
Three Peptide Modulators of the Human Voltage-Gated Sodium Channel 1.7, an Important Analgesic Target, from the Venom of an Australian Tarantula

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
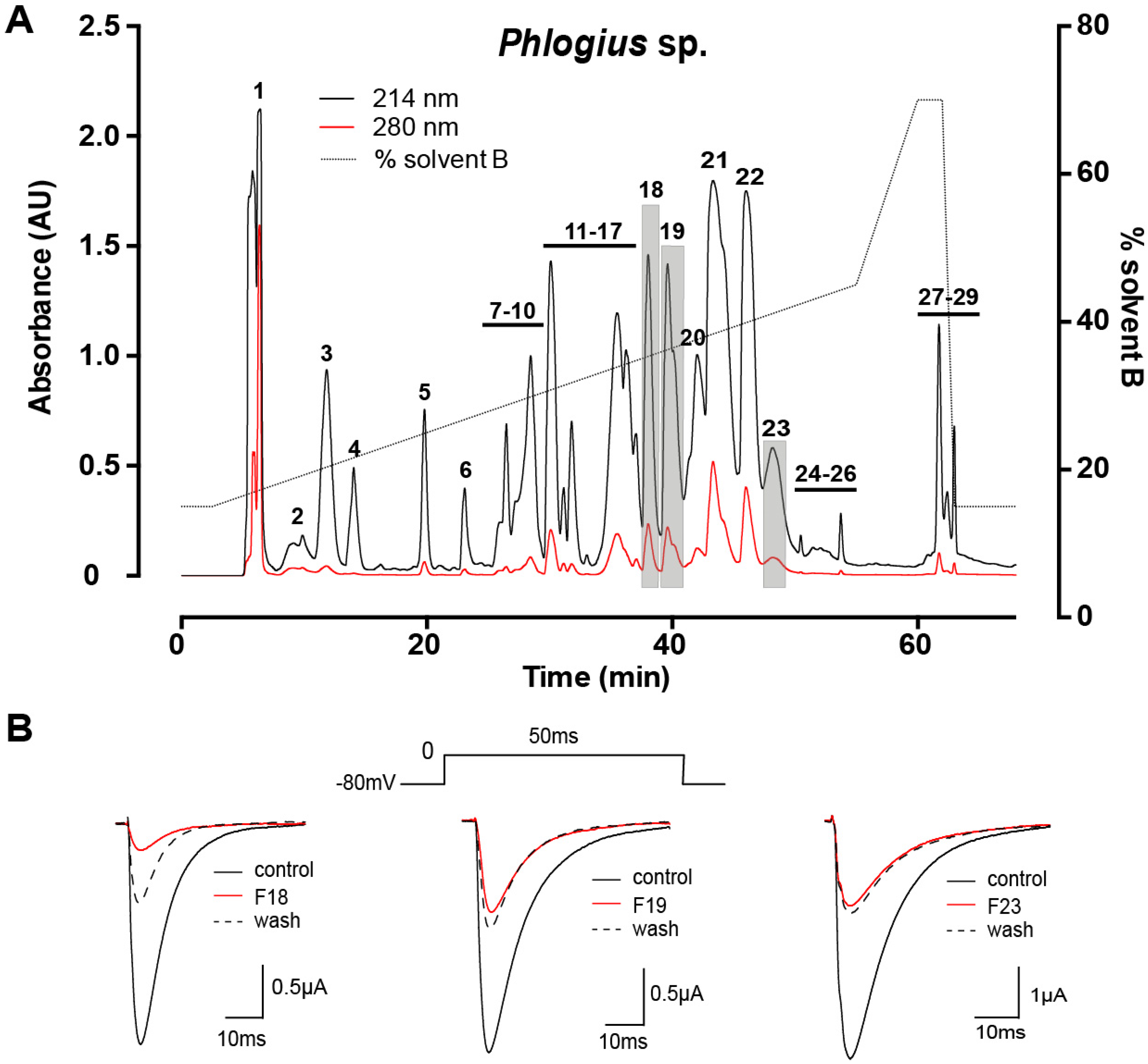
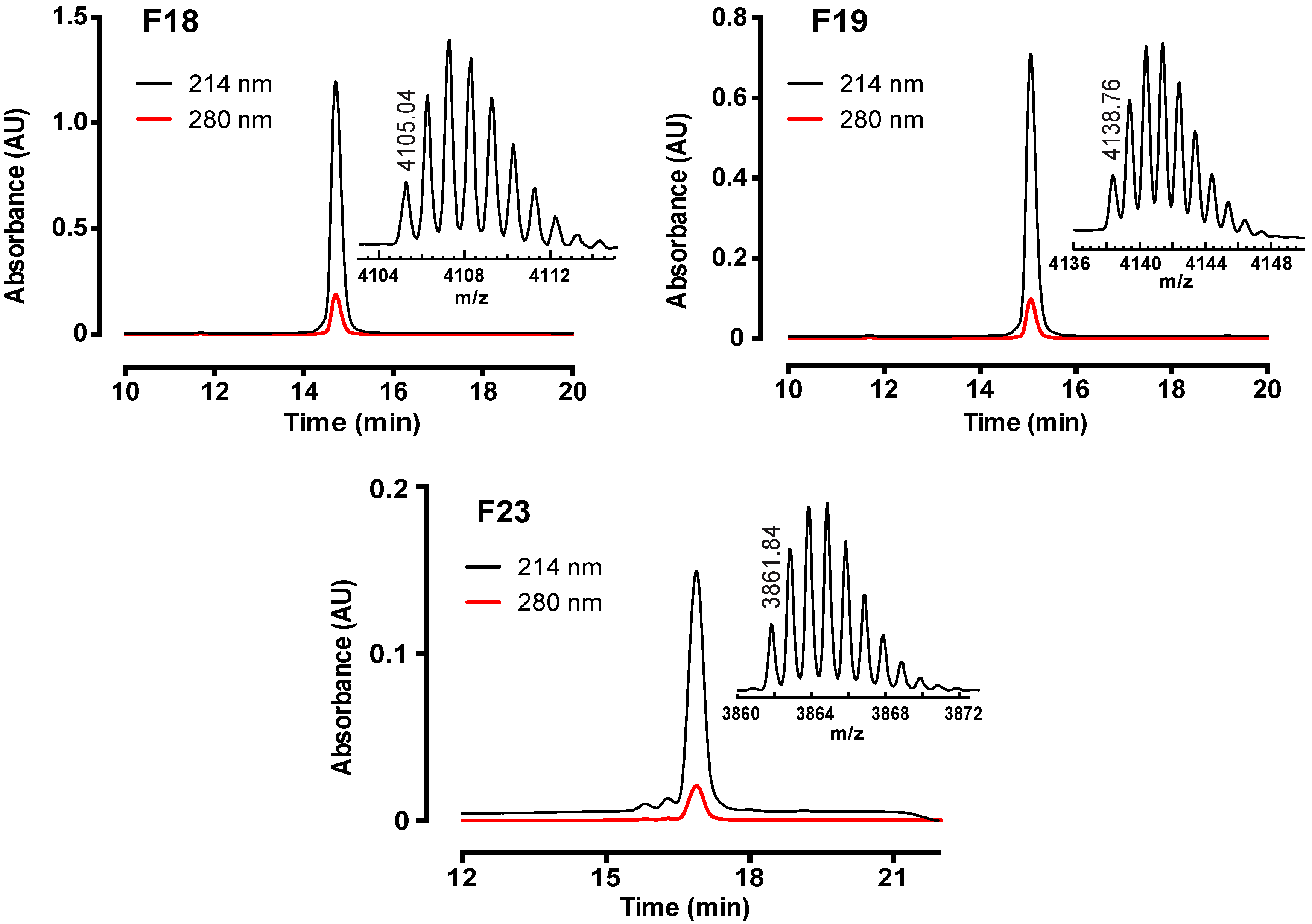
2.1. Assay-Guided Fractionation and Peptide Purification


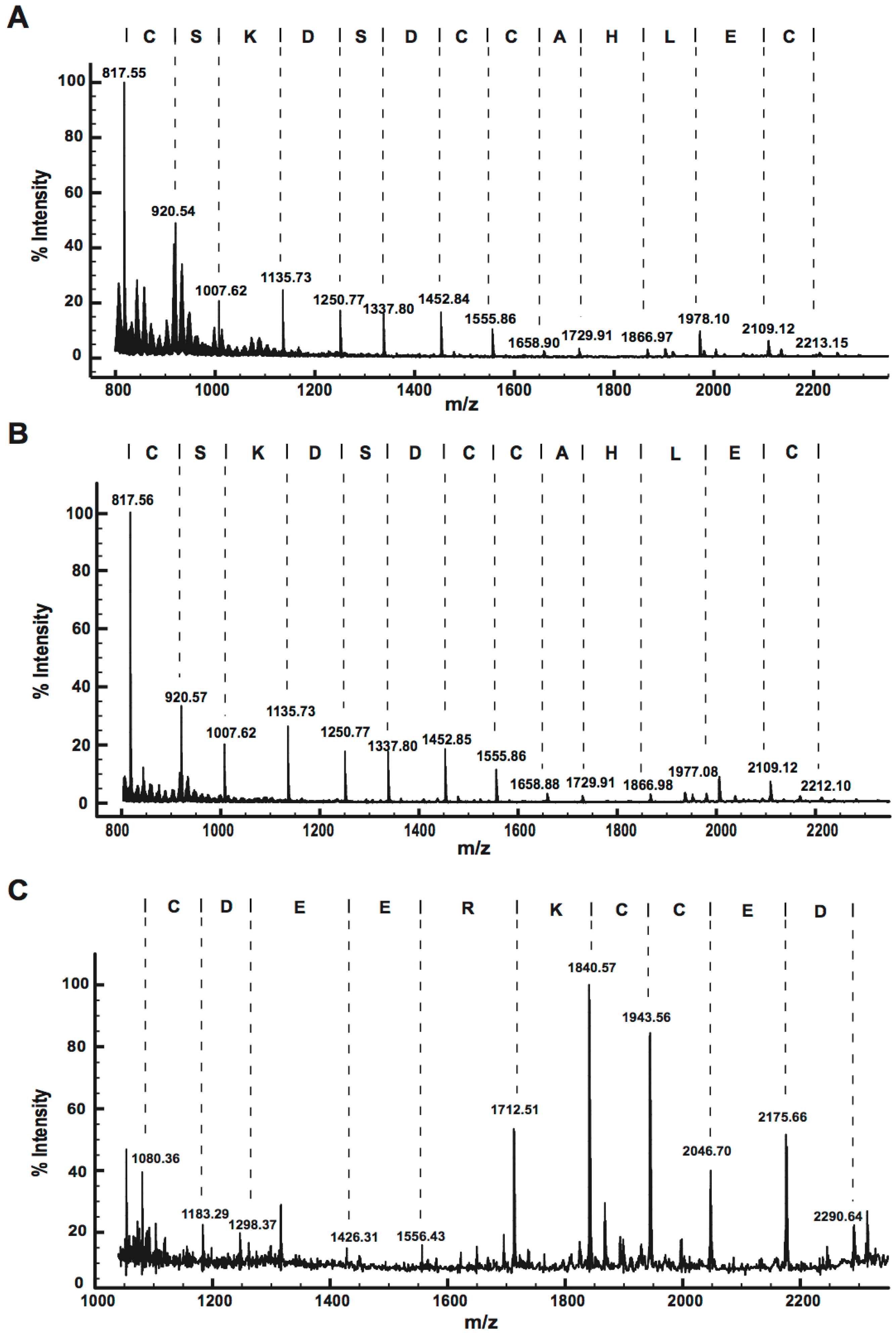
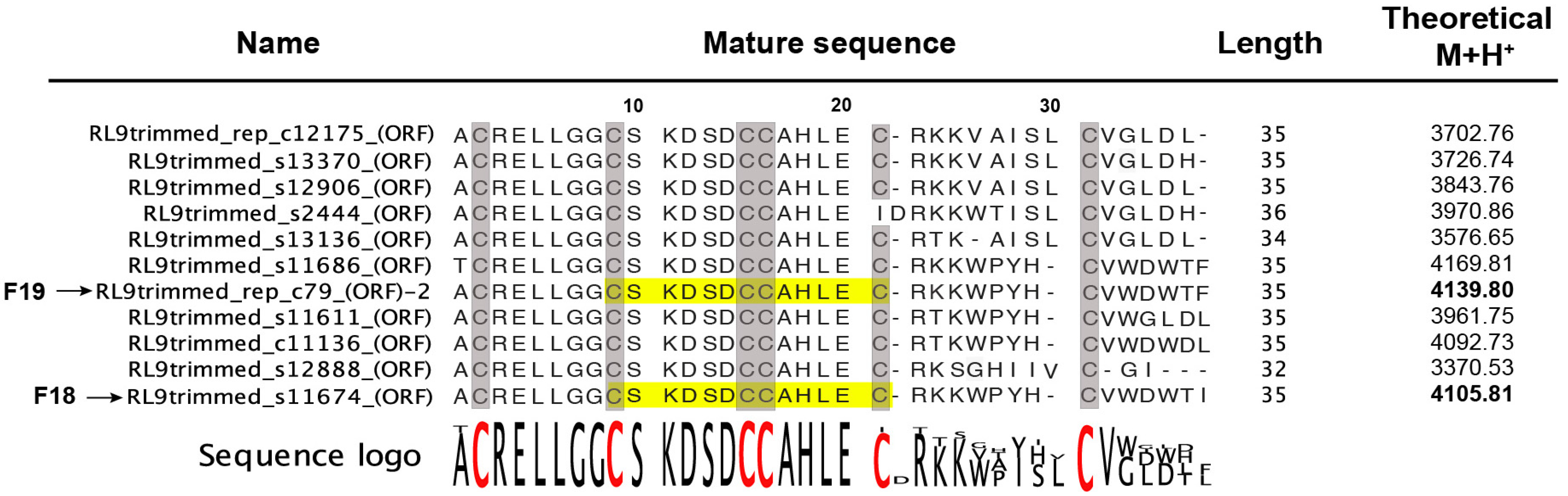
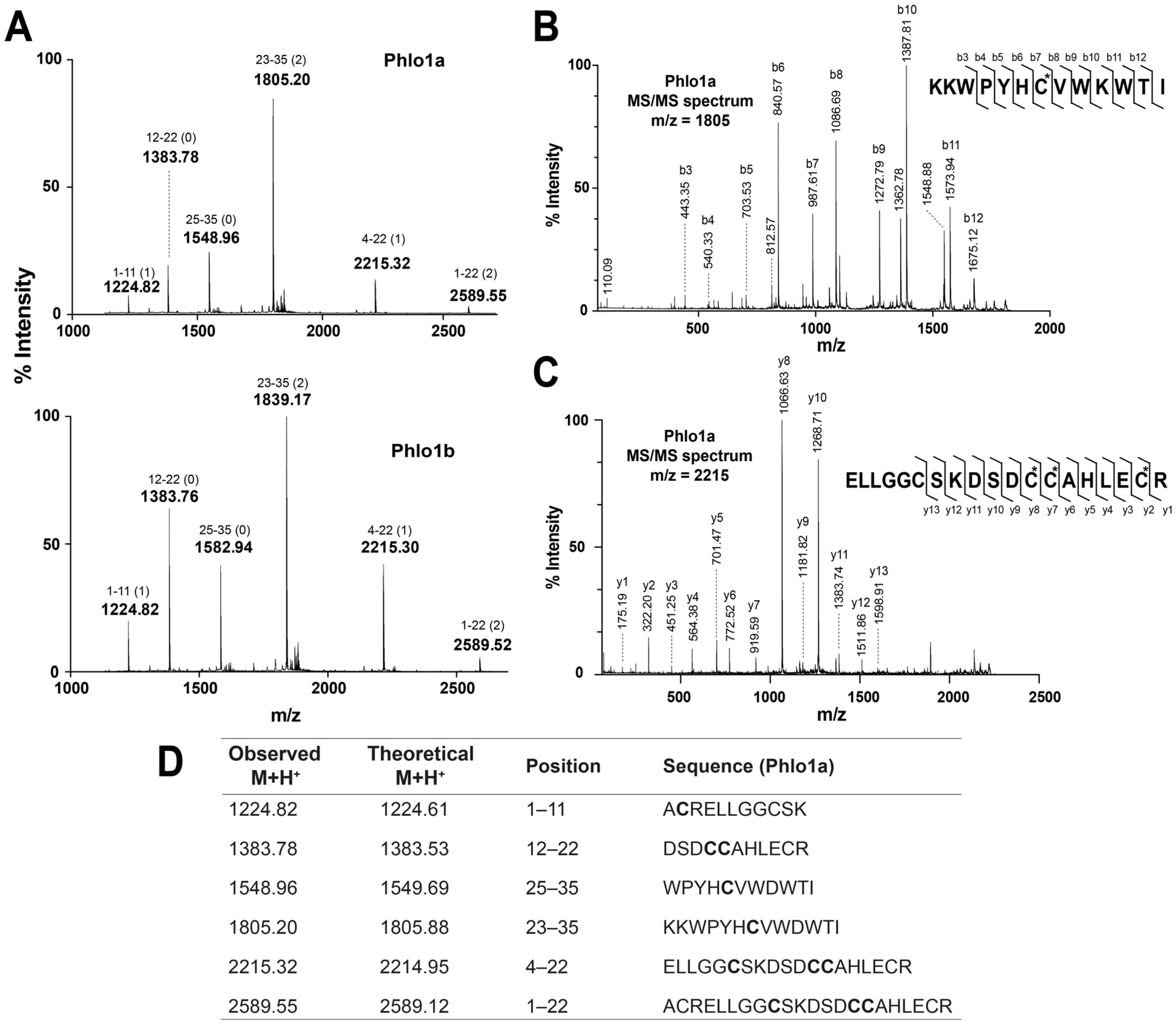
2.2. Peptide Sequence Determination
2.2.1. Venom-Gland Transcriptome
2.2.2. MALDI-TOF MS Using 1,5-DAN Matrix


2.2.3. MALDI-TOF MS Analysis of Tryptic Peptides


2.2.4. Ladder Sequencing Using Carboxypeptidase Y


2.3. Electrophysiological Characterisation of Phlogius Peptides
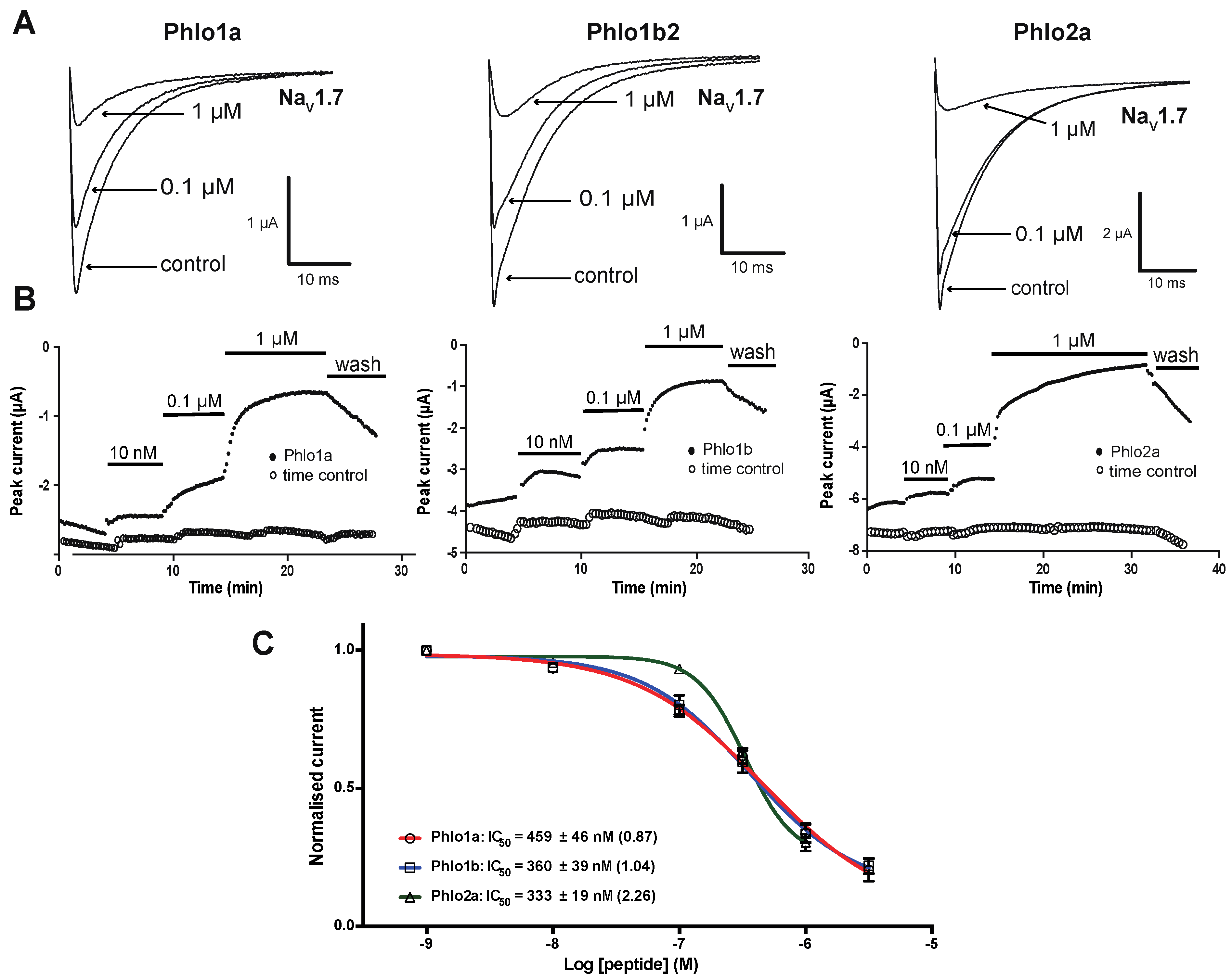
2.3.1. Effects of Phlo1a, Phlo1b and Phlo2a on hNaV1.7 Currents

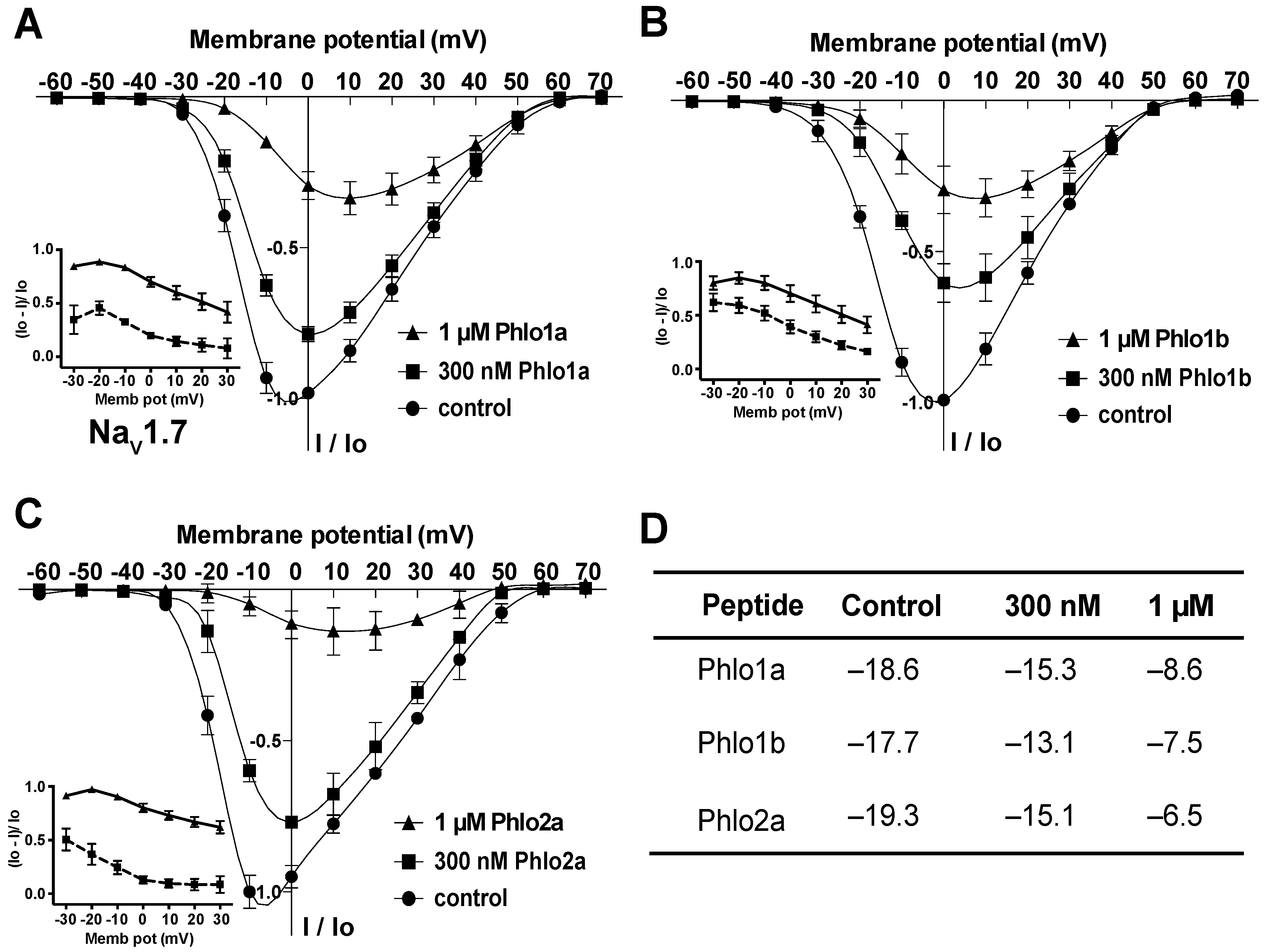
2.3.2. Effect of Phlo1a, Phlo1b and Phlo2a on the Current-Voltage Relationship for hNaV1.7

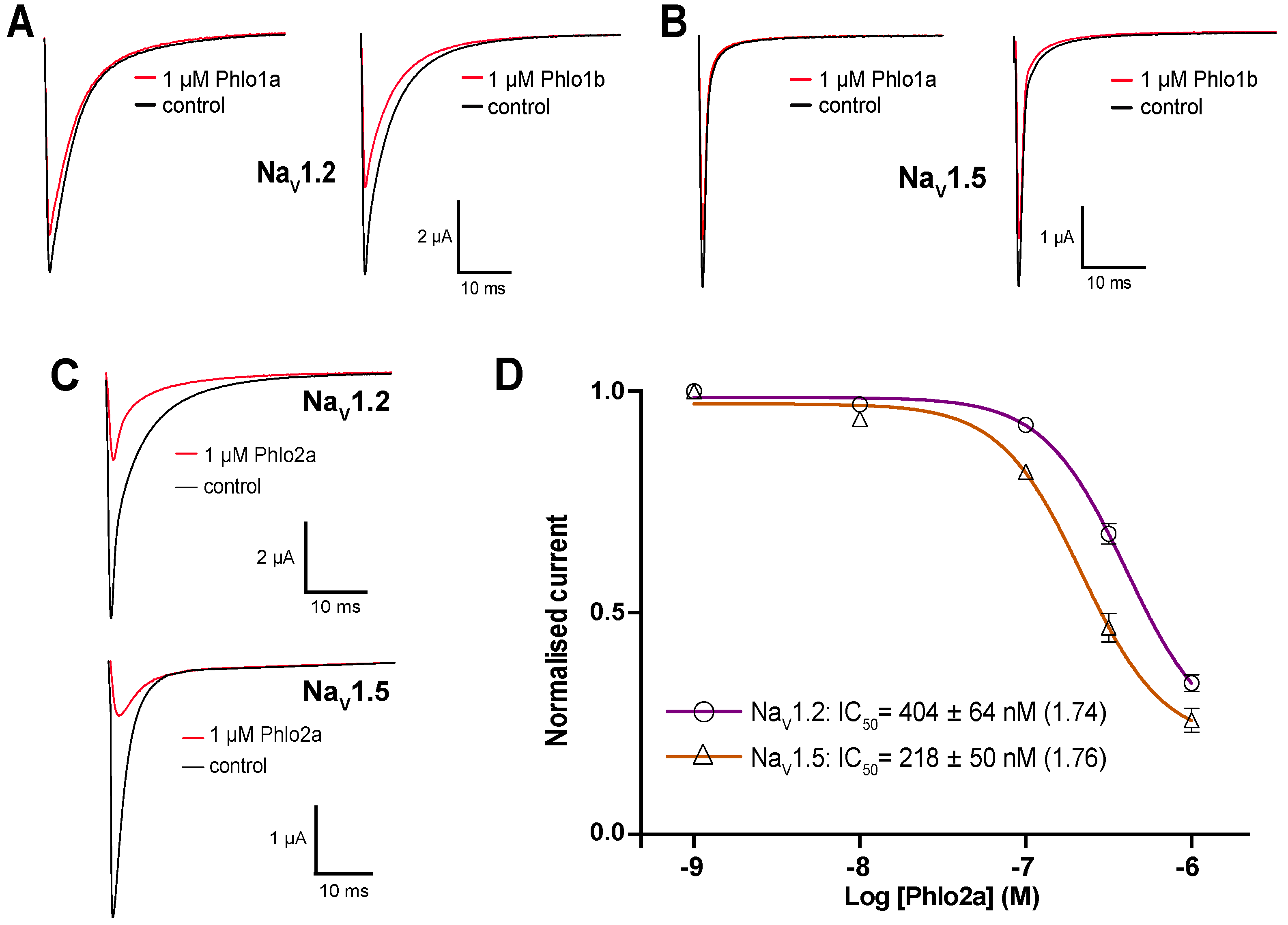
2.3.3. Subtype Selectivity of Phlogius Toxins

3. Experimental Section
3.1. Venom Fractionation and Peptide Purification
3.2. MALDI-TOF Mass Spectrometry
3.3. Reduction/Alkylation of Cysteine Residues
3.4. Tryptic Digestion
3.5. Carboxypeptidase Y Digestion
3.6. Preparation and Analysis of Venom-Gland Transcriptome
3.7. Heterologous Expression of Vertebrate NaV Channels in Frog Oocytes
3.8. Two-Electrode Voltage-Clamp Electrophysiology
3.9. Deposition of Protein and cDNA Sequence Information
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Clare, J.J.; Tate, S.N.; Nobbs, M.; Romanos, M.A. Voltage-gated sodium channels as therapeutic targets. Drug Discov. Today 2000, 5, 506–520. [Google Scholar] [CrossRef]
- England, S.; de Groot, M.J. Subtype-selective targeting of voltage-gated sodium channels. Br. J. Pharmacol. 2009, 158, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Dib-Hajj, S.D.; Tyrrell, L.; Te Morsche, R.H.; Drenth, J.P.; Waxman, S.G. Deletion mutation of sodium channel NaV1.7 in inherited erythromelalgia: Enhanced slow inactivation modulates dorsal root ganglion neuron hyperexcitability. Brain 2011, 134, 1972–1986. [Google Scholar] [CrossRef] [PubMed]
- Estacion, M.; Dib-Hajj, S.D.; Benke, P.J.; Te Morsche, R.H.; Eastman, E.M.; Macala, L.J.; Drenth, J.P.; Waxman, S.G. NaV1.7 gain-of-function mutations as a continuum: A1632E displays physiological changes associated with erythromelalgia and paroxysmal extreme pain disorder mutations and produces symptoms of both disorders. J. Neurosci. 2008, 28, 11079–11088. [Google Scholar] [CrossRef] [PubMed]
- Theile, J.W.; Cummins, T.R. Recent developments regarding voltage-gated sodium channel blockers for the treatment of inherited and acquired neuropathic pain syndromes. Front. Pharmacol. 2011, 2, 54. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Li, S.; Xu, Z.; Li, H.; Ma, L.; Fan, J.; Bu, D.; Liu, B.; Fan, Z.; et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004, 41, 171–174. [Google Scholar] [PubMed]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xiao, Y.; Kang, D.; Liu, J.; Li, Y.; Undheim, E.A.; Klint, J.K.; Rong, M.; Lai, R.; King, G.F. Discovery of a selective NaV1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models. Proc. Natl. Acad. Sci. USA 2013, 110, 17534–17539. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Vetter, I. No gain, no pain: NaV1.7 as an analgesic target. ACS Chem. Neurosci. 2014, 5, 749–751. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Escoubas, P.; Nicholson, G.M. Peptide toxins that selectively target insect NaV and CaV channels. Channels 2008, 2, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Kalia, J.; Milescu, M.; Salvatierra, J.; Wagner, J.; Klint, J.K.; King, G.F.; Olivera, B.M.; Bosmans, F. From foe to friend: Using animal toxins to investigate ion channel function. J. Mol. Biol. 2015, 427, 158–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Smith, J.J.; Vetter, I.; Rupasinghe, D.B.; Er, S.Y.; Senff, S.; Herzig, V.; Mobli, M.; Lewis, R.J.; Bosmans, F.; et al. Seven novel modulators of the analgesic target NaV1.7 uncovered using a high-throughput venom-based discovery approach. Br. J. Pharmacol. 2015, 172, 2445–2458. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; Rash, L. Tarantulas: Eight-legged pharmacists and combinatorial chemists. Toxicon 2004, 43, 555–574. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Hardy, M.C. Spider-venom peptides: Structure, pharmacology, and potential for control of insect pests. Annu. Rev. Entomol. 2013, 58, 475–496. [Google Scholar] [CrossRef] [PubMed]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-venom peptides as therapeutics. Toxins 2010, 2, 2851–2871. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Quinton, L.; Demeure, K.; Dobson, R.; Gilles, N.; Gabelica, V.; de Pauw, E. New method for characterizing highly disulfide-bridged peptides in complex mixtures: Application to toxin identification from crude venoms. J. Proteome Res. 2007, 6, 3216–3223. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Herzig, V.; Wood, D.L.; Newell, F.; Chaumeil, P.A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. ArachnoServer 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2011, 39, D653–D657. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Shu, Q.; Liu, Z.; Liang, S. Function and solution structure of huwentoxin-IV, a potent neuronal tetrodotoxin (TTX)-sensitive sodium channel antagonist from Chinese bird spider Selenocosmia huwena. J. Biol. Chem. 2002, 277, 47564–47571. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Deng, M.; He, Q.; Meng, E.; Jiang, L.; Liao, Z.; Rong, M.; Liang, S. Molecular diversity and evolution of cystine knot toxins of the tarantula Chilobrachys jingzhao. Cell. Mol. Life Sci. 2008, 65, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Middleton, R.E.; Warren, V.A.; Kraus, R.L.; Hwang, J.C.; Liu, C.J.; Dai, G.; Brochu, R.M.; Kohler, M.G.; Gao, Y.D.; Garsky, V.M.; et al. Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry 2002, 41, 14734–14747. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Martin-Eauclaire, M.F.; Swartz, K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 2008, 456, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Rash, L.; Zhu, S.; Diochot, S.; Lazdunski, M.; Escoubas, P.; Tytgat, J. Four novel tarantula toxins as selective modulators of voltage-gated sodium channel subtypes. Mol. Pharmacol. 2006, 69, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Priest, B.T.; Blumenthal, K.M.; Smith, J.J.; Warren, V.A.; Smith, M.M. ProTx-I and ProTx-II: Gating modifiers of voltage-gated sodium channels. Toxicon 2007, 49, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Swartz, K.J. Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 2010, 31, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, W.A.; Calhoun, J.; Burrows, R.; Bailey, T.; Kohler, M.G.; Weinglass, A.B.; Kaczorowski, G.J.; Garcia, M.L.; Koltzenburg, M.; Priest, B.T. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 2008, 74, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Cummins, T.R.; Alphy, S.; Blumenthal, K.M. Molecular interactions of the gating modifier toxin ProTx-II with NaV1.5: Implied existence of a novel toxin binding site coupled to activation. J. Biol. Chem. 2007, 282, 12687–12697. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, S.; Kraus, R.L.; Scheuer, T.; Catterall, W.A. Inhibition of sodium channel gating by trapping the domain II voltage sensor with protoxin II. Mol. Pharmacol. 2008, 73, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, Y.; Iwamoto, S.; Tanaka, K. Rapid sequencing and disulfide mapping of peptides containing disulfide bonds by using 1,5-diaminonaphthalene as a reductive matrix. J. Mass Spectrom. 2006, 41, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.E.; Butler, J.P.; Gelfanova, V.; You, J.S.; Knierman, M.D. A simplified procedure for the reduction and alkylation of cysteine residues in proteins prior to proteolytic digestion and mass spectral analysis. Anal. Biochem. 2004, 333, 174–181. [Google Scholar] [CrossRef] [PubMed]
- USCF, Protein Prospector. Available online: http://prospector.ucsf.edu/prospector/mshome.htm (accessed on 29 June 2015).
- Schroeder, C.I.; Rash, L.D.; Vila-Farres, X.; Rosengren, K.J.; Mobli, M.; King, G.F.; Alewood, P.F.; Craik, D.J.; Durek, T. Chemical synthesis, 3D structure, and ASIC binding site of the toxin mambalgin-2. Angew. Chem. Int. Ed. Engl. 2014, 53, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.L.; Miljenovic, T.; Cai, S.; Raven, R.J.; Kaas, Q.; Escoubas, P.; Herzig, V.; Wilson, D.; King, G.F. ArachnoServer: A database of protein toxins from spiders. BMC Genomics 2009, 10, 375. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chow, C.Y.; Cristofori-Armstrong, B.; Undheim, E.A.B.; King, G.F.; Rash, L.D. Three Peptide Modulators of the Human Voltage-Gated Sodium Channel 1.7, an Important Analgesic Target, from the Venom of an Australian Tarantula. Toxins 2015, 7, 2494-2513. https://doi.org/10.3390/toxins7072494
Chow CY, Cristofori-Armstrong B, Undheim EAB, King GF, Rash LD. Three Peptide Modulators of the Human Voltage-Gated Sodium Channel 1.7, an Important Analgesic Target, from the Venom of an Australian Tarantula. Toxins. 2015; 7(7):2494-2513. https://doi.org/10.3390/toxins7072494
Chicago/Turabian StyleChow, Chun Yuen, Ben Cristofori-Armstrong, Eivind A. B. Undheim, Glenn F. King, and Lachlan D. Rash. 2015. "Three Peptide Modulators of the Human Voltage-Gated Sodium Channel 1.7, an Important Analgesic Target, from the Venom of an Australian Tarantula" Toxins 7, no. 7: 2494-2513. https://doi.org/10.3390/toxins7072494