
Investigation of Salmonella Phage–Bacteria Infection Profiles: Network Structure Reveals a Gradient of Target-Range from Generalist to Specialist Phage Clones in Nested Subsets

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phages and Bacterial Strains
2.2. Antibiotic Susceptibility Tests
2.3. Bacteriophage Susceptibility Test
2.4. Representation of In Vitro Infection Data in a Phage–Bacterium Infection Matrix
2.5. Assignment of Infection Profiles and Clustering
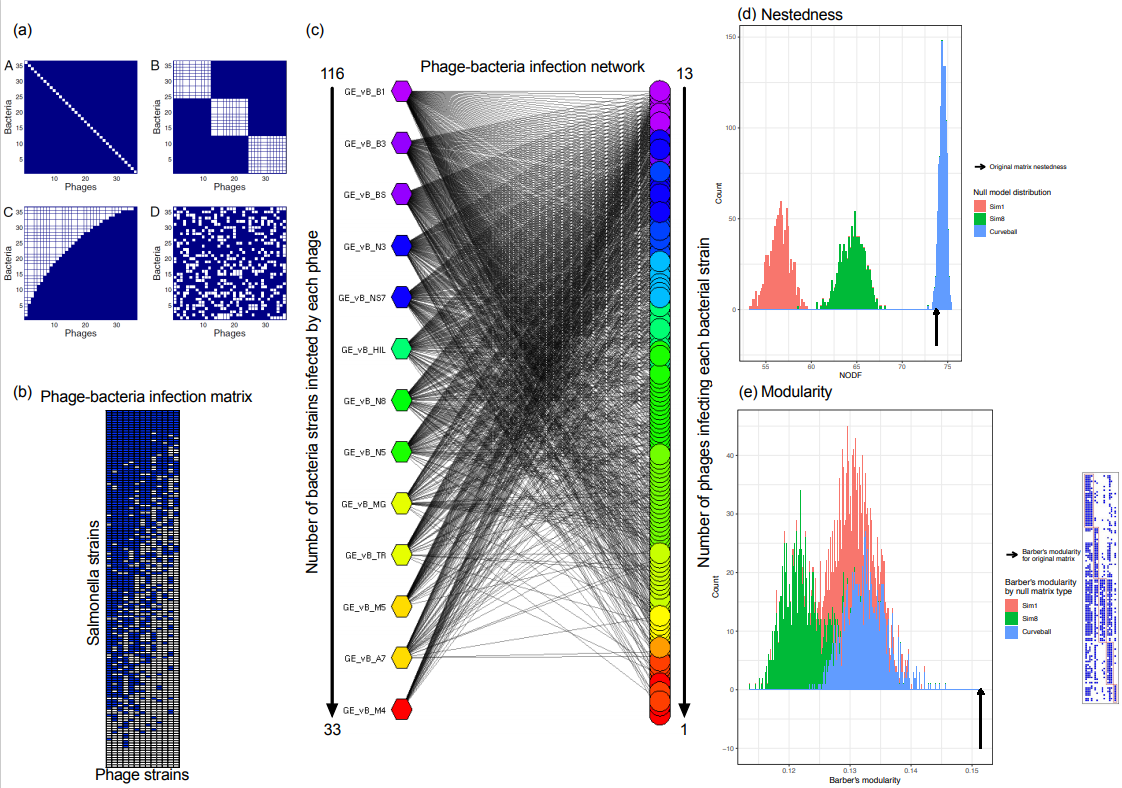
2.6. Construction of Salmonella Phage–Bacterium Infection Network
2.7. Calculating Nestedness and Modularity
2.8. Statistical Analysis
3. Results
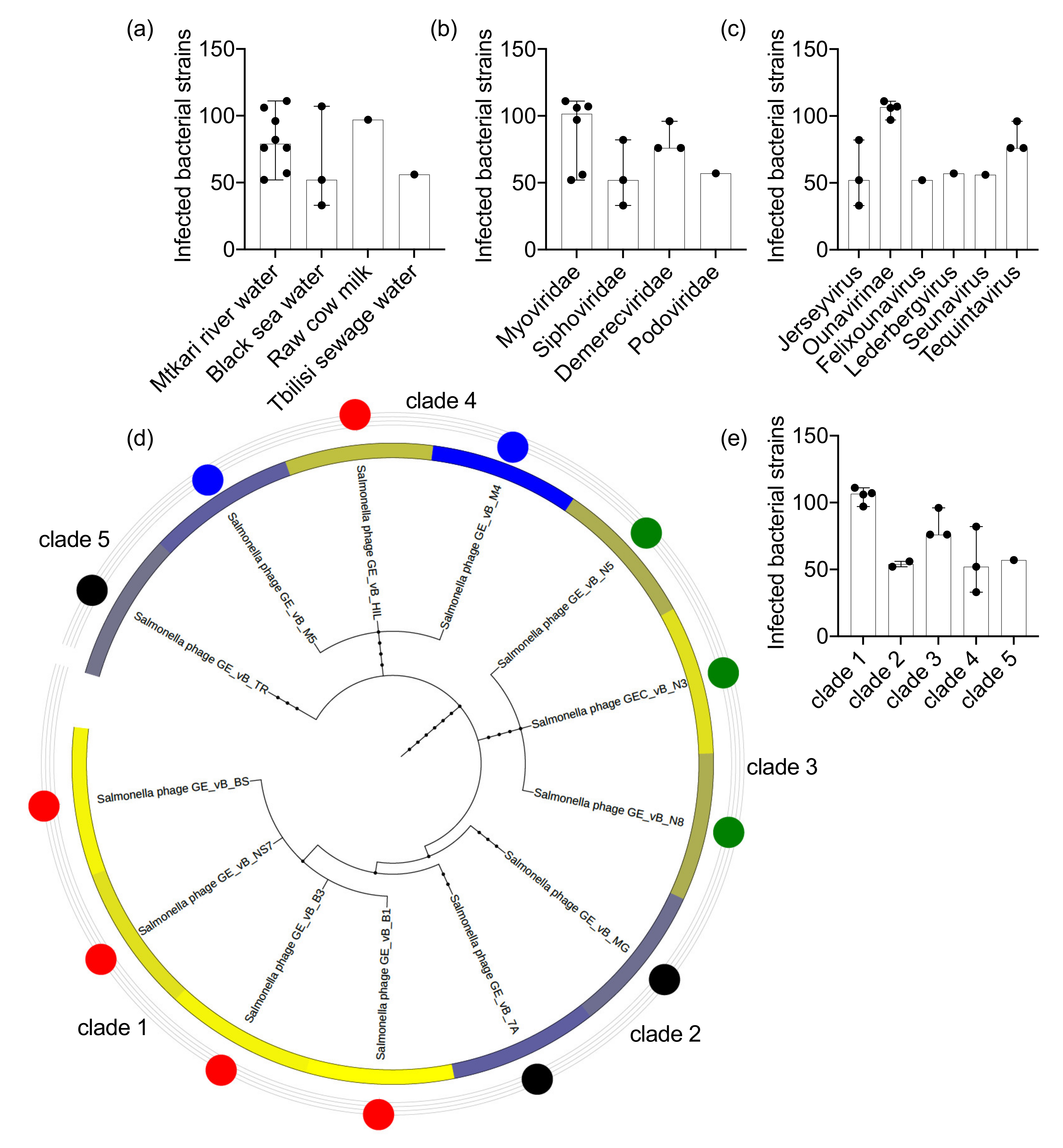
3.1. The Salmonella Phage Target-Range Varies between Clones of Different Taxonomic Lineages and Isolated from Different Environmental Sites
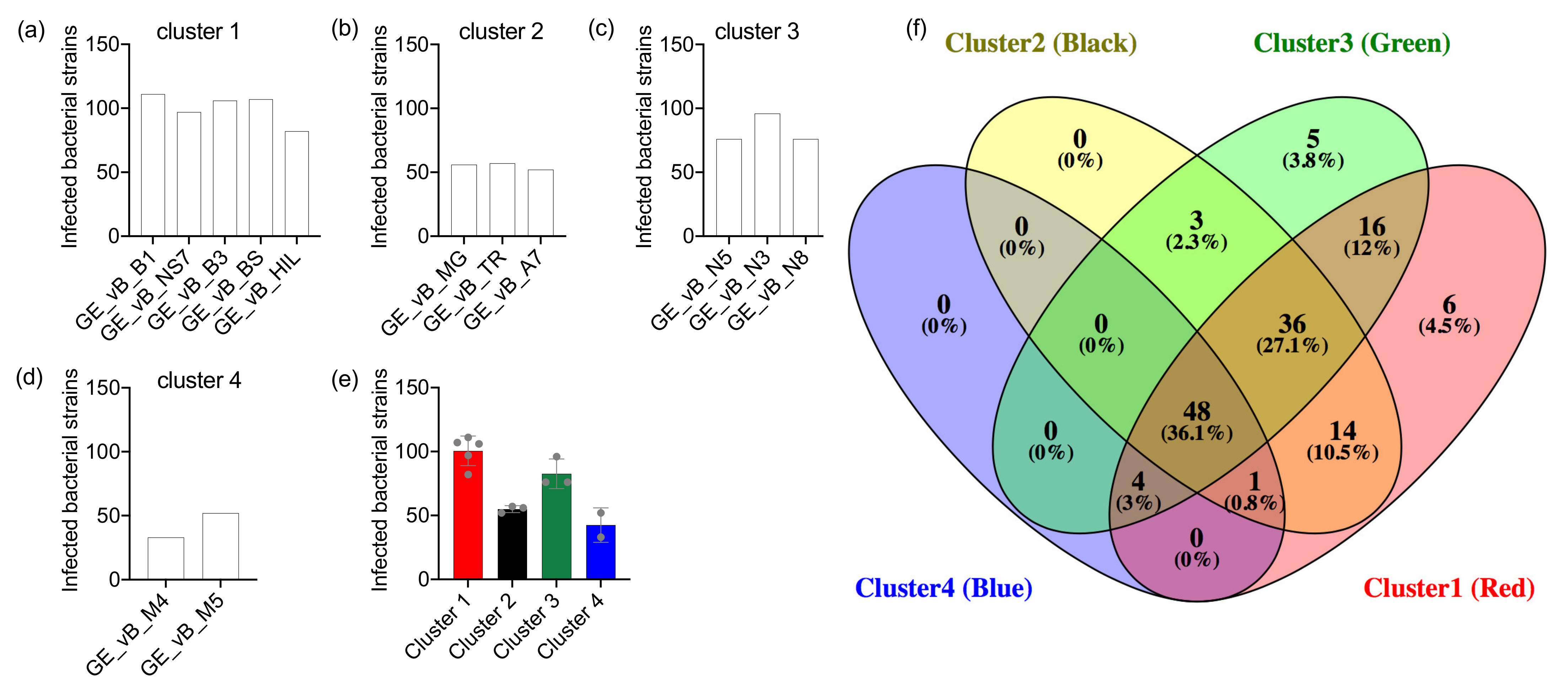
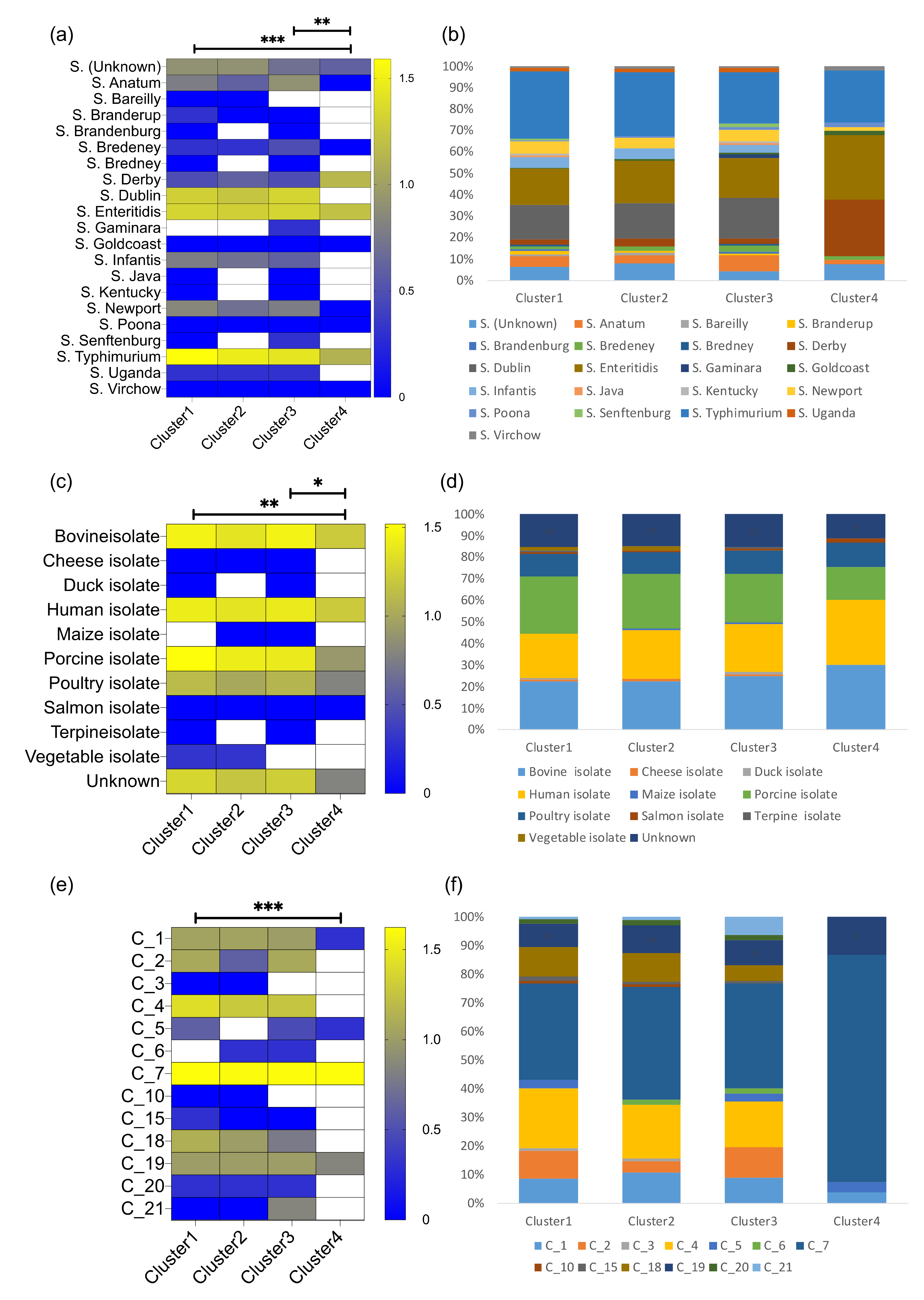
3.2. The Salmonella Phages Are Grouped into Clusters with Different Infection Profiles
3.3. The Genetic Distances of the Salmonella Phages Are Positively Associated to Their Infection Profiles
3.4. The Salmonella Phage–Bacteria Infection Network Is Nested
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Salmonella (Non-Typhoidal). Newsroom. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/salmonella-(non-typhoidal) (accessed on 6 April 2021).
- Centers for Disease Control and Prevention. Salmonella. Available online: https://www.cdc.gov/salmonella/index.html (accessed on 6 May 2021).
- Wang, X.; Biswas, S.; Paudyal, N.; Pan, H.; Li, X.; Fang, W.; Yue, M. Antibiotic Resistance in Salmonella Typhimurium Isolates Recovered from the Food Chain Through National Antimicrobial Resistance Monitoring System Between 1996 and 2016. Front. Microbiol. 2019, 10, 985. [Google Scholar] [CrossRef] [Green Version]
- Chanishvili, N. Literature Review of the Practical Application of Bacteriophage Research; Bacteriology Research Developments, Virology Research Progress; George Eliava Institute of Bacteriophage, Microbiology and Virology: Tbilisi, Georgia; Nova Science Publishers: Hauppauge, NY, USA, 2012. [Google Scholar]
- Chanishvili, N. Bacteriophages as Therapeutic and Prophylactic Means: Summary of the Soviet and Post Soviet Experiences. Curr. Drug Deliv. 2016, 13, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Chanishvili, N. Phage Therapy—History from Twort and d’Herelle Through Soviet Experience to Current Approaches. Adv. Virus Res. 2012, 83, 3–40. [Google Scholar] [CrossRef]
- Michalska-Smith, M.J.; Allesina, S. Telling ecological networks apart by their structure: A computational challenge. PLoS Comput. Biol. 2019, 15, e1007076. [Google Scholar] [CrossRef] [PubMed]
- Flores, C.O.; Meyer, J.R.; Valverde, S.; Farr, L.; Weitz, J.S. Statistical structure of host-phage interactions. Proc. Natl. Acad. Sci. USA 2011, 108, E288–E297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckling, A.; Rainey, P.B. Antagonistic coevolution between a bacterium and a bacteriophage. Proc. R. Soc. B Boil. Sci. 2002, 269, 931–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitz, J.S.; Poisot, T.; Meyer, J.R.; Flores, C.O.; Valverde, S.; Sullivan, M.B.; Hochberg, M.E. Phage-bacteria infection networks. Trends Microbiol. 2013, 21, 82–91. [Google Scholar] [CrossRef]
- Valverde, S.; Elena, S.F.; Solé, R. Spatially induced nestedness in a neutral model of phage-bacteria networks. Virus Evol. 2017, 3, vex021. [Google Scholar] [CrossRef]
- Mestres, J.; Gregori-Puigjané, E.; Valverde, S.; Solé, R.V. Data completeness—the Achilles heel of drug-target networks. Nat. Biotechnol. 2008, 26, 983–984. [Google Scholar] [CrossRef]
- Fortunato, S.; Barthelemy, M. Resolution limit in community detection. Proc. Natl. Acad. Sci. USA 2007, 104, 36–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, W.; Almeida-Neto, M.; Gotelli, N.J. A consumer’s guide to nestedness analysis. Oikos 2009, 118, 3–17. [Google Scholar] [CrossRef]
- Beckett, S.J.; Williams, H. Coevolutionary diversification creates nested-modular structure in phage–bacteria interaction networks. Interface Focus 2013, 3, 20130033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makalatia, K.; Kakabadze, E.; Wagemans, J.; Grdzelishvili, N.; Bakuradze, N.; Natroshvili, G.; Macharashvili, N.; Sedrakyan, A.; Arakelova, K.; Ktsoyan, Z.; et al. Characterization of Salmonella Isolates from Various Geographical Regions of the Caucasus and Their Susceptibility to Bacteriophages. Viruses 2020, 12, 1418. [Google Scholar] [CrossRef] [PubMed]
- Kakabadze, E.; Makalatia, K.; Merabishvili, M.; Grdzelishvili, N.; Bakuradze, N.; Mkrtchyan, M.; Goderdzishvili, M.; Pirnay, J.-P.; Chanishvili, N.; Sedrykyan, A. Phage typing, antibiotic resistance and genomic rep-PCR fingerprinting of clinical Salmonella isolates from the Caucasus region. Int. J. Infect. Dis. 2018, 73, 148. [Google Scholar] [CrossRef]
- Kakabadze, E.; Makalatia, K.; Grdzelishvili, N.; Bakuradze, N.; Goderdzishvili, M.; Kusradze, I.; Phoba, M.F.; Lunguya, O.; Lood, C.; Lavigne, R.; et al. Selection of Potential Therapeutic Bacteriophages that Lyse a CTX-M-15 Ex-tended Spectrum beta-Lactamase Producing Salmonella enterica Serovar Typhi Strain from the Democratic Republic of the Congo. Viruses 2018, 10, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achtman, M.; Zhou, Z.; Alikhan, N.-F.; Tyne, W.; Parkhill, J.; Cormican, M.; Chiou, C.-S.; Torpdahl, M.; Litrup, E.; Prendergast, D.M.; et al. Genomic diversity of Salmonella enterica—The UoWUCC 10K genomes project. Wellcome Open Res. 2020, 5, 223. [Google Scholar] [CrossRef] [PubMed]
- Wayne, P. Performance Standards for Antimicrobial Susceptibility Testing; Clinical and Laboratory Standards Institute: Annapolis Junction, MD, USA, 1992. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef]
- Ciccarelli, F.D.; Doerks, T.; von Mering, C.; Creevey, C.; Snel, B.; Bork, P. Toward Automatic Reconstruction of a Highly Resolved Tree of Life. Science 2006, 311, 1283–1287. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.; Otasek, D.; Ali, M.; McGuffin, M.J.; Xie, W.; Devani, B.; Van Toch, I.L.; Jurisica, I. NAViGaTOR: Network Analysis, Visualization and Graphing Toronto. Bioinformatics 2009, 25, 3327–3329. [Google Scholar] [CrossRef] [Green Version]
- Almeida-Neto, M.; Guimaraes, P.; Guimaraes, P.R., Jr.; Loyola, R.D.; Ulrich, W. A consistent metric for nestedness analysis in ecological systems: Reconciling concept and measurement. Oikos 2008, 117, 1227–1239. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. Ordination Methods, Diversity Analysis and Other Functions for Community and Vegetation Ecologists. 2020. Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 1 January 2021).
- Barber, M.J. Modularity and community detection in bipartite networks. Phys. Rev. E 2007, 76, 066102. [Google Scholar] [CrossRef] [Green Version]
- Dormann, C.F.; Gruber, B.; Fruend, J. Introducing the bipartite package: Analysing ecological networks. R News 2008, 8, 8–11. [Google Scholar]
- Beckett, S.J. Improved community detection in weighted bipartite networks. R. Soc. Open Sci. 2016, 3, 140536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotelli, N.J.H.; Edmund, M.; Ellison, A.M. EcoSimR-Alpha; Early Development/Alpha Release of EcoSimR. 2015. Available online: http://www.uvm.edu/~ngotelli/EcoSim/EcoSim.html. (accessed on 1 January 2021).
- Strona, G.; Nappo, D.; Boccacci, F.; Fattorini, S.; San-Miguel-Ayanz, J. A fast and unbiased procedure to randomize ecological binary matrices with fixed row and column totals. Nat. Commun. 2014, 5, 4114. [Google Scholar] [CrossRef] [PubMed]
- Wallig, M.; Weston, S.; Tenenbaum, D.; Microsoft Corporation. doParallel: Foreach Parallel Adaptor for the ’Parallel’ Package. Provides a Parallel Backend for the %dopar% Function Using the Parallel Package. 2020. Available online: https://cran.r-project.org/web/packages/doParallel/index.html. (accessed on 1 January 2021).
- Gencay, Y.E.; Gambino, M.; Prüssing, T.F.; Brøndsted, L. The genera of bacteriophages and their receptors are the major determinants of host range. Environ. Microbiol. 2019, 21, 2095–2111. [Google Scholar] [CrossRef]
- Sedrakyan, A.M.; Ktsoyan, Z.A.; Arakelova, K.A.; Zakharyan, M.K.; Hovhannisyan, A.I.; Gevorgyan, Z.U.; Mnatsakanyan, A.A.; Kakabadze, E.G.; Makalatia, K.B.; Chanishvili, N.A.; et al. Extended-Spectrum beta-Lactamases in Human Isolates of Multi-drug-Resistant Non-typhoidal Salmonella enterica. Front. Microbiol. 2020, 11, 592223. [Google Scholar] [CrossRef] [PubMed]
- Makalatia, K.H.; Kakabadze, E.; Bakuradze, N.; Grdzelishvili, N.; Natroshvili, G.; Kusradze, I.; Goderdzishvili, M.; Sedrakyan, A.; Arakelova, K.; Mkrtchyan, M.; et al. Activity of Bacteriophages to Multiply Resistant Strains of Salmonella and Their Various Serotypes. Vet. Biotechnol. 2018, 32, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Leiman, P.G.; Shneider, M.M. Contractile Tail Machines of Bacteriophages. In Viral Molecular Machines; Springer: Boston, MA, USA, 2012; pp. 93–114. [Google Scholar]
- Stone, E.; Campbell, K.; Grant, I.; McAuliffe, O. Understanding and Exploiting Phage–Host Interactions. Viruses 2019, 11, 567. [Google Scholar] [CrossRef] [Green Version]
- Rivera, D.; Hudson, L.K.; Denes, T.G.; Hamilton-West, C.; Pezoa, D.; Moreno-Switt, A.I. Two Phages of the Genera Felixounavirus Subjected to 12 Hour Challenge on Salmonella Infantis Showed Distinct Genotypic and Phenotypic Changes. Viruses 2019, 11, 586. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, A.A. Studies of a receptor for felix O-1 phage in Salmonella Minnesota. J. Gen. Microbiol. 1967, 48, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindberg, A.A.; Holme, T. Influence of O side chains on the attachment of the Felix O-1 bacteriophage to Salmonella bacteria. J. Bacteriol. 1969, 99, 513–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macphee, D.G.; Krishnapillai, V.; Roantree, R.J.; Stocker, B.A.D. Mutations in Salmonella typhimurium Conferring Resistance to Felix O Phage without Loss of Smooth Character. J. Gen. Microbiol. 1975, 87, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Kim, K.P.; Heu, S.; Lee, S.J.; Adhya, S.; Ryu, S. Identification of host receptor and receptor-binding module of a newly sequenced T5-like phage EPS7. FEMS Microbiol. Lett. 2008, 289, 202–209. [Google Scholar] [CrossRef]
- Kim, M.; Ryu, S. Characterization of a T5-Like Coliphage, SPC35, and Differential Development of Resistance to SPC35 in Salmonella enterica Serovar Typhimurium and Escherichia coli. Appl. Environ. Microbiol. 2011, 77, 2042–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Jiang, Y.; Vincent, M.; Sun, Y.; Yu, H.; Wang, J.; Bao, Q.; Kong, H.; Hu, S. Complete genome sequence of bacteriophage T5. Virology 2005, 332, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Heller, K.; Braun, V. Polymannose O-antigens of Escherichia coli, the binding sites for the reversible adsorption of bacteriophage T5+ via the L-shaped tail fibers. J. Virol. 1982, 41, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskella, B.; Brockhurst, M.A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef] [Green Version]
- Flores, C.O.; Valverde, S.; Weitz, J.S. Multi-scale structure and geographic drivers of cross-infection within marine bacteria and phages. ISME J. 2012, 7, 520–532. [Google Scholar] [CrossRef] [Green Version]
- Lenski, R.; Levin, B.R. Constraints on the Coevolution of Bacteria and Virulent Phage: A Model, Some Experiments, and Predictions for Natural Communities. Am. Nat. 1985, 125, 585–602. [Google Scholar] [CrossRef]
- Agrawal, A.F.; Lively, C.M. Modelling infection as a two-step process combining gene-for-gene and matching-allele genetics. Proc. R. Soc. B Boil. Sci. 2003, 270, 323–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendling, C.C.; Goehlich, H.; Roth, O. The structure of temperate phage–bacteria infection networks changes with the phylogenetic distance of the host bacteria. Biol. Lett. 2018, 14, 20180320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wichels, A.; Biel, S.S.; Gelderblom, H.R.; Brinkhoff, T.; Muyzer, G.; Schütt, C. Bacteriophage diversity in the North Sea. Appl. Environ. Microbiol. 1998, 64, 4128–4133. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.N. The Geographic Mosaic Theory of Coevolution. Am. Nat. 1999, 153. [Google Scholar] [CrossRef] [Green Version]
- Gomez, P.; Buckling, A. Bacteria-phage antagonistic coevolution in soil. Science 2011, 332, 106–109. [Google Scholar] [CrossRef] [Green Version]
- Métris, A.; Sudhakar, P.; Fazekas, D.; Demeter, A.; Ari, E.; Olbei, M.; Branchu, P.; Kingsley, R.; Baranyi, J.; Korcsmáros, T. SalmoNet, an integrated network of ten Salmonella enterica strains reveals common and distinct pathways to host adaptation. npj Syst. Biol. Appl. 2017, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Fong, K.; Tremblay, D.M.; Delaquis, P.; Goodridge, L.; Levesque, R.C.; Moineau, S.; Suttle, C.A.; Wang, S. Diversity and Host Specificity Revealed by Biological Characterization and Whole Genome Sequencing of Bacteriophages Infecting Salmonella enterica. Viruses 2019, 11, 854. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Turner, P.E.; Burch, C.L. Pleiotropic costs of niche expansion in the RNA bacteriophage phi 6. Genetics 2006, 172, 751–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, R.; Friman, V.-P.; Smith, M.; Brockhurst, M.A. Cross-resistance is modular in bacteria–phage interactions. PLoS Biol. 2018, 16, e2006057. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, M.A.; Barbour, M.A.; Zaman, L.; Hall, A.R.; Buckling, A.; Bascompte, J. Coevolutionary dynamics shape the structure of bacteria-phage infection networks. Evolution 2019, 73, 1001–1011. [Google Scholar] [CrossRef]
- Gurney, J.; Aldakak, L.; Betts, A.; Gougat-Barbera, C.; Poisot, T.; Kaltz, O.; Hochberg, M.E. Network structure and local adaptation in co-evolving bacteria-phage interactions. Mol. Ecol. 2017, 26, 1764–1777. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Name | Study id | Tax id | Family | Genus | Taxonomic Clade | Infection Profile | Infected Hosts |
|---|---|---|---|---|---|---|---|---|
| Mtkari river water | Salmonella phage GEC_vB_B1 | GE_vB_B1 | 2108164 | Myoviridae | Unclassified Ounavirinae | Clade 1 | Cluster 1 | 111 |
| Salmonella phage GEC_vB_B3 | GE_vB_B3 | 2108165 | Myoviridae | Unclassified Ounavirinae | Clade 1 | Cluster 1 | 106 | |
| Salmonella phage GEC_vB_HIL | GE_vB_HIL | 2108167 | Siphoviridae | Unclassified Jerseyvirus | Clade 4 | Cluster 1 | 82 | |
| Salmonella phage GEC_vB_7A | GE_vB_7A | 2108163 | Myoviridae | Unclassified Felixounavirus | Clade 2 | Cluster 2 | 52 | |
| Salmonella phage GEC_vB_TR | GE_vB_TR | 2108174 | Podoviridae | Unclassified Lederbergvirus | Clade 5 | Cluster 2 | 57 | |
| Salmonella phage GEC_vB_N5 | GE_vB_N5 | 2108171 | Demerecviridae | Unclassified Tequintavirus | Clade 3 | Cluster 3 | 76 | |
| Salmonella phage GEC_vB_N8 | GE_vB_N8 | 2108172 | Demerecviridae | Unclassified Tequintavirus | Clade 3 | Cluster 3 | 76 | |
| Salmonella phage GEC_vB_N3 | GEC_vB_N3 | 2777377 | Demerecviridae | Unclassified Tequintavirus | Clade 3 | Cluster 3 | 96 | |
| Black sea water | Salmonella phage GEC_vB_BS | GE_vB_BS | 2108166 | Myoviridae | Unclassified Ounavirinae | Clade 1 | Cluster 1 | 107 |
| Salmonella phage GEC_vB_M4 | GE_vB_M4 | 2108168 | Siphoviridae | Unclassified Jerseyvirus | Clade 4 | Cluster 4 | 33 | |
| Salmonella phage GEC_vB_M5 | GE_vB_M5 | 2108169 | Siphoviridae | Unclassified Jerseyvirus | Clade 4 | Cluster 4 | 52 | |
| Raw cow milk | Salmonella phage GEC_vB_NS7 | GE_vB_NS7 | 2108173 | Myoviridae | Unclassified Ounavirinae | Clade 1 | Cluster 1 | 97 |
| Tbilisi sewage water | Salmonella phage GCE_vB_MG | GE_vB_MG | 2108170 | Myoviridae | Unclassified Seunavirus | Clade 2 | Cluster 2 | 56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makalatia, K.; Kakabadze, E.; Bakuradze, N.; Grdzelishvili, N.; Stamp, B.; Herman, E.; Tapinos, A.; Coffey, A.; Lee, D.; Papadopoulos, N.G.; et al. Investigation of Salmonella Phage–Bacteria Infection Profiles: Network Structure Reveals a Gradient of Target-Range from Generalist to Specialist Phage Clones in Nested Subsets. Viruses 2021, 13, 1261. https://doi.org/10.3390/v13071261
Makalatia K, Kakabadze E, Bakuradze N, Grdzelishvili N, Stamp B, Herman E, Tapinos A, Coffey A, Lee D, Papadopoulos NG, et al. Investigation of Salmonella Phage–Bacteria Infection Profiles: Network Structure Reveals a Gradient of Target-Range from Generalist to Specialist Phage Clones in Nested Subsets. Viruses. 2021; 13(7):1261. https://doi.org/10.3390/v13071261
Chicago/Turabian StyleMakalatia, Khatuna, Elene Kakabadze, Nata Bakuradze, Nino Grdzelishvili, Ben Stamp, Ezra Herman, Avraam Tapinos, Aidan Coffey, David Lee, Nikolaos G. Papadopoulos, and et al. 2021. "Investigation of Salmonella Phage–Bacteria Infection Profiles: Network Structure Reveals a Gradient of Target-Range from Generalist to Specialist Phage Clones in Nested Subsets" Viruses 13, no. 7: 1261. https://doi.org/10.3390/v13071261