THO Complex Subunit 7 Homolog Negatively Regulates Cellular Antiviral Response against RNA Viruses by Targeting TBK1

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, Antibodies, and Reagents
2.2. Plasmids
2.3. Yeast Two-Hybrid Screening Assay and Dual-Luciferase Reporter Assay
2.4. Coimmunoprecipitation, Immunoblotting, and Native PAGE Assays
2.5. RNA Purification and Fluorescent Quantitative PCR
2.6. Statistical Analysis
3. Results
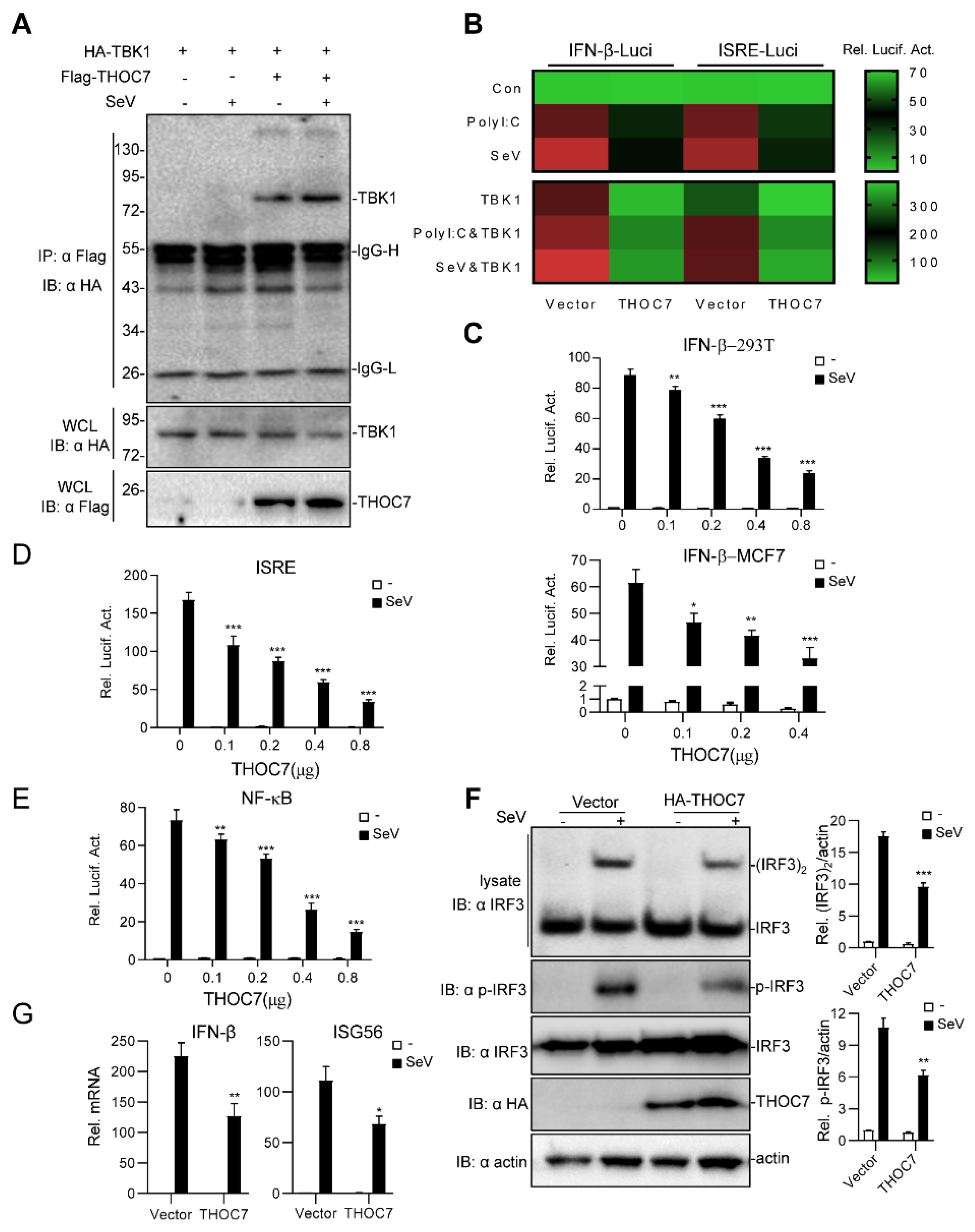
3.1. THOC7 Overexpression Negatively Regulates Type I IFN Production
3.2. THOC7 Knockdown Upregulates Type I IFN Production
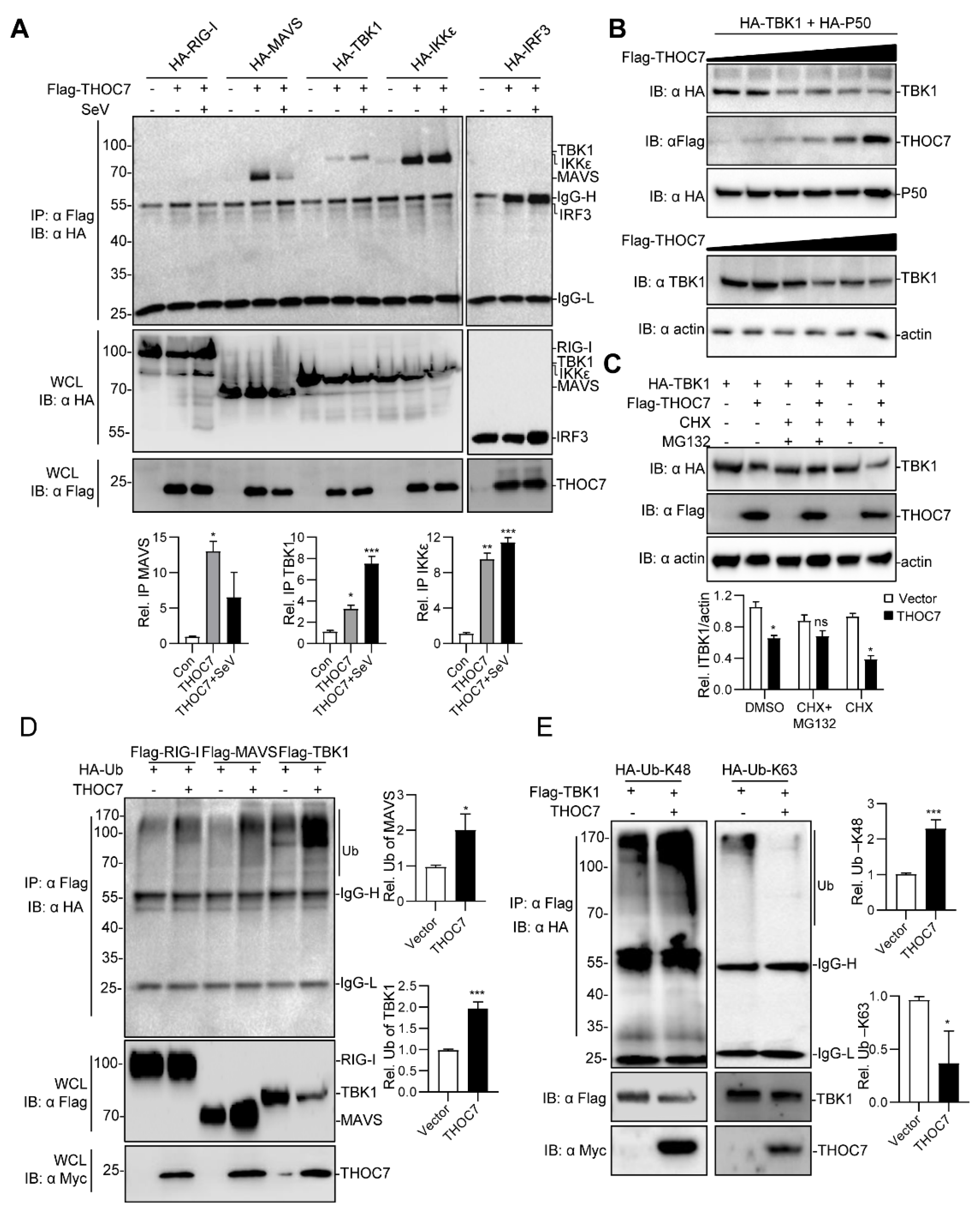
3.3. THOC7 Regulates the RLR Signaling Pathway by Targeting TBK1
3.4. THOC7 Is Involved in MAVS Signalosome and Promotes the Proteasomal Degradation of TBK1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Benko, S.; Philpott, D.J.; Girardin, S.E. The microbial and danger signals that activate Nod-like receptors. Cytokine 2008, 43, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, J.H.; Ferrero, R.L.; Philpott, D.J.; Girardin, S.E. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 2006, 7, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr. Opin. Immunol. 2011, 23, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Jacobs, J.L.; Coyne, C.B. Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol. 2013, 425, 5009–5019. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Soares, F.; Tattoli, I.; Girardin, S.E. Mitochondria in innate immunity. EMBO Rep. 2011, 12, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helgason, E.; Phung, Q.T.; Dueber, E.C. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: The emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 2013, 587, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Zhang, H.Y.; Piao, Z.F.; Zheng, S.J.; Chen, Y.; Chen, D.X.; Duan, Z.P. Inhibition of glycogen synthase kinase 3b activity regulates Toll-like receptor 4-mediated liver inflammation. Chin. J. Hepatol. 2012, 20, 693–697. [Google Scholar]
- Gabhann, J.N.; Higgs, R.; Brennan, K.; Thomas, W.; Damen, J.E.; Ben Larbi, N.; Krystal, G.; Jefferies, C.A. Absence of SHIP-1 results in constitutive phosphorylation of tank-binding kinase 1 and enhanced TLR3-dependent IFN-beta production. J. Immunol. 2010, 184, 2314–2320. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liang, L.; Fan, Y.; Sun, S.; An, L.; Shi, Z.; Cheng, J.; Jia, W.; Sun, W.; Mori-Akiyama, Y.; et al. PPM1B negatively regulates antiviral response via dephosphorylating TBK1. Cell. Signal. 2012, 24, 2197–2204. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, L.; Berman, M.; Kong, Y.Y.; Dorf, M.E. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 2011, 35, 426–440. [Google Scholar] [CrossRef]
- Liu, D.; Sheng, C.; Gao, S.; Yao, C.; Li, J.; Jiang, W.; Chen, H.; Wu, J.; Pan, C.; Chen, S.; et al. SOCS3 Drives Proteasomal Degradation of TBK1 and Negatively Regulates Antiviral Innate Immunity. Mol. Cell. Boil. 2015, 35, 2400–2413. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, T.; Zhang, J.; Yang, M.; Li, N.; Xu, X.; Cao, X. The E3 ubiquitin ligase Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon. Nat. Immunol. 2009, 10, 744–752. [Google Scholar] [CrossRef]
- Charoenthongtrakul, S.; Gao, L.; Parvatiyar, K.; Lee, D.; Harhaj, E.W. RING finger protein 11 targets TBK1/IKKi kinases to inhibit antiviral signaling. PLoS ONE 2013, 8, e53717. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, Y.; Zhu, L.; Liu, D.; Songyang, Z.; Wang, H.Y.; Wang, R.F. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat. Immunol. 2012, 13, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Saul, V.V.; Niedenthal, R.; Pich, A.; Weber, F.; Schmitz, M.L. SUMO modification of TBK1 at the adaptor-binding C-terminal coiled-coil domain contributes to its antiviral activity. Biochim. Biophys. Acta 2015, 1853, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Hakimi, M.A.; Baillat, D.; Chen, X.; Farber, M.J.; Klein-Szanto, A.J.; Cooch, N.S.; Godwin, A.K.; Shiekhattar, R. Linking transcriptional elongation and messenger RNA export to metastatic breast cancers. Cancer Res. 2005, 65, 3011–3016. [Google Scholar] [CrossRef]
- Tascou, S.; Kang, T.W.; Trappe, R.; Engel, W.; Burfeind, P. Identification and characterization of NIF3L1 BP1, a novel cytoplasmic interaction partner of the NIF3L1 protein. Biochem. Biophys. Res. Commun. 2003, 309, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Das, R.; Cheng, H.; Hurt, E.; Dorman, N.; Reed, R. Recruitment of the human TREX complex to mRNA during splicing. Genes Dev. 2005, 19, 1512–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, T.S.; Chen, T.; Wang, D.D.; Xu, L.G. HAUS8 regulates RLRVISA antiviral signaling positively by targeting VISA. Mol. Med. Rep. 2018, 18, 2458–2466. [Google Scholar]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Robitaille, A.C.; Mariani, M.K.; Fortin, A.; Grandvaux, N. A High Resolution Method to Monitor Phosphorylation-dependent Activation of IRF3. J. Vis. Exp. JoVE 2016, 107, e53723. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, T.; Yoneyama, M.; Yamaguchi, K.; Suhara, W.; Mori, W.; Shiota, K.; Okabe, Y.; Namiki, H.; Fujita, T. Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: Common and unique pathways. Genes Cells 2001, 6, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009, 227, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, J.; Cai, X.; Wu, J.; Chen, X.; Wu, Y.T.; Sun, L.; Chen, Z.J. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife 2013, 2, e00785. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Hertrich, C.; Wassermann, R.; Hornung, V. Nucleic acid driven sterile inflammation. Clin. Immunol. 2013, 147, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W. Negative regulation of TBK1-mediated antiviral immunity. FEBS Lett. 2013, 587, 542–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehwinkel, J.; Herold, A.; Gari, K.; Kocher, T.; Rode, M.; Ciccarelli, F.L.; Wilm, M.; Izaurralde, E. Genome-wide analysis of mRNAs regulated by the THO complex in Drosophila melanogaster. Nat. Struct. Mol. Boil. 2004, 11, 558–566. [Google Scholar] [CrossRef]
- Dominguez-Sanchez, M.S.; Barroso, S.; Gomez-Gonzalez, B.; Luna, R.; Aguilera, A. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011, 7, e1002386. [Google Scholar] [CrossRef]
- Colgan, K.J.; Boyne, J.R.; Whitehouse, A. Uncoupling of hTREX demonstrates that UAP56 and hTHO-complex recruitment onto herpesvirus saimiri intronless transcripts is required for replication. J. Gen. Virol. 2009, 90 Pt 6, 1455–1460. [Google Scholar] [CrossRef] [Green Version]
- Read, E.K.; Digard, P. Individual influenza A virus mRNAs show differential dependence on cellular NXF1/TAP for their nuclear export. J. Gen. Virol. 2010, 91 Pt 5, 1290–1301. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, T.; Tonne, J.M.; Ikeda, Y. Murine leukemia virus uses TREX components for efficient nuclear export of unspliced viral transcripts. Viruses 2014, 6, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Boyne, J.R.; Colgan, K.J.; Whitehouse, A. Recruitment of the complete hTREX complex is required for Kaposi’s sarcoma-associated herpesvirus intronless mRNA nuclear export and virus replication. PLoS Pathog. 2008, 4, e1000194. [Google Scholar] [CrossRef]
- Zhao, Q.; Liang, D.; Sun, R.; Jia, B.; Xia, T.; Xiao, H.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded replication and transcription activator impairs innate immunity via ubiquitin-mediated degradation of myeloid differentiation factor 88. J. Virol. 2015, 89, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Reed, R.; Cheng, H. TREX, SR proteins and export of mRNA. Curr. Opin. Cell Biol. 2005, 17, 269–273. [Google Scholar] [CrossRef] [PubMed]
- El Bounkari, O.; Guria, A.; Klebba-Faerber, S.; Claussen, M.; Pieler, T.; Griffiths, J.R.; Whetton, A.D.; Koch, A.; Tamura, T. Nuclear localization of the pre-mRNA associating protein THOC7 depends upon its direct interaction with Fms tyrosine kinase interacting protein (FMIP). FEBS Lett. 2009, 583, 13–18. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, T.-S.; Xie, T.; Li, J.; Yang, Y.-X.; Li, C.; Wang, W.; Cao, L.; Rao, H.; Ju, C.; Xu, L.-G. THO Complex Subunit 7 Homolog Negatively Regulates Cellular Antiviral Response against RNA Viruses by Targeting TBK1. Viruses 2019, 11, 158. https://doi.org/10.3390/v11020158
He T-S, Xie T, Li J, Yang Y-X, Li C, Wang W, Cao L, Rao H, Ju C, Xu L-G. THO Complex Subunit 7 Homolog Negatively Regulates Cellular Antiviral Response against RNA Viruses by Targeting TBK1. Viruses. 2019; 11(2):158. https://doi.org/10.3390/v11020158
Chicago/Turabian StyleHe, Tian-Sheng, Tao Xie, Jing Li, Ya-Xian Yang, Changsheng Li, Weiying Wang, Lingzhen Cao, Hua Rao, Cynthia Ju, and Liang-Guo Xu. 2019. "THO Complex Subunit 7 Homolog Negatively Regulates Cellular Antiviral Response against RNA Viruses by Targeting TBK1" Viruses 11, no. 2: 158. https://doi.org/10.3390/v11020158