3.1. Grid Search
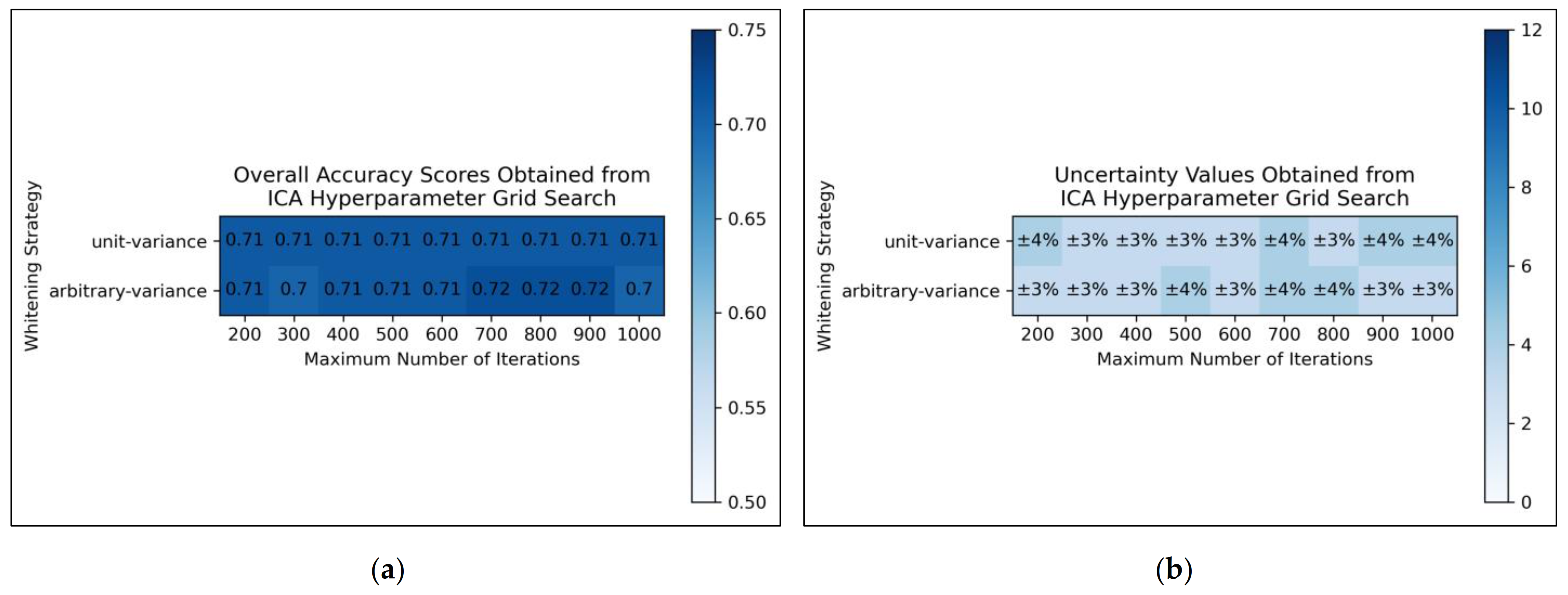
From the results of the grid search, it was deemed that for ICA, arbitrary variance performed marginally better than unit variance in terms of overall accuracy scores, with the most optimal scores occurring between 700 and 900 maximum iterations, although this was possibly a mere statistical fluctuation in otherwise considerably uniform data (
Figure 2a). Uncertainty values appeared uniform and trivially fluctuating, although use of arbitrary variance overall gave rise to lower uncertainties than use of unit variance (
Figure 2b). It was decided from this grid search that the ICA algorithm would use arbitrary variance and a maximum number of 800 iterations (a natural middle ground between 700 and 900).
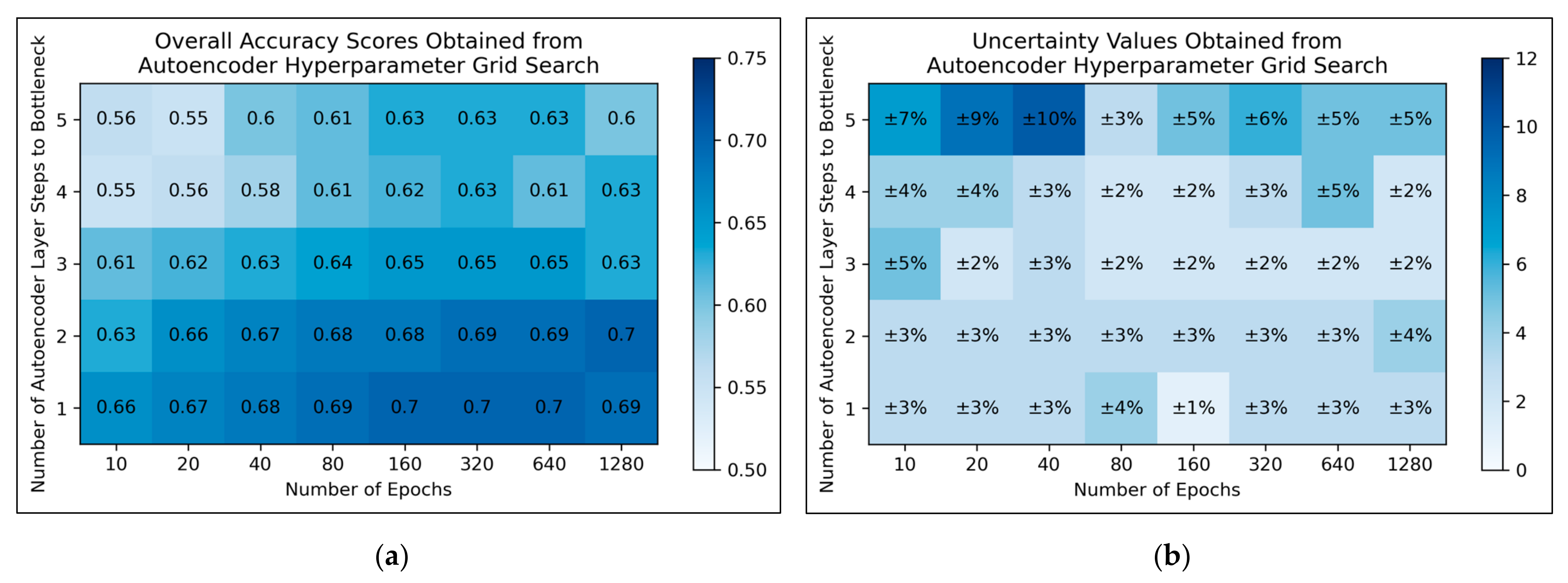
For autoencoders, a tendency for improved model performance was observed as hyperparameter states progressed to the lower-right corner of the heat map, i.e., minimal autoencoder complexity and larger numbers of epochs were optimal (
Figure 3a). Affirming this, an approximate trend of lower uncertainty values was apparent along this direction in hyperparameter space (
Figure 3b). Nonetheless, a slight decline in overall accuracy was present when increasing the number of epochs from 640 to 1280, for the simplest autoencoder architecture; the most optimal performances instead occurred between 160-640 epochs. Thereby, 320 epochs were selected for further use, as a natural middle ground of this optimal range, whereas the selected architecture would follow just 1 step to the bottleneck (i.e., an autoencoder with 3 layers in total).
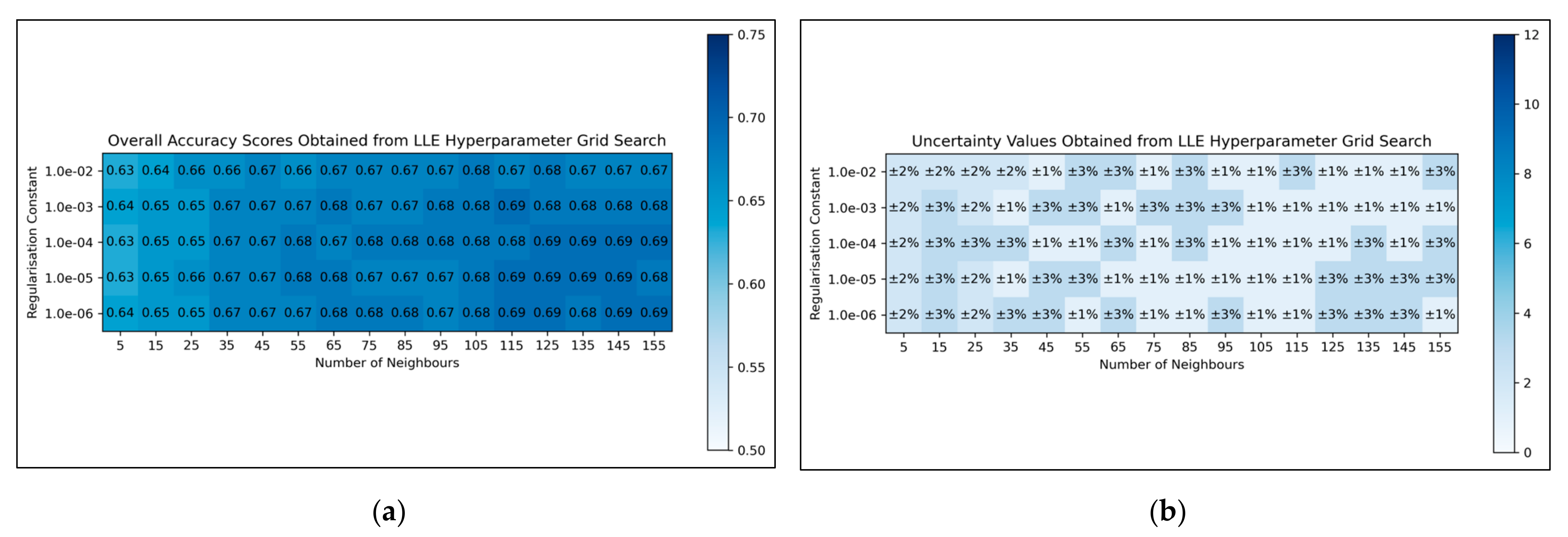
For LLE, a general trend was uncovered, of more optimal performances for increased numbers of neighbours and decreased regularisation constants (
Figure 4a). This was less clearly reflected in uncertainty values, which appeared overall more uniform and to have otherwise varied in a random manner (
Figure 4b). Although the overall trend in LLE performance is clear, it does not perfectly hold for every hyperparameter state. It was decided from these results that the regularisation constant would be controlled at 10
−6, whereas the number of neighbours would be controlled at 115 (as this was the lowest number of neighbours, with the lowest uncertainty value, which resulted in the most optimal performance when using a regularisation constant of 10
−6).
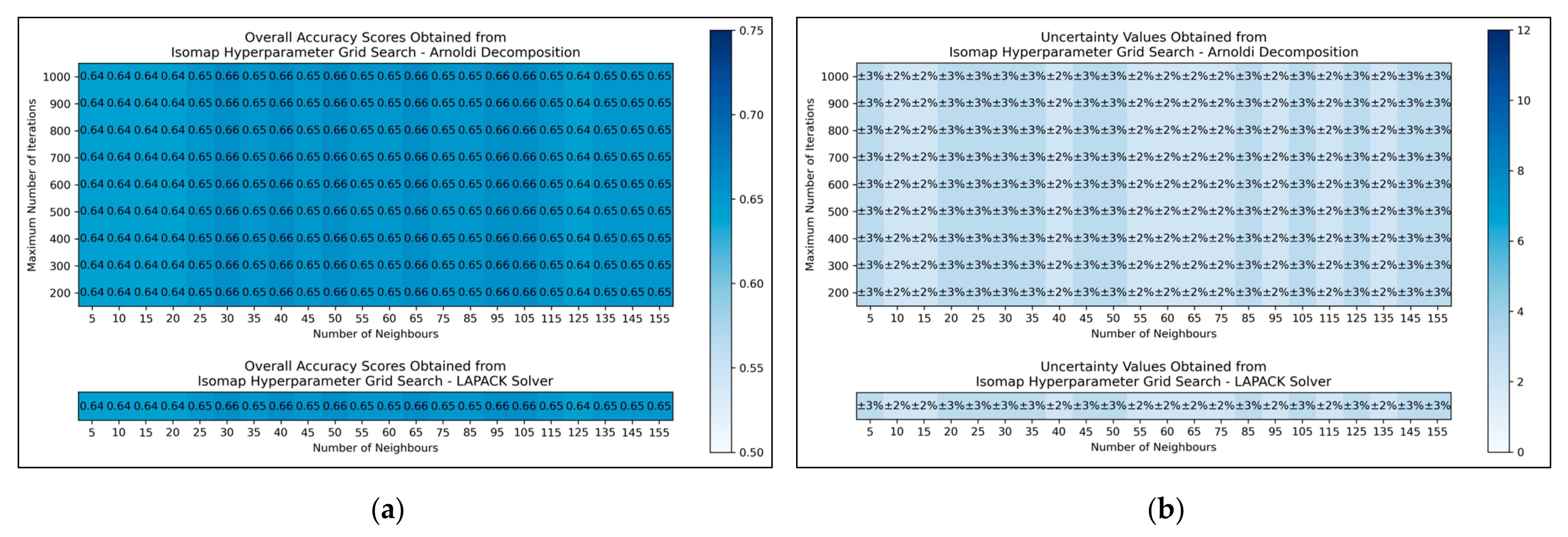
The results for the grid search for isomap shows negligible variation across varied numbers of neighbours considered, albeit with a slight improvement in average performance as numbers were increased beyond 20 (
Figure 5a,b). No variation in performance or corresponding uncertainty occurred across different eigenvalue decomposition algorithms, nor for different maximum iteration values when using Arnoldi decomposition. Although the success of the isomap parameter optimisation was limited, the available evidence suggested that the number of neighbours would be optimally configured at 65. This configuration not only lead to the tied most optimal performance, along with lowest uncertainty, but also served as a natural middle ground between other values that performed comparably well. The eigenvalue decomposition algorithm was set as “auto” to enable the software to choose the most efficient algorithm of the two (both appeared to be identical in performance, through the grid search).
Overall, the grid search stage of this study was of mixed success. Clear and coherent optimisations were reached for autoencoders (
Figure 3) and LLE (
Figure 4), demonstrable via their heatmaps, although their optimal states were notably within proximity of the lower-left bounds of the heat maps. Hence a more optimal state may exist outside the bounds of the search space, which could be explored in future research. A less distinct optimised state was found for ICA (
Figure 2), which may indicate that the search space was not wide or precise enough, or that alternatively other hyperparameters should have been varied instead. Despite this, some level of optimisation was still possible via the ICA grid search. The least success from the grid search however occurred for isomap (
Figure 5), where the varied eigenvalue decomposition algorithms and maximum number of iterations (when using Arnoldi decomposition) had no observed impact on QSAR model performance. This indicates that the choice of including these hyperparameters for the isomap grid search was not optimal. While the search space for numbers of neighbours did have some level of variance in overall accuracy (
Figure 5a), no clear trend was discernable and the variance was furthermore not clearly distinguishable from possible naturally occurring statistical noise in the data. This indicates that the search space for the number of neighbours hyperparameter was insufficient, even if it was sufficient for the LLE grid search (
Figure 4).
Overall, grid search offers an appropriate approach for this study, as it would not have been feasible to choose hyperparameters, search spaces and required levels of resolution through any exact theoretical
a priori deduction, for such a complex dataset containing 11,268 dimensions; instead a level of starting assumption was necessary. Nonetheless, a potential future research direction could be to expand considered hyperparameters, their respective search spaces and respective levels of resolution, based on the outcomes of an initial grid search (particularly based on whether sufficient optimisations could be found from the initial grid search). The uneven and arbitrary nature of the resolutions between different grid searches could also be more coherently standardised via a clear methodology, for a future study. Additionally, the inherent limitations of grid searches as a hyperparameter optimisation method (particularly the limitation of resolution of the search space and potentially missing more optimal states between chosen search values) could be addressed via choosing another optimisation approach such as random searches and Bayesian optimisation, perhaps to be ran in parallel to grid searches, followed by a final comparison of optimised hyperparameters found [
26].
The grid search stage of the study provides an early level of insight into the feasibility of the explored dimensionality reduction algorithms and their respective chosen hyperparameters, as overall accuracy scores approximately between 60–70% were obtained, with comparatively insignificant levels of uncertainty. This indicates that all configured dimensionality reduction techniques were able to reduce the dimensionality of the feature space, while conserving sufficient information to empower simple MLP classifiers to perform measurably more accurately than a random guess and comparable to performance metrics obtained through our previous research [
8]. While the controlled dimensionality of 100 may have inhibited certain techniques from producing more optimal feature spaces, the later stage of subsequent comparison of performances over numerous ascending dimensionalities would help alleviate this limitation, while the controlled value of 100 dimensions is deemed as having maintained an appropriate and feasible set of conditions for a valid prior grid search.
3.2. Comparative Performances of Dimensionality Reduction Techniques
Direct comparison of dimensionality reduction technique performances, using optimised hyperparameters, is illustrated in
Figure 6:
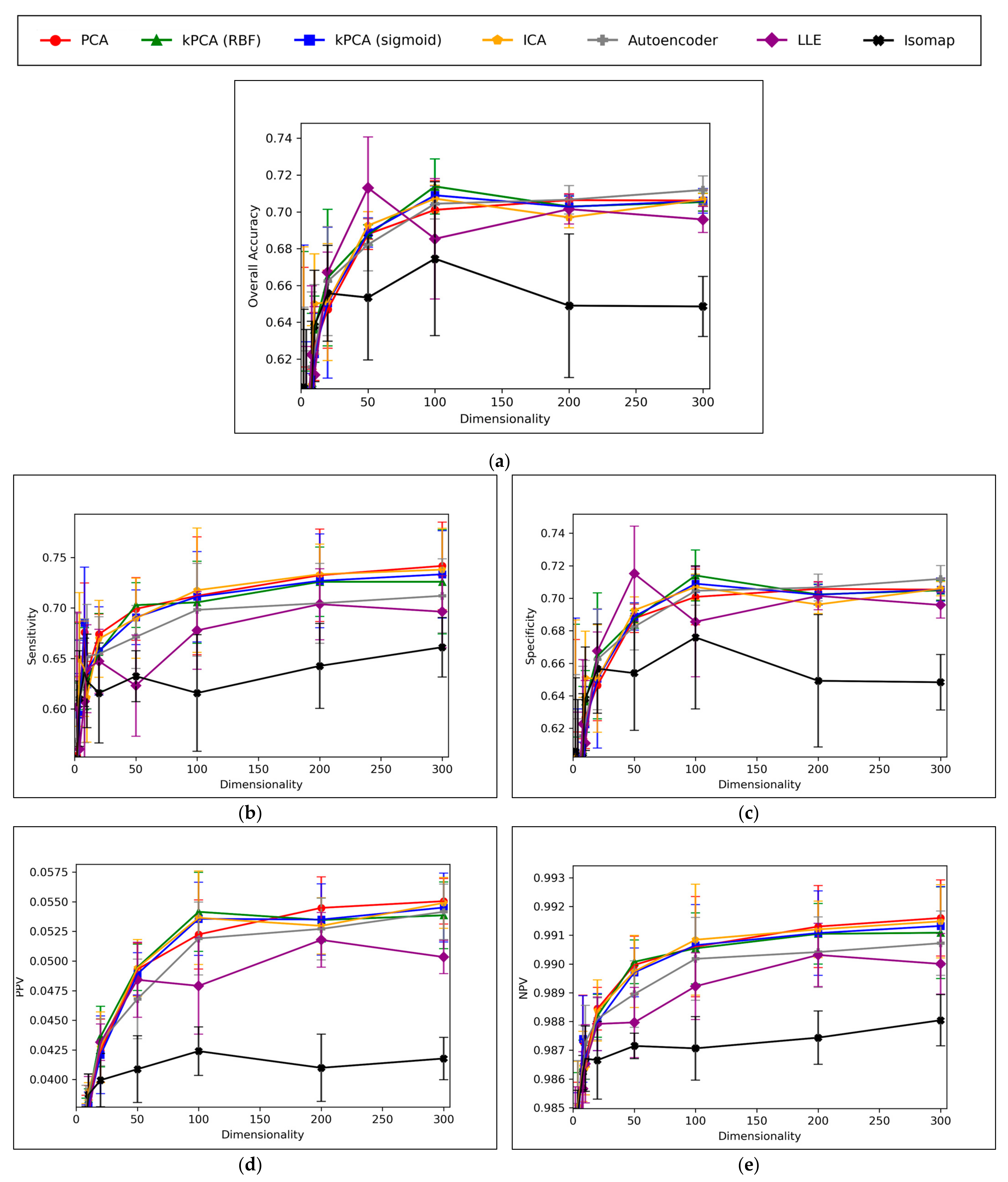
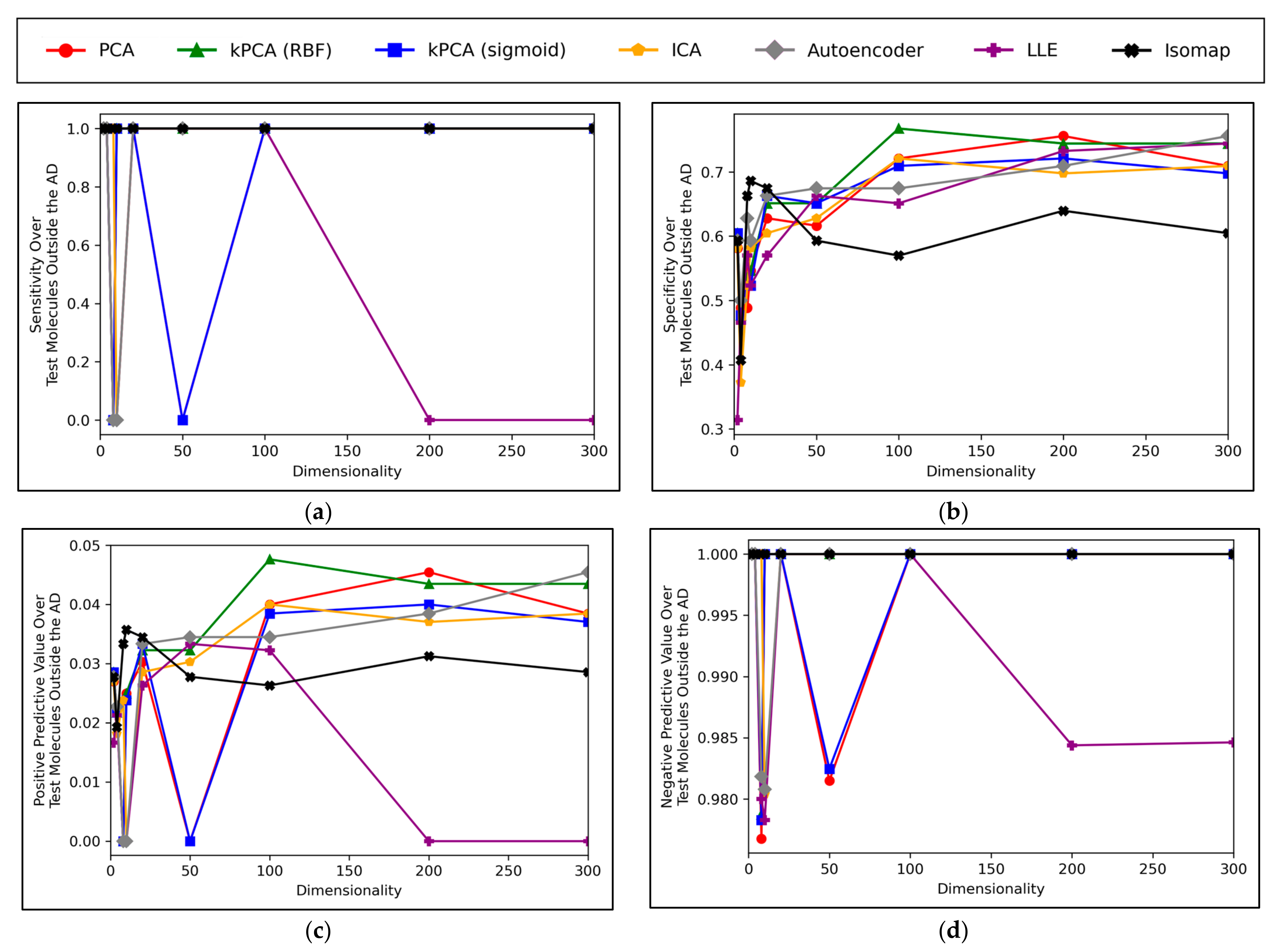
A convergence of overall accuracy scores was observed, for the majority of models, at 100 dimensions and beyond (
Figure 6a). However, the sensitivity scores showed a gradually increasing trend for the highest performing models, even beyond 100 dimensions (
Figure 6b). For specificity scores, similar trends and findings were found to theose of overall accuracy scores, due to the imbalanced nature of classes in the data (
Figure 6c). Furthermore, similar trends were present for PPV and NPV values, however the PPV values were significantly smaller than the NPV values, naturally due to the data imbalance between mutagenic and non-mutagenic molecules (
Figure 6d,e).
The overall convergence displayed in
Figure 6 (especially
Figure 6a,c) suggested that optimal dimensionality was approximately reached via the dimensionality reduction techniques and hence no further useful information could be provided through increased dimensionality. This also implies that the search space and resolution of dimensionalities was sufficient. Although sensitivity scores in
Figure 6b do not show an absolute convergence, the gradient of improvement for dimensionalities increased beyond 100 is notably more gradual and hence convergence is approximately inferable. Additionally, the imbalance of mutagenic molecules (significantly outnumbered by non-mutagenic molecules) may call into question the exact accuracy of any trends discerned solely in terms of sensitivity.
Overall, the results indicate that accuracy scores peaked within range of ~70%, even for the best performing dimensionality reduction techniques, which is also in line with the results of our previous study [
8]. It is however possible that autoencoders may have been capable of reaching higher peak overall accuracy scores, at higher dimensionalities, as evidenced by a less visibly converged gradient of improvement, as well as reaching a peak above all other techniques at the maximum tested dimensionality of 300. The best performing technique is not absolute or conclusive from the results, as PCA, both forms of kPCA, ICA and autoencoders performed with closely comparable average overall accuracies, furthermore with considerable overlap between margins of error. At certain dimensionalities, LLE also performed at comparable levels. PCA, both forms of kPCA and ICA demonstrated more robust sensitivity scores than any of the other methods, however more equivalent performance was observed from autoencoders and LLE in terms of specificity.
The consistently high performance of linear dimensionality reduction techniques, often greater than the performance of non-linear techniques, strongly suggests that the original 11,268-dimensional dataset was linearly separable, or at least approximately so. This is in line with Cover’s theorem and the underlying hypothesis of this study [
12]. Although non-linear techniques such as kPCA, autoencoders and LLE overall performed comparably to PCA and ICA (which further affirms the hypothesis), PCA and ICA as linear techniques offer generally simpler and often more efficient techniques for processing high-dimensionality datasets into more manageable feature spaces for deep learning driven QSAR models. That said, molecules in toxicological space are not randomly distributed and hence potential non-linearly separable relationships in higher dimensional toxicological datasets may be more appropriately handled with autoencoders and LLE, in the absence of any
a priori knowledge, as they in any case can perform in a comparable manner to linear techniques for data that are linearly separable. It is hence a more robust strategy to use generally applicable non-linear dimensionality reduction techniques, such as autoencoders, not just for datasets where
N > D + 1 (and hence with increased likelihood of non-linear separability), but also when
a priori knowledge of the dataset’s separability is lacking and/or difficult to characterize.
Isomap significantly underperformed throughout the different graphs of
Figure 6, in comparison to the other dimensionality reduction techniques explored. This however could be expected, given the failure of the isomap hyperparameter grid search to uncover any clear optimal state; isomap has demonstrated effectiveness in numerous other QSAR modelling studies and may have performed measurably better, given a more robust hyperparameter Optimisation and resulting final hyperparameter configuration. On this note, other dimensionality reduction techniques may have also been hindered by the limitations of the grid search stage of this study, hence while some level of conclusions may be drawn from the results of this study, it is also important to note the limitations of these findings. Similarly, the simplicity of the MLP classifiers with only 2 hidden layers of 500 neurons, may have capped the potential of the results to reach higher peak accuracy scores, although the controlled nature of the MLP classifiers nonetheless serves as a valid means for drawing comparison between competing dimensionality reduction techniques. Nonetheless, it is possible that the peak overall accuracy scores within range of ~70% from
Figure 6a may have been due to the limitations of the MLP classifier stage of the QSAR model, rather than any of the employed dimensionality reduction techniques, hence a future expansion to the study may benefit from varying the complexity of the DNNs used for classification.
It was deemed that the chosen selection of PCA, kPCA (using two different non-linear kernels), ICA, autoencoders, LLE and isomap, was sufficiently diverse in providing a valid basis for drawing a limited comparison between different dimensionality reduction techniques, especially in terms of linear techniques vs non-linear, for their suitability in building deep learning based QSAR models of mutagenicity and other toxicological endpoints. However 6 dimensionality reduction techniques compared in this study is not an absolute representation of all dimensionality reduction techniques, especially with regards to the diverse space of non-linear methods; wider comparison could be explored in future research.
While it was intended that while all findings of this study would naturally be directly applicable to the specific dataset used, these results were also aimed to provide insight into the general suitability of the algorithms explored, for similar QSAR modelling applications in future. Nonetheless it may be appropriate to extend future studies to include additional datasets.
3.3. Analysis of Applicability Domain
The AD was calculated across all iterations and is irrespective of dimensionality reduction technique used.
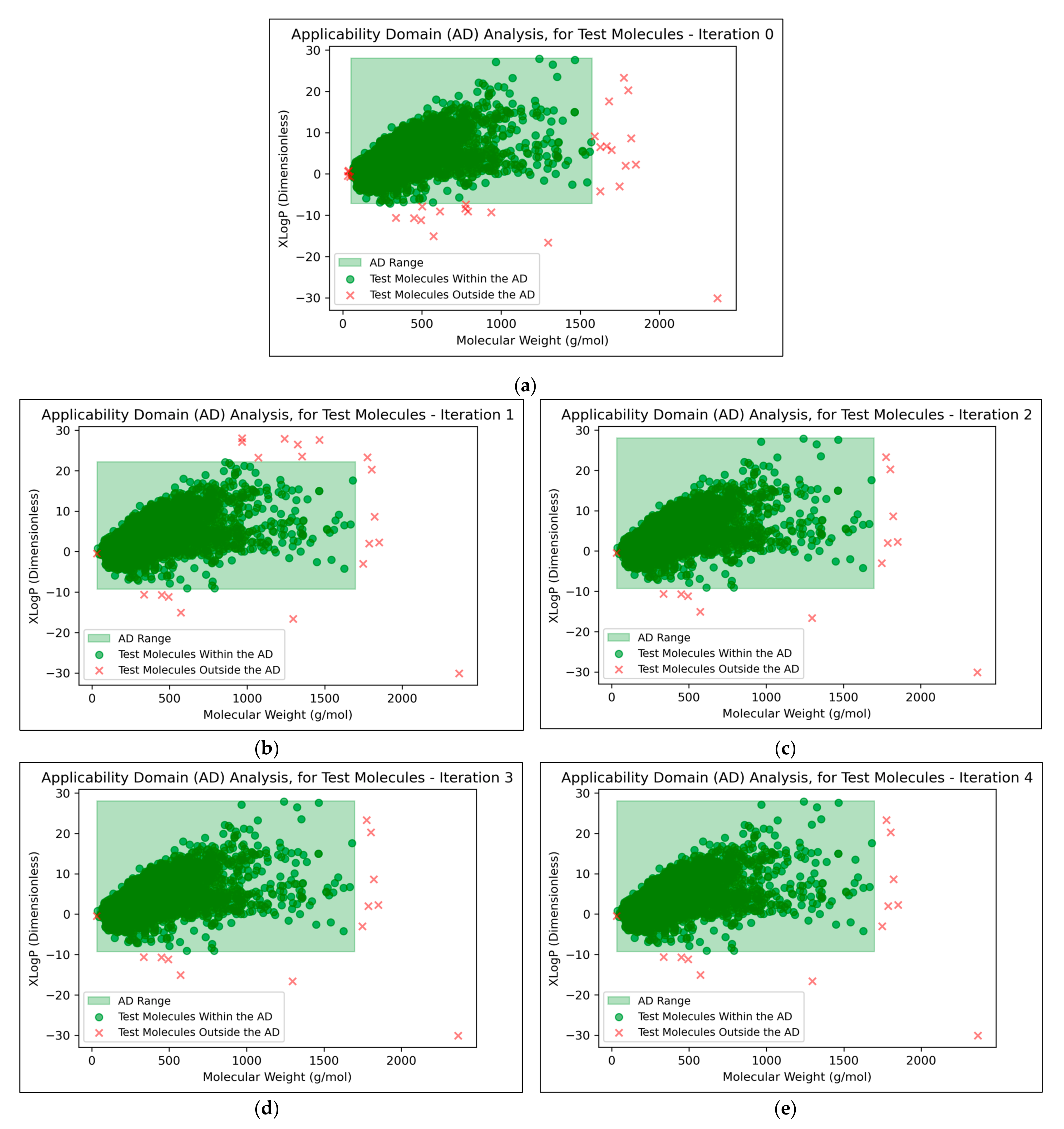
It was found that an average of 0.15% (2 sf, over all iterations) of molecules were located outside the AD (
Figure 7). This comparatively low level indicated that the QSAR models used data that were sufficiently spaced and stratified, although it may additionally suggest that the definition of the AD used was limited and insufficient for explaining the ~30% of testing data that were misclassified at peak QSAR model performances (see
Figure 6). It may also be observed from
Figure 7 that some of the more prominent and/or eccentric datapoints outside of the AD are repeated in the testing data across different iterations; these datapoints hence represent non-mutagenic molecules that were assigned to the permanent testing data pool (and comparable outliers did not occur in the training data, as these would have widened the boundaries of the AD accordingly).
It is generally expected that QSAR models are at risk of underperforming when used to test molecular data that exists outside of their AD, hence the performances of the QSAR models of this study, for classifying testing molecules outside of the AD, were investigated (
Figure 8):
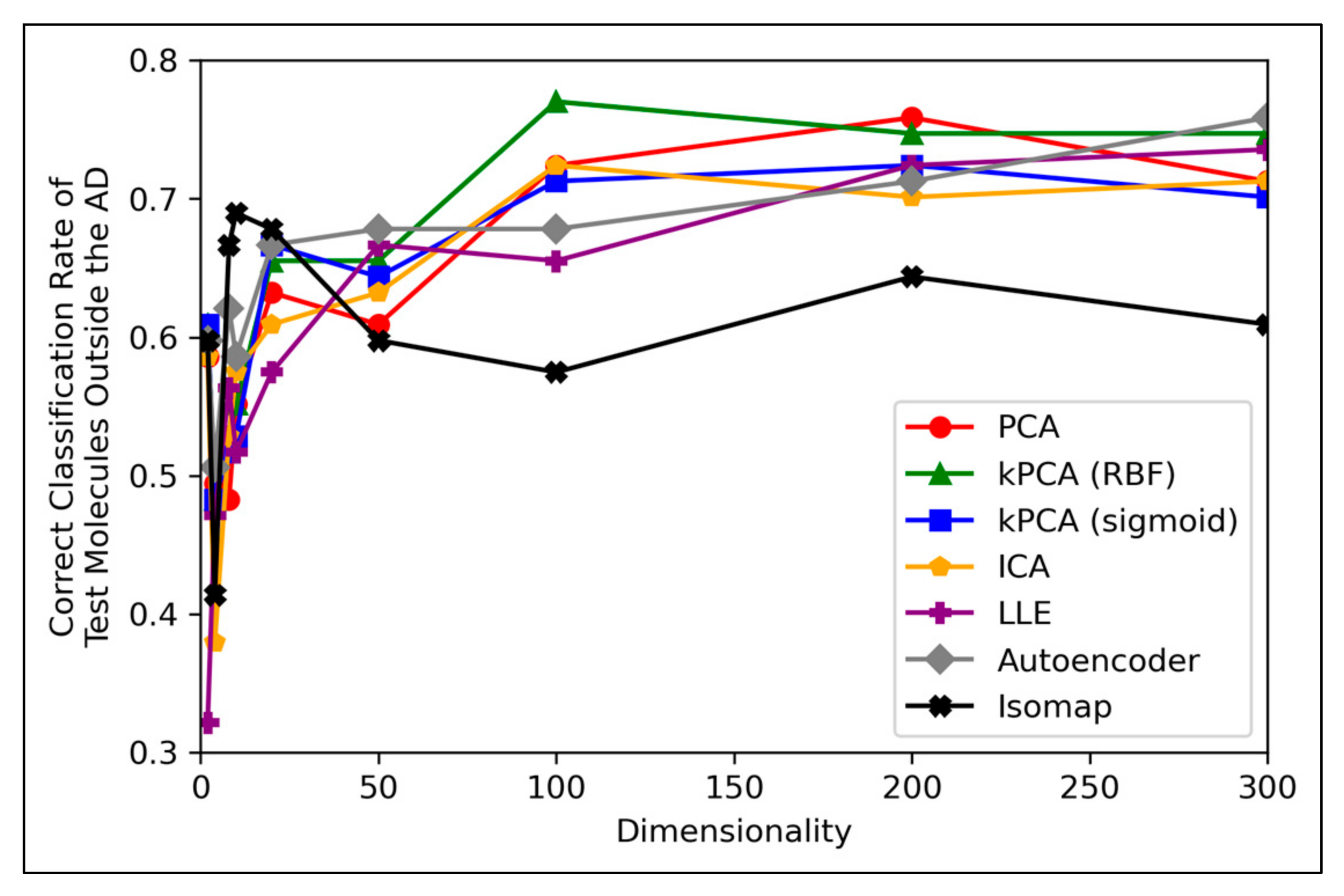
Figure 8 displays peak performances that were notably higher than the ~70% overall accuracy scores observed in
Figure 6a, for all dimensionality reduction techniques except for isomap (although even isomap approached ~70% correct classification rate, at lower dimensionalities). Closely comparable trends were further found to hold in terms of specificity, however analysis in terms of sensitivity scores was limited, due to a scarcity of mutagenic molecules outside of the AD (see
Appendix B). These findings are unexpected, as it demonstrates that the majority of QSAR models on average performed more optimally at classifying molecules outside the AD than at classifying molecules inside the AD. This could be due to the adopted definition of the AD for this study being insufficient. Although this is difficult to verify, as any conclusions drawn from comparisons between
Figure 6 and
Figure 8 may be inherently limited, due to the comparatively much lower amount of molecular datapoints that were available for constructing
Figure 8 (0.15% of molecules, compared to 100% of molecules used to construct
Figure 6). Nonetheless, another interesting finding from
Figure 8 is that while techniques such as PCA, kPCA and ICA remained largely stagnant in performance at 100 dimensions and beyond, autoencoders and LLE continued to gradually improve in classification rate (with autoencoders outperforming all other techniques at 300 dimensions). This may indicate that non-linear dimensionality reduction techniques, particularly autoencoders, at a larger number of dimensions, could perform more optimally than linear techniques for processing data that contains a larger number of statistical outliers. The fact that a similar indication was also present from
Figure 6a is reaffirming of the possibility that autoencoders may have reached more optimal overall accuracy scores at higher dimensionalities. It should however be noted that PCA, kPCA and ICA outcompeted autoencoders and LLE at lower dimensionalities of 100, despite PCA and ICA being linear techniques, as well as that PCA and kPCA underwent no grid search hyperparameter optimisation stage. kPCA, when using the RBF function as a kernel, surpassed 75% correct classification rate at 100 dimensions and was the most consistent top-performing technique in
Figure 8. Similar findings for kPCA (RBF) at 100 dimensions are also observable from
Figure 6a. The frequently more optimal performance of kPCA (RBF), compared to PCA and even kPCA (sigmoid), suggests that a RBF kernel was indeed more optimal for navigating the initial feature space.
Considering the above findings, it was deemed beneficial to obtain further insight into the classes of molecules which were outside the AD, as well as generally how the classes of molecules were distributed according to the chemical space used to the define the AD.
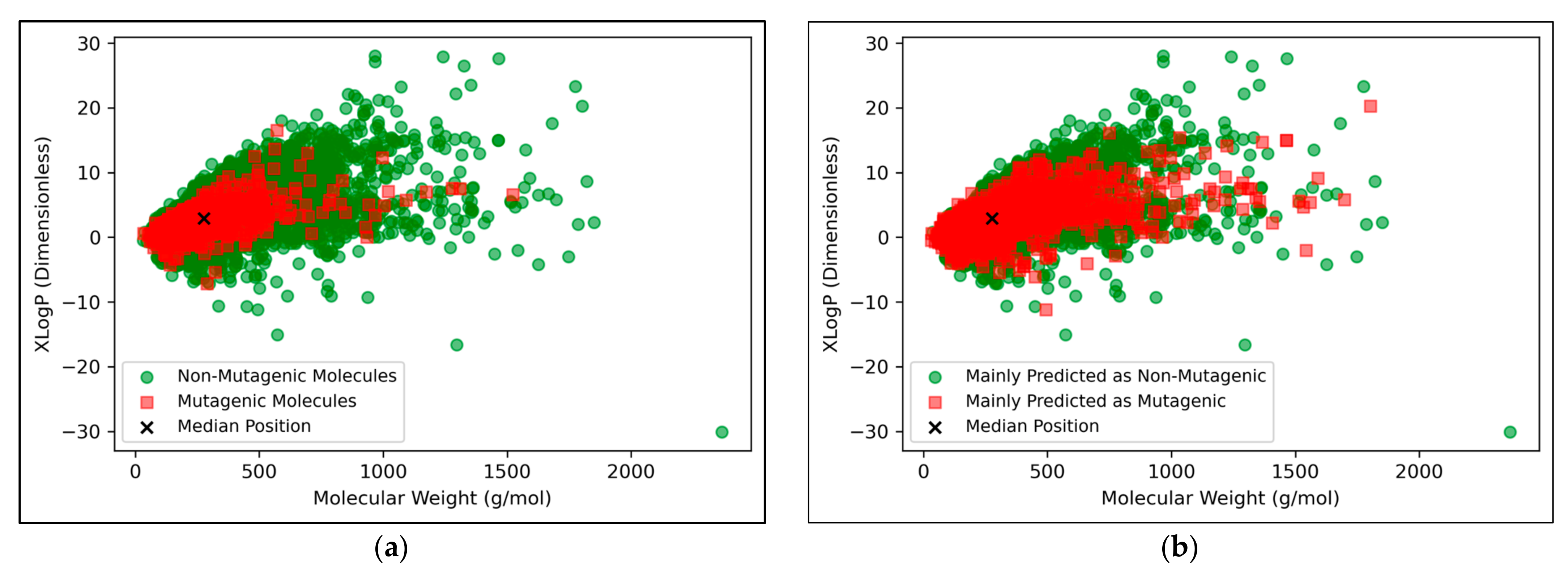
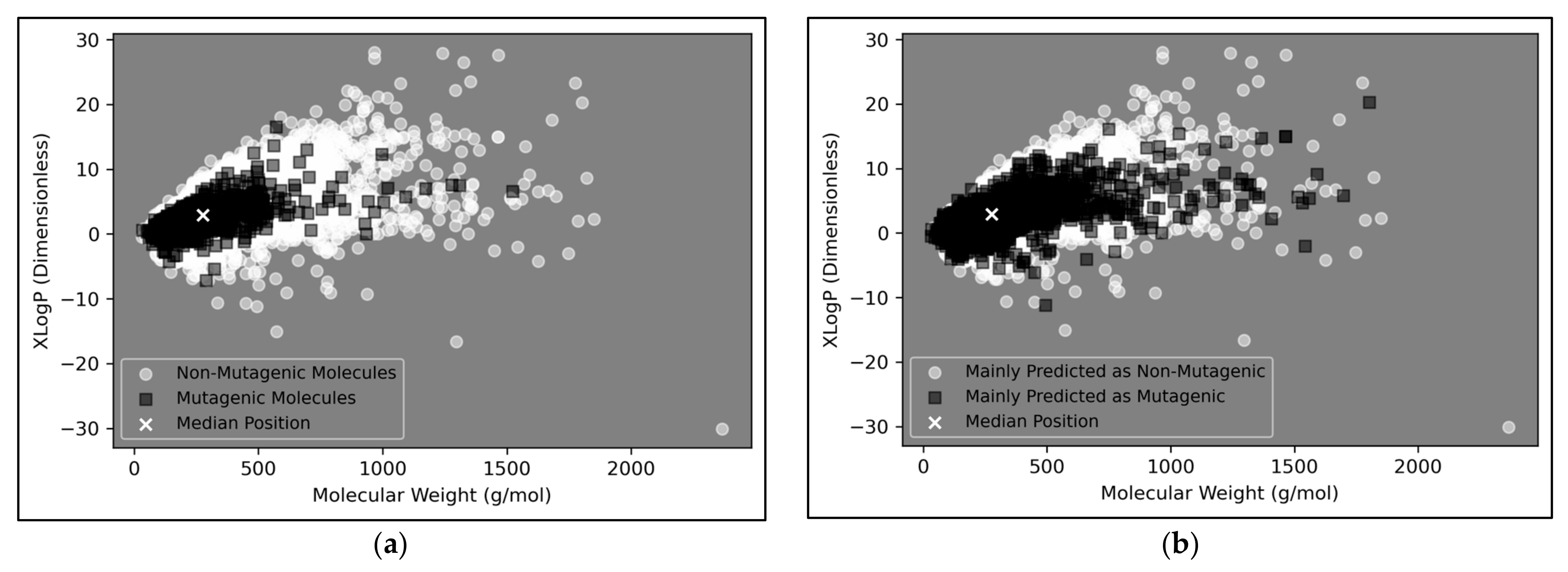
The vast majority of molecules outside the AD were non-mutagenic, along with a general majority of statistical outliers from the main cluster body of the visualised chemical space (
Figure 9a). This may be a purely probabilistic phenomenon, as a significantly larger number of non-mutagenic molecules occurred in the curated dataset, compared to mutagenic molecules, which is supported by the observation that a minority of mutagenic molecules also appear as visible outliers, to a certain extent. This suggested that a minority percentage of both classes naturally were outliers in terms of their distribution in XLogP/MW chemical space, but that non-mutagenic cases were simply more frequent due to the inherent data imbalance. Regardless of this, the significance of the findings of
Figure 8 may be called into question, given the extremity of the data imbalance of the molecules outside the AD, implying that the QSAR models were
de-facto only being compared by their ability to classify non-mutagenic outliers; their overall improved performances may suggest that the MLP classifier simply learned to classify the majority of statistical outliers as non-mutagenic (an explanation which is supported by
Figure 9a). While initial stratification of the training data attempted to remove any bias occurring in favour of any particular class, inherent limitations of the distribution of the classes in chemical space would have been less easily overcome and appears as a possible fundamental limitation of the 2014 AQICP dataset. Additionally, the success of the QSAR models in identifying statistical outliers of the physicochemical XLogP/MW chemical space as non-mutagenic, is an affirmation of the suitability of the techniques used. This is because the QSAR models demonstrated predictions that were directly interpretable via the physicochemical space used for defining the AD, despite having used feature vectors that were more abstract quantifications of toxicological space, via matrices of TCs between SMILES descriptors.
Figure 9b appears to display a considerably wider or less dense distribution of mutagenic predictions than that of the true distribution of
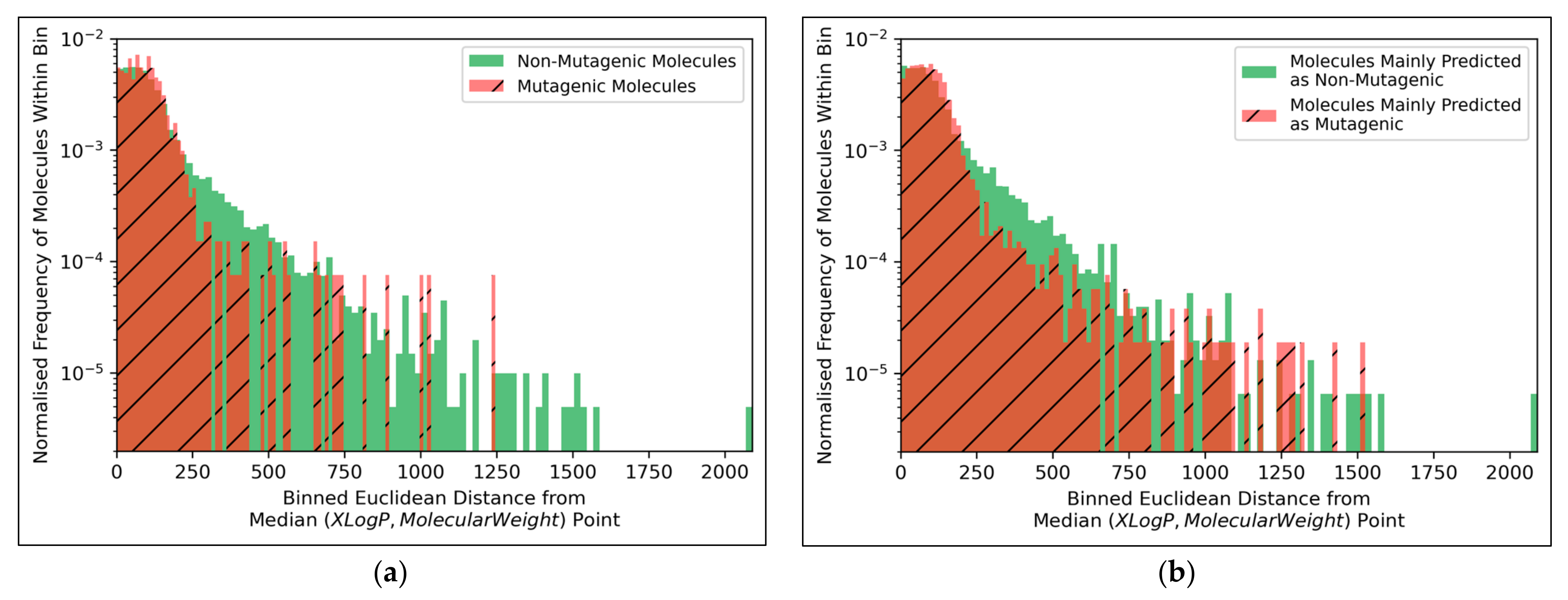
Figure 9a, but the large density of datapoints makes this difficult to compare. Normalised histograms of distances from the median point were hence plotted in
Figure 10.
The distributions of both classes of molecules, in terms of distance from the median point in XLogP/MW chemical space, were exponential distributions and approximately equivalent, except for the greater definition of the non-mutagenic exponential distribution, especially across further distance bins, owing to the inherent data imbalance (
Figure 10a). This points to the main limitation of the 2014 AQICP dataset as concerning the inherent data imbalance, rather than any unequal distribution of the classes. The results also demonstrated that the MLP classifier mostly followed a balanced prediction of mutagenic and non-mutagenic molecules, over the combined distribution, with the exception of extreme outliers (
Figure 10b). Certain characteristics of each distribution (such as the lower normalized frequencies of mutagenic molecules, compared to non-mutagenic molecules, spanning over bins ~200 to ~500) were captured by the MLP classifier. While this may indicate sufficient ability of the simple MLP classifiers for capturing subtle differences between distributions of mutagenic and non-mutagenic molecules in chemical space, it may also be a sign of overfitting.
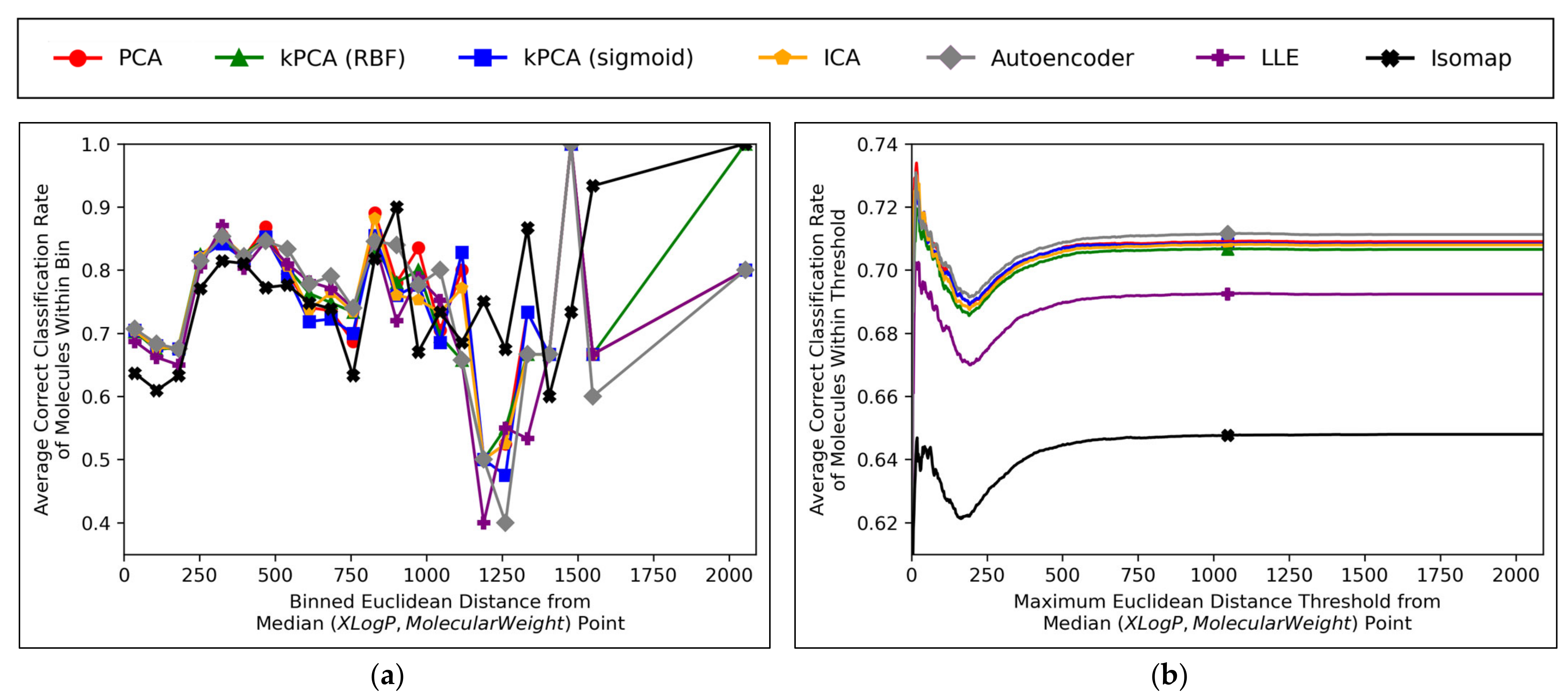
Further insight into the performances of the different dimensionality reduction techniques, over differing Euclidean distances from the median point in XLogP/MW chemical space, were obtained via the graphical plots of
Figure 11.
A universal dip in performance occurred, for molecules closest to the median point in XLogP/MW chemical space (
Figure 11a), although it is suggested that this dip occurred later (
Figure 11b) and hence the earlier positioning of the dip in
Figure 11a appears to have been due to the imprecise nature of the 30 bins used. An additional dip in performance is visible in
Figure 11a, having occurred for all techniques except for isomap, between
x-axis values of ~1100 and ~1300. While this additional dip was also apparent in the raw data used to construct
Figure 11b, the thresholded nature of
Figure 11b (where averages are drawn over the entire dataset
within a certain distance) means that higher average values were maintained from a denser pool of higher-performance molecules at closer Euclidean distances, with negligible impact from any lower performances over less densely populated farther regions. Isomap generally performed with local correct classification rates of 70–80% (
Figure 11a), but this figure was lowered by initially poorer performance over the significantly more densely populated region of chemical space surrounding the median point. This nonetheless may suggest that isomap, despite having underperformed in terms of all other measures in this study and having seemingly not undergone a successful grid search for hyperparameter optimisation, could possibly offer unique advantages in coverage of sparser regions of chemical space. This would further suggest that perhaps the hyperparameter optimisation was more successful than previously realised, but that merely the performance metrics used for comparison were inherently flawed and biased towards favouring other algorithms which naturally performed more optimally over the densest central region of the XLogP/MW chemical space.
The initial performance dip region of the chemical space is most coherently displayed by
Figure 11b, which affected all QSAR models to a closely comparable extent, regardless of dimensionality reduction technique used. This suggests that the particular region of chemical space in question was fundamentally problematic. Overall accuracy score shown in
Figure 11 was significantly hindered by this performance dip over a considerably more dense region of the chemical space (see
Figure 10); overall accuracy scores more within 80% may have otherwise been achieved, as per the more successful QSAR model performances observed over other regions in
Figure 11a. It is nonetheless unclear why such a dip occurred over a distinctly problematic region, with such extensive data availability, nor how the region is spatially distributed (it is so far only characterised by a directionless distance range from the median point, but may be concentrated in a more specific location of an XLogP/MW plot). A future expansion of this study may hence entail a deeper investigation into more problematic regions of the considered chemical space, as well as potentially exploring models that can identify and exclude (or otherwise assign lower reported confidence levels on predictions from) molecules within such regions.
Aside from the above observations,
Figure 11b demonstrates that the performances of PCA, kPCA (both types of kernel), ICA and autoencoders were closely comparable, which further affirms the hypothesis that non-linear dimensionality reduction techniques would be sufficient for navigating
N ≤ D + 1 training data (as per Cover’s theorem) [
12], but that certain non-linear techniques would perform sufficiently too (while inherently being more widely applicable to any non-linearly separable datasets used in future QSAR modelling applications). Although autoencoders mildly outperformed all other techniques in
Figure 11b, this is deemed as having been due to the use of results arising from 300-dimensional data, which (as per
Figure 6a and
Figure 8) represents a particular point where autoencoders transiently outperformed other techniques; 300 dimensions was merely chosen as an arbitrary control, where some extent of valid further analysis over the XLogP/MW chemical space, as used for defining the AD, would be possible.
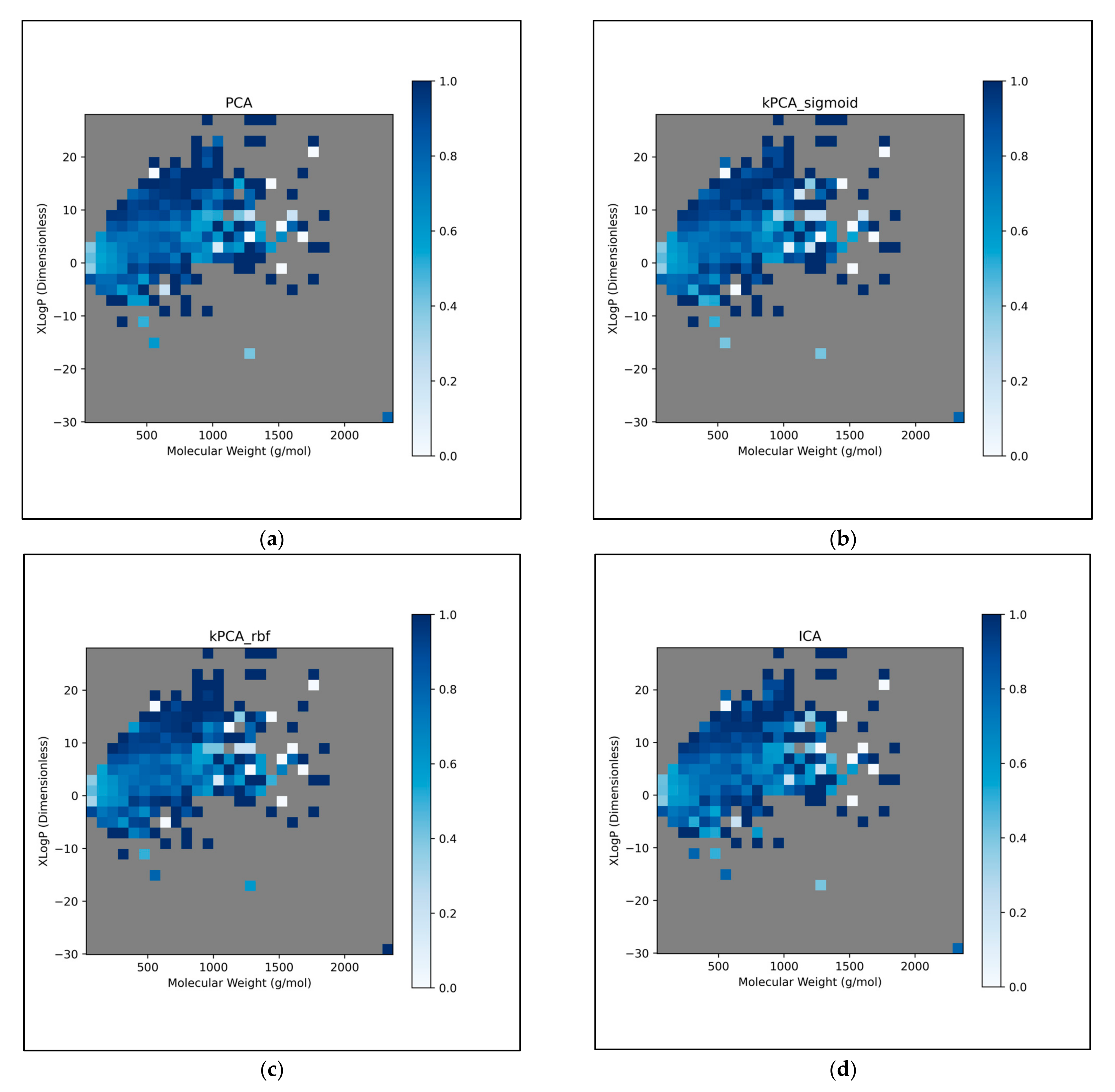
The trends of
Figure 11 were further explored, by directly analysing performances over different discretised regions of the XLogP/MW chemical space:
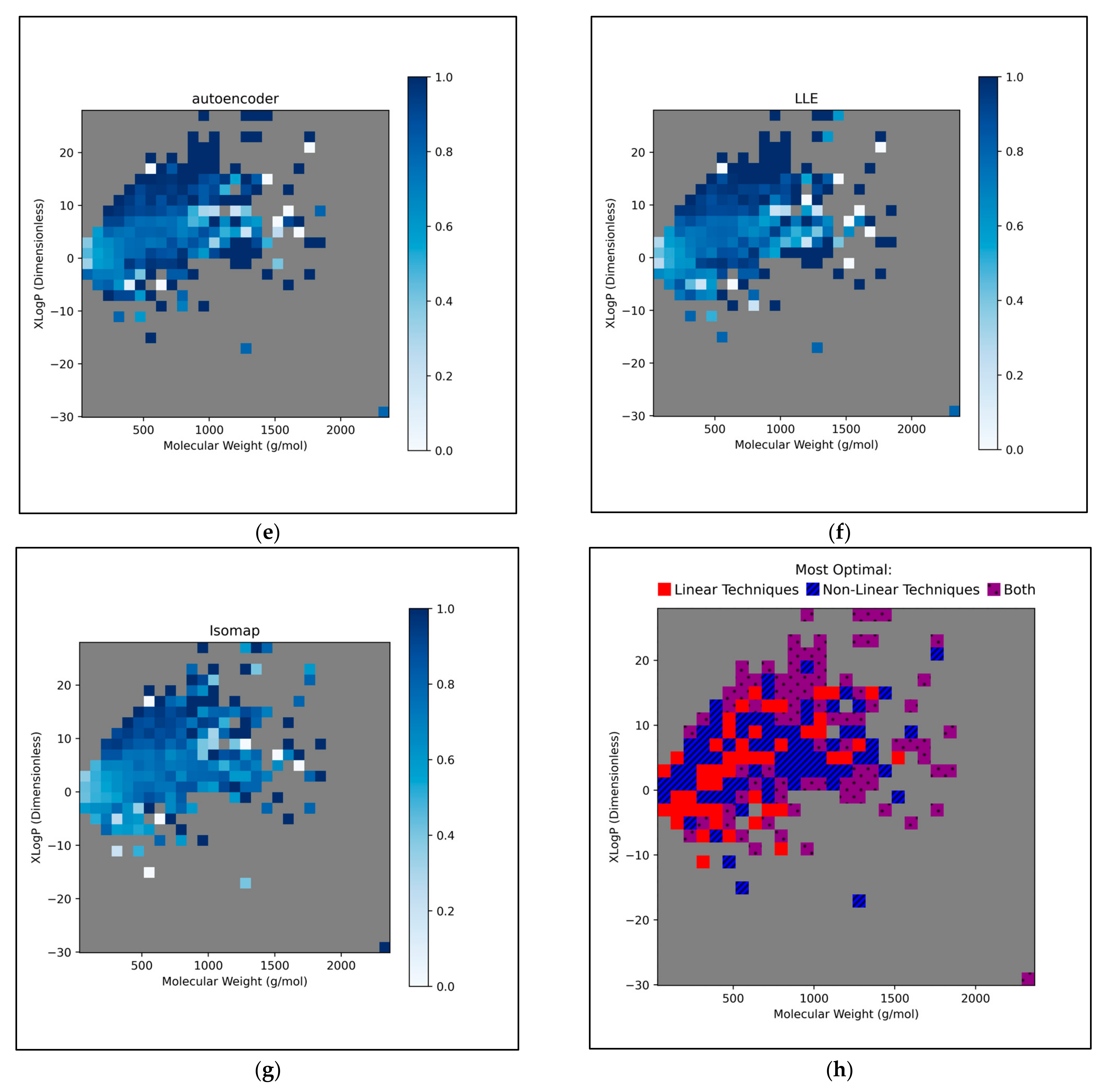
This further exploration of QSAR model performance, over all chemical space, uncovered compatible trends with those of
Figure 11 (
Figure 12). For all dimensionality reduction techniques, a certain region, within the densely populated region the median point, underperformed compared to similarly close and dense regions (
Figure 12a–g). This region occurred directly to the left of the median point and appears to be responsible for the universal dips in performance observed in
Figure 11b. In terms of differences in performances over different regions, between different dimensionality reduction techniques, distinct differences were found that were supportive of
Figure 11a. Isomap in particular was found to have performed more uniformly than other techniques, albeit in most cases at a lower overall accuracy, however this held advantages over certain regions of chemical space (mostly to the right of the median point) that were problematic or of mixed performance, for other techniques. Although the idea that each technique could offer a unique insight into toxicological space was demonstrated to a certain extent, many wider trends concerning problematic regions were found to be universal. Further investigation revealed an overall balance in superiority of linear and non-linear techniques, over the studied discretised regions of the chemical space (
Figure 12h).


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















