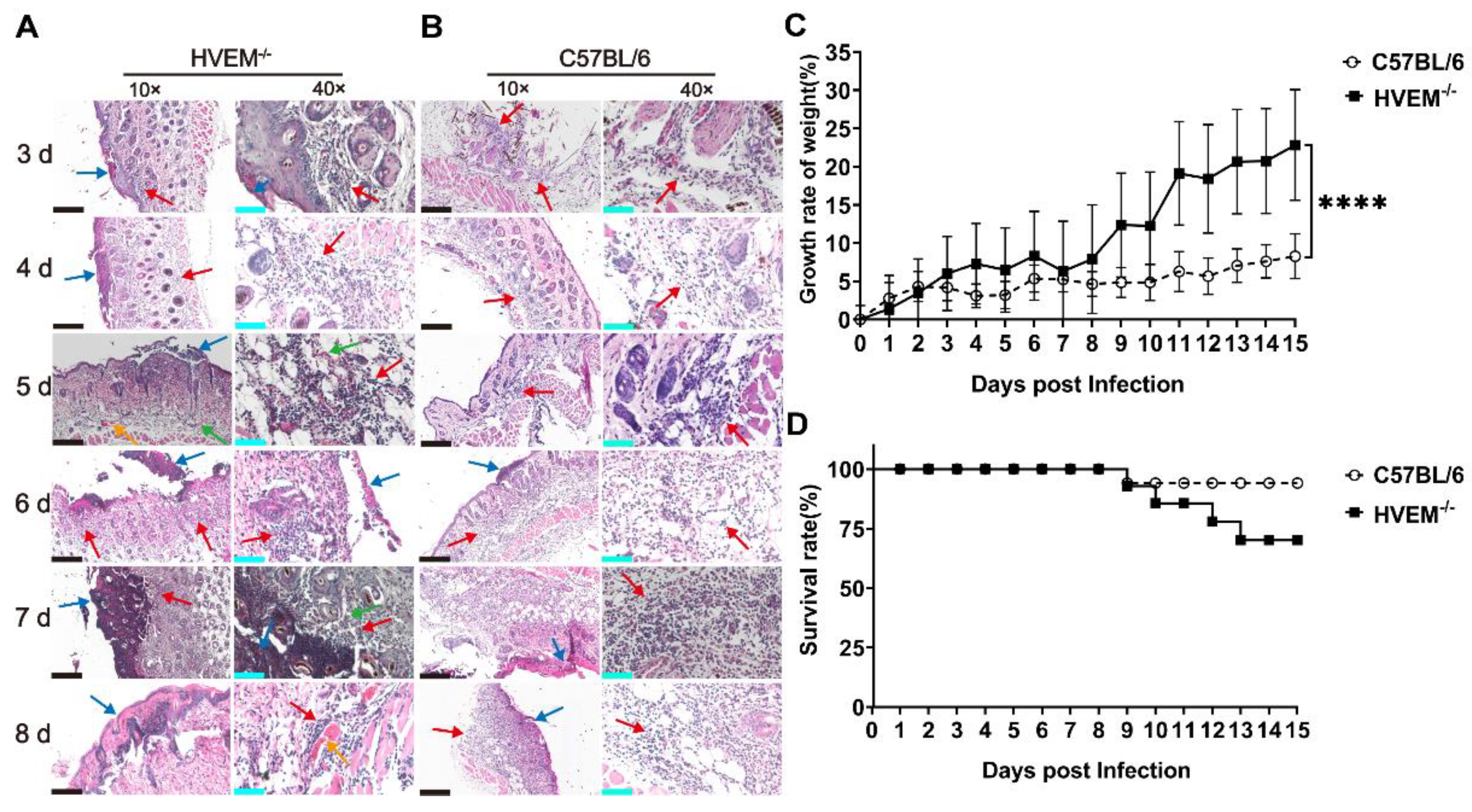
3.4. HSV-1 Infection in Dendritic Cells Leads to Alleviation of Infectious Manifestations in the Acute Phase
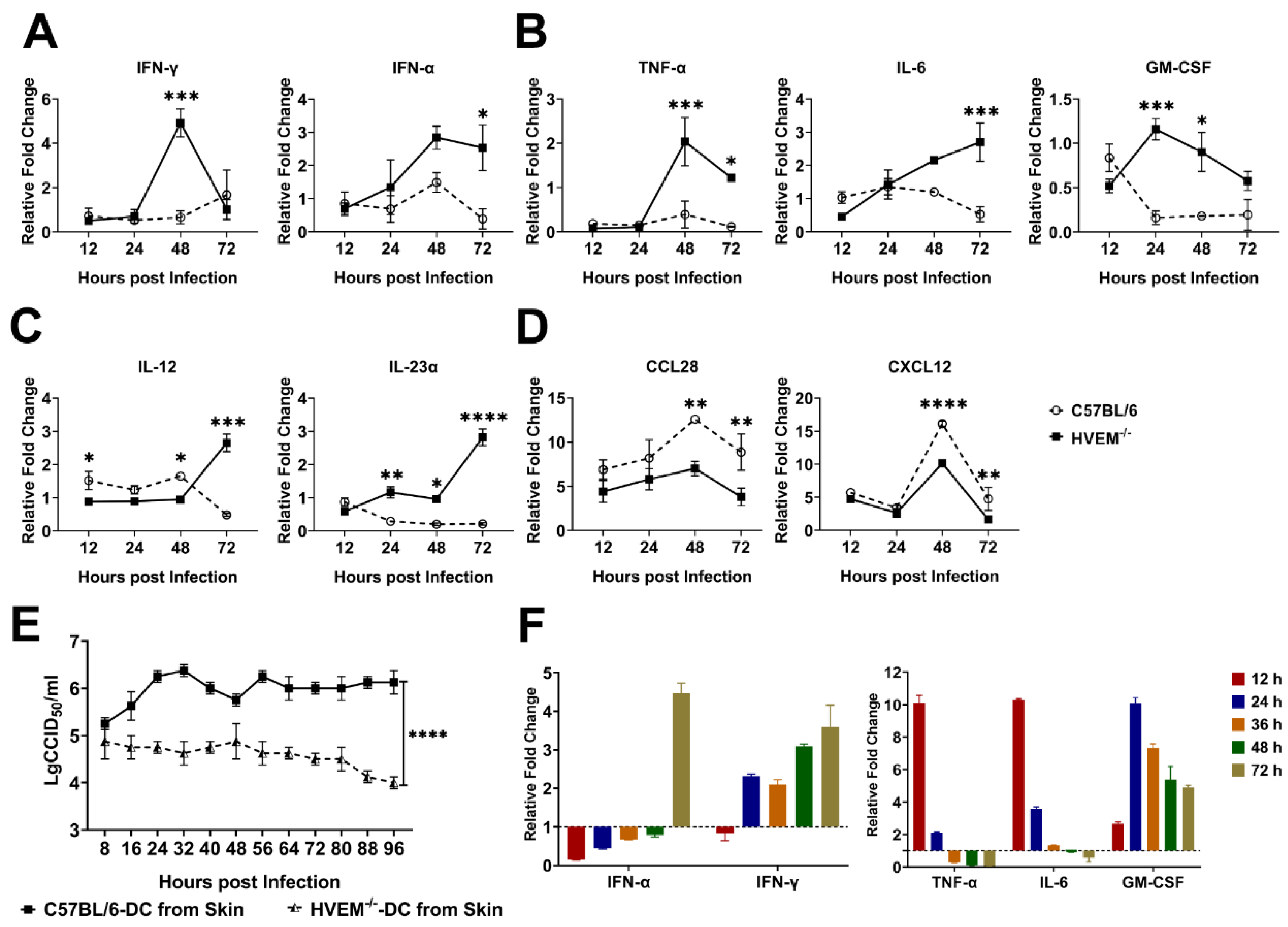
3.5. HSV-1 Infection Interferes with the Roles of Dendritic Cells in the Lymph Nodes
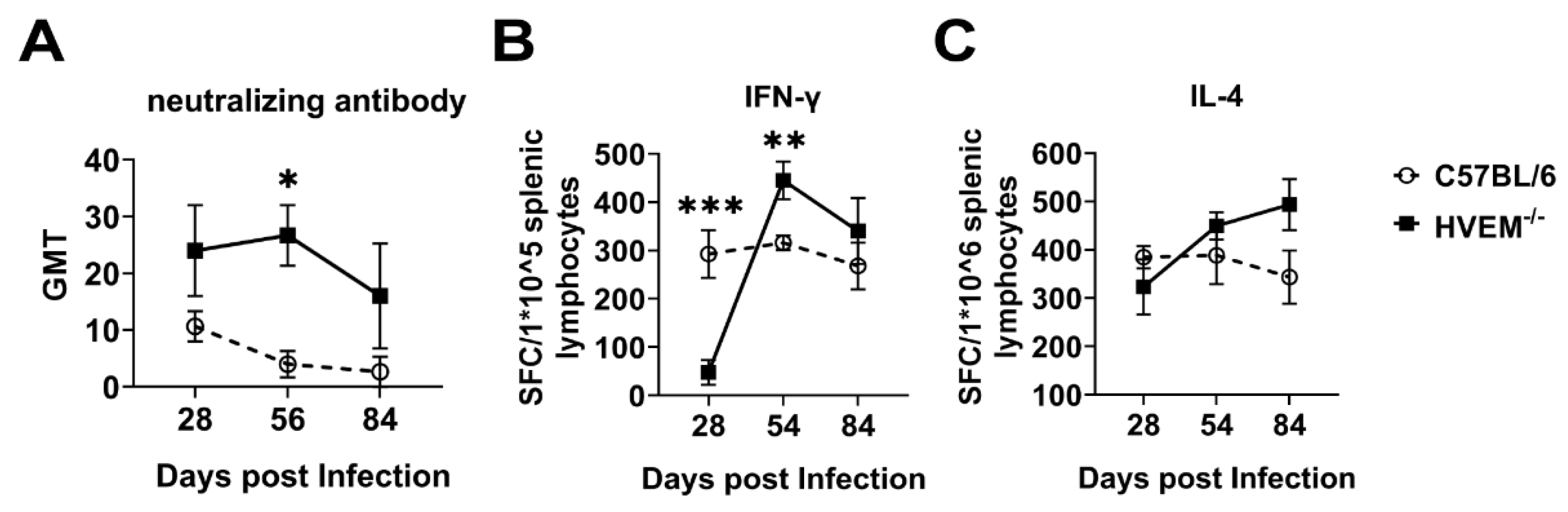
3.6. HSV-1 Infection of Dendritic Cells Followed by Suppression of Innate Immunity Leads to a Weakened Specific Antiviral Immune Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, V.; Mobeen, F.; Prakash, T. Comparative Genomics of Herpesviridae Family to Look for Potential Signatures of Human Infecting Strains. Int. J. Genom. 2016, 2016, 9543274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukhanova, M.K.; Korovina, A.N.; Kochetkov, S.N. Human herpes simplex virus: Life cycle and development of inhibitors. Biochemistry 2014, 79, 1635–1652. [Google Scholar] [CrossRef]
- Baldwin, K.J.; Cummings, C.L. Herpesvirus Infections of the Nervous System. Continuum Lifelong Learn. Neurol. 2018, 24, 1349–1369. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, G.; Kruszon-Moran, D.; Flagg, E.W.; Paulose-Ram, R. Prevalence of Herpes Simplex Virus Type 1 and Type 2 in Persons Aged 14–49: United States, 2015–2016. NCHS Data Brief 2018. pp. 1–8. Available online: https://www.cdc.gov/nchs/products/databriefs/db304.htm (accessed on 10 November 2021).
- Lamers, S.L.; Newman, R.M.; Laeyendecker, O.; Tobian, A.A.; Colgrove, R.C.; Ray, S.C.; Koelle, D.M.; Cohen, J.; Knipe, D.M.; Quinn, T.C. Global Diversity within and between Human Herpesvirus 1 and 2 Glycoproteins. J. Virol. 2015, 89, 8206–8218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jambunathan, N.; Clark, C.M.; Musarrat, F.; Chouljenko, V.N.; Rudd, J.; Kousoulas, K.G. Two Sides to Every Story: Herpes Simplex Type-1 Viral Glycoproteins gB, gD, gH/gL, gK, and Cellular Receptors Function as Key Players in Membrane Fusion. Viruses 2021, 13, 1849. [Google Scholar] [CrossRef] [PubMed]
- Tognarelli, E.I.; Palomino, T.F.; Corrales, N.; Bueno, S.M.; Kalergis, A.M.; Gonzalez, P.A. Herpes Simplex Virus Evasion of Early Host Antiviral Responses. Front. Cell. Infect. Microbiol. 2019, 9, 127. [Google Scholar] [CrossRef]
- Paludan, S.R.; Bowie, A.G.; Horan, K.A.; Fitzgerald, K.A. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011, 11, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.S.; Liu, H.M. The Molecular Basis of Viral Inhibition of IRF- and STAT-Dependent Immune Responses. Front. Immunol. 2018, 9, 3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.; Bai, Q.; Baek, H.J.; Felmet, K.; Burton, E.A.; Goins, W.F.; Cohen, J.B.; Glorioso, J.C. Soluble V domain of Nectin-1/HveC enables entry of herpes simplex virus type 1 (HSV-1) into HSV-resistant cells by binding to viral glycoprotein D. J. Virol. 2006, 80, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.; Bourque, J.; Kuehm, L.; Opejin, A.; Teague, R.M.; Gross, C.; Hawiger, D. Immunomodulatory Functions of BTLA and HVEM Govern Induction of Extrathymic Regulatory T Cells and Tolerance by Dendritic Cells. Immun. 2016, 45, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, M.W.; Cheung, T.C.; Ware, C.F. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol. Rev. 2011, 244, 169–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prechtel, A.T.; Turza, N.M.; Kobelt, D.J.; Eisemann, J.I.; Coffin, R.S.; McGrath, Y.; Hacker, C.; Ju, X.; Zenke, M.; Steinkasserer, A. Infection of mature dendritic cells with herpes simplex virus type 1 dramatically reduces lymphoid chemokine-mediated migration. J. Gen. Virol. 2005, 86, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.; Turza, N.M.; Muhl-Zurbes, P.; Lechmann, M.; Boutell, C.; Coffin, R.S.; Everett, R.D.; Steinkasserer, A.; Prechtel, A.T. Herpes simplex virus type 1 induces CD83 degradation in mature dendritic cells with immediate-early kinetics via the cellular proteasome. J. Virol. 2007, 81, 6326–6338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Liao, Y.; Lian, Y.; Jiang, G.; Jiang, L.; Dong, C.; Yang, E.; Wang, L.; Xu, X.; Feng, M.; et al. Role of innate lymphoid cells and dendritic cells in intradermal immunization of the enterovirus antigen. NPJ Vaccines 2019, 4, 14. [Google Scholar] [CrossRef]
- Inaba, K.; Inaba, M.; Romani, N.; Aya, H.; Deguchi, M.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992, 176, 1693–1702. [Google Scholar] [CrossRef]
- Tormanen, K.; Wang, S.; Jaggi, U.; Ghiasi, H.A.-O. Restoring Herpesvirus Entry Mediator (HVEM) Immune Function in HVEM(−/−) Mice Rescues Herpes Simplex Virus 1 Latency and Reactivation Independently of Binding to Glycoprotein, D. J. Virol. 2020, 94, e00700-20. [Google Scholar] [CrossRef]
- Pasero, C.; Speiser, D.E.; Derré, L.; Olive, D. The HVEM network: New directions in targeting novel costimulatory/co-inhibitory molecules for cancer therapy. Curr. Opin. Pharmacol. 2012, 12, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Stiles, K.M.; Whitbeck, J.C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. Herpes simplex virus glycoprotein D interferes with binding of herpesvirus entry mediator to its ligands through downregulation and direct competition. J. Virol. 2010, 84, 11646–11660. [Google Scholar] [CrossRef] [Green Version]
- Prechtel, A.T.; Steinkasserer, A. CD83: An update on functions and prospects of the maturation marker of dendritic cells. Arch. Dermatol. Res. 2007, 299, 59–69. [Google Scholar] [CrossRef]
- Del Balzo, D.; Capmany, A.; Cebrian, I.; Damiani, M.T. Chlamydia trachomatis Infection Impairs MHC-I Intracellular Trafficking and Antigen Cross-Presentation by Dendritic Cells. Front. Immunol. 2021, 12, 662096. [Google Scholar] [CrossRef]
- Bedoui, S.; Whitney, P.G.; Waithman, J.; Eidsmo, L.; Wakim, L.; Caminschi, I.; Allan, R.S.; Wojtasiak, M.; Shortman, K.; Carbone, F.R.; et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat. Immunol. 2009, 10, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Zamora, M.; Linehan, M.M.; Iijima, N.; Gonzalez, D.; Haberman, A.; Iwasaki, A. Differential roles of migratory and resident DCs in T cell priming after mucosal or skin HSV-1 infection. J. Exp. Med. 2009, 206, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Whitley, R.; Baines, J. Clinical management of herpes simplex virus infections: Past, present, and future. F1000Research 2018, 7, 1000–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiecki, M.; Wang, Y.; Gilfillan, S.; Colonna, M. Plasmacytoid dendritic cells contribute to systemic but not local antiviral responses to HSV infections. PLoS Pathog. 2013, 9, e1003728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucinda, N.; Figueiredo, M.M.; Pessoa, N.L.; Santos, B.S.; Lima, G.K.; Freitas, A.M.; Machado, A.M.; Kroon, E.G.; Antonelli, L.R.; Campos, M.A. Dendritic cells, macrophages, NK and CD8(+) T lymphocytes play pivotal roles in controlling HSV-1 in the trigeminal ganglia by producing IL1-beta, iNOS and granzyme B. Virol. J. 2017, 14, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurt-Jones, E.A.; Orzalli, M.H.; Knipe, D.M. Innate Immune Mechanisms and Herpes Simplex Virus Infection and Disease. Adv. Anat. Embryol. Cell. Biol. 2017, 223, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.S.; Danaher, R.J.; Jacob, R.J. Molecular aspects of herpes simplex virus I latency, reactivation, and recurrence. Crit. Rev. Oral Biol. Med. 1998, 9, 541–562. [Google Scholar] [CrossRef]
- Fatahzadeh, M.; Schwartz, R.A. Human herpes simplex virus infections: Epidemiology, pathogenesis, symptomatology, diagnosis, and management. J. Am. Acad. Dermatol. 2007, 57, 737–763. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, S.; Wang, Y.; Pan, S.; Lan, B.; Liu, Y.; Zhang, L.; Leng, Q.; Chen, D.; Zhang, C.; et al. The Us3 Protein of Herpes Simplex Virus 1 Inhibits T Cell Signaling by Confining Linker for Activation of T Cells (LAT) Activation via TRAF6 Protein. J. Biol. Chem. 2015, 290, 15670–15678. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Su, C.; Zheng, C. Herpes Simplex Virus 1 Serine Protease VP24 Blocks the DNA-Sensing Signal Pathway by Abrogating Activation of Interferon Regulatory Factor 3. J. Virol. 2016, 90, 5824–5829. [Google Scholar] [CrossRef] [Green Version]
- Biron, C.A.; Brossay, L. NK cells and NKT cells in innate defense against viral infections. Curr. Opin. Immunol. 2001, 13, 458–464. [Google Scholar] [CrossRef]
- Chew, T.; Taylor, K.E.; Mossman, K.L. Innate and adaptive immune responses to herpes simplex virus. Viruses 2009, 1, 979–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourque, J.; Hawiger, D. Immunomodulatory Bonds of the Partnership between Dendritic Cells and T Cells. Crit. Rev. Immunol. 2018, 38, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Kobelt, D.; Lechmann, M.; Steinkasserer, A. The interaction between dendritic cells and herpes simplex virus-1. Dendritic Cells Virus Infect. 2003, 276, 145–161. [Google Scholar] [CrossRef]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Kvansakul, M.; Caria, S.; Hinds, M.G. The Bcl-2 Family in Host-Virus Interactions. Viruses 2017, 9, 290. [Google Scholar] [CrossRef] [Green Version]
- Lechmann, M.; Krooshoop, D.J.; Dudziak, D.; Kremmer, E.; Kuhnt, C.; Figdor, C.G.; Schuler, G.; Steinkasserer, A. The extracellular domain of CD83 inhibits dendritic cell-mediated T cell stimulation and binds to a ligand on dendritic cells. J. Exp. Med. 2001, 194, 1813–1821. [Google Scholar] [CrossRef]
- Heilingloh, C.S.; Muhl-Zurbes, P.; Steinkasserer, A.; Kummer, M. Herpes simplex virus type 1 ICP0 induces CD83 degradation in mature dendritic cells independent of its E3 ubiquitin ligase function. J. Gen. Virol. 2014, 95, 1366–1375. [Google Scholar] [CrossRef]
- Heilingloh, C.S.; Kummer, M.; Muhl-Zurbes, P.; Drassner, C.; Daniel, C.; Klewer, M.; Steinkasserer, A. L Particles Transmit Viral Proteins from Herpes Simplex Virus 1-Infected Mature Dendritic Cells to Uninfected Bystander Cells, Inducing CD83 Downmodulation. J. Virol. 2015, 89, 11046–11055. [Google Scholar] [CrossRef] [Green Version]
- Aranda, A.M.; Epstein, A.L. Herpes simplex virus type 1 latency and reactivation: An update. Med. Sci. M/S 2015, 31, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Weir, J.P. Regulation of herpes simplex virus gene expression. Gene 2001, 271, 117–130. [Google Scholar] [CrossRef]
- Royer, D.J.; Hendrix, J.F.; Larabee, C.M.; Reagan, A.M.; Sjoelund, V.H.; Robertson, D.M.; Carr, D.J.J. Vaccine-induced antibodies target sequestered viral antigens to prevent ocular HSV-1 pathogenesis, preserve vision, and preempt productive neuronal infection. Mucosal. Immunol. 2019, 12, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Maruzuru, Y.; Ichinohe, T.; Sato, R.; Miyake, K.; Okano, T.; Suzuki, T.; Koshiba, T.; Koyanagi, N.; Tsuda, S.; Watanabe, M.; et al. Herpes Simplex Virus 1 VP22 Inhibits AIM2-Dependent Inflammasome Activation to Enable Efficient Viral Replication. Cell Host Microbe 2018, 23, 254–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef]
- Šedý, J.R.; Ramezani-Rad, P. HVEM network signaling in cancer. Adv. Cancer Res. 2019, 142, 145–186. [Google Scholar] [CrossRef]
- Ware, C.F. Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol. Rev. 2008, 223, 186–201. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C57BL/6 | HVEM−/− | |
|---|---|---|
| 1 dpi | −/+ | + |
| 2 dpi | + | −/+ |
| 3 dpi | − | + |
| 4 dpi | + | ++ |
| 5 dpi | + | +++ |
| 6 dpi | ++ | ++++ |
| 7 dpi | +++ | ++ |
| 8 dpi | + | + |
| 9 dpi | −/+ | −/+ |
| 10 dpi | − | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Cheng, J.; Xu, X.; Li, X.; Zhang, J.; Ma, D.; Jiang, G.; Liao, Y.; Fan, S.; Niu, Z.; et al. HSV-1 Infection of Epithelial Dendritic Cells Is a Critical Strategy for Interfering with Antiviral Immunity. Viruses 2022, 14, 1046. https://doi.org/10.3390/v14051046
Gao Y, Cheng J, Xu X, Li X, Zhang J, Ma D, Jiang G, Liao Y, Fan S, Niu Z, et al. HSV-1 Infection of Epithelial Dendritic Cells Is a Critical Strategy for Interfering with Antiviral Immunity. Viruses. 2022; 14(5):1046. https://doi.org/10.3390/v14051046
Chicago/Turabian StyleGao, Yang, Jishuai Cheng, Xingli Xu, Xueqi Li, Jingjing Zhang, Danjing Ma, Guorun Jiang, Yun Liao, Shengtao Fan, Zhenye Niu, and et al. 2022. "HSV-1 Infection of Epithelial Dendritic Cells Is a Critical Strategy for Interfering with Antiviral Immunity" Viruses 14, no. 5: 1046. https://doi.org/10.3390/v14051046