

Making and Breaking—Insight into the Symmetry of Salen Analogues

Faculty of Chemistry, University of Wrocław, ul. F. Joliot-Curie 14, 50-383 Wrocław, Poland
*
Author to whom correspondence should be addressed.
Symmetry 2023, 15(2), 424; https://doi.org/10.3390/sym15020424
Submission received: 28 December 2022
/
Revised: 25 January 2023
/
Accepted: 31 January 2023
/
Published: 5 February 2023
(This article belongs to the Special Issue Structural Symmetry and Asymmetry Implications in Development of Recent Pharmacy and Medicine)
Abstract
:This study focuses on selected members of the general salen-analogues family possessing two O-H⋯N hydrogen bonds, namely three isomers of N,N’-bis(salicylidene)-X-phenylenediamine, denoted as ortho, meta and para. Two of the isomers are not planar in the published crystal structures. The current study tackles the problem of symmetry and interactions within the molecules, as well as in the crystal lattice. The aromaticity of the phenyl rings is evaluated using the Harmonic Oscillator Model of Aromaticity (HOMA) index. Intra- and inter-molecular non-covalent interactions are studied via Hirshfeld surface analysis, Independent Gradient Model (IGM), Quantum Theory of Atoms in Molecules (QTAIM), Non-Covalent Interaction (NCI) index, Electron Localisation Function (ELF), Core-Valence Bifurcation (CVB) index and Symmetry-Adapted Perturbation Theory (SAPT). Density Functional Theory (DFT) simulations were carried out in vacuo and with solvent reaction field based on Polarisable Continuum Model (IEF-PCM formulation) at the B97XD/6-311+G(2d,2p) level. Crystal structure analyses were performed for the data reported previously in the literature. The obtained results demonstrate that the three isomers differ greatly in their structural properties (molecular symmetry is broken for the ortho and meta isomers in the solid state) and ability to form intermolecular interactions, while retaining overall similar physico-chemical characteristics, e.g., aromaticity of the phenyl rings. It was found that the presence of the polar solvent does not significantly affect the structure of the studied compounds. An application of the Hirshfeld surface analysis revealed the nature of the non-covalent interactions present in the investigated crystals. The SAPT results showed that the stability of the dimers extracted from the crystals of the Schiff base derivatives arises from electrostatics and dispersion.
Keywords:
Schiff base; salen analog; salophen; intramolecular hydrogen bond; DFT; Hirshfeld surface; IGM; PCM; HOMA; QTAIM; ELF; NCI; CVB; SAPT
1. Introduction
Schiff bases are a group of organic compounds for which the defining characteristic is the presence of a double bond between nitrogen and carbon atoms, RRN = CR [1]. This feature places them in the broader imine group. According to the IUPAC definition, the specific attribute of Schiff bases is that the carbon and nitrogen atoms are attached to alkyl or aryl groups, not to the hydrogen atoms (R ≠ H) [2].
The history of Schiff bases discovery began in Italy, where Hugo Schiff conducted numerous condensation reactions of aniline with aldehydes, e.g., acetaldehyde, valeraldehyde, benzaldehyde and cinnamaldehyde. The formation of new compounds, imines, was described for the first time in the paper entitled ‘Eine neue Reihe organischer Basen (A New Series of Organic Bases)’ [1,3].
Over the years, Schiff bases have gained significance in various areas of science such as organic synthesis, material science, catalysis and biochemistry [4,5,6,7,8,9,10,11]. An interesting example of applications development is provided by the macrocyclic Schiff bases, which were accidentally discovered by Curtis in 1961 while studying nickel ions and diamines [12]. Yellow crystals formed in the experiment were confirmed to be complex compounds of nickel ions coordinated by macrocycles. The reaction was possible due to the presence of acetone which undergoes the condensation with 1,2-ethanediamine [12]. To this day, the macrocyclic chemistry of Schiff bases is in constant development [13] due to their ability to coordinate a large number of different species, such as metal ions or small chemical compounds. It is also possible to design host compounds, which are able to selectively coordinate guest molecules [14], by use of chiral components (e.g., chiral diamines) or by controlling the size of the macrocycles [13].
Schiff base ligands are able to coordinate metal ions either by the imine nitrogen, another atom (which usually comes from the aldehyde fragment), or even both. Schiff bases retain their relevance to this day, especially are considered the so called ’privileged ligands’ [15]. In fact, they are able to stabilise many different metals in various oxidation states, controlling their performance in a variety of useful catalytic transformations [16,17,18]. Dendrimeric complexes of iron coordinated by Schiff bases can exhibit mesophase behaviour and variable high vs. low spin composition [19]. Azomethines, a specific subset of Schiff bases (often considered synonymous) [2], are also known for their ability to catalyse redox reactions, e.g., copper(I) complexes for the selective oxidation of primary alcohols to aldehydes under ambient conditions [20] or the electrocatalytic reduction of CO to formate by an iron complex [21]. Both macrocyclic and multidentate Schiff base ligands and their derivatives may also find applications in the field of medicine and drug development. Their complexes with metal ions (mostly Co, Ni, Zn) show promising properties as, e.g., anticancer agents [22]. In addition, the aforementioned ability of enantioselective Schiff bases synthesis is crucial in drug development. Their bioactivity and specificity can be improved through changes in their three-dimensional conformation (by, e.g., introduction of suitable substituents), which can result in increasingly effective and safer drugs. The chirality of the compounds translates into greater complexity, which in turn results in a lower probability of interaction with inappropriate molecular targets [23].
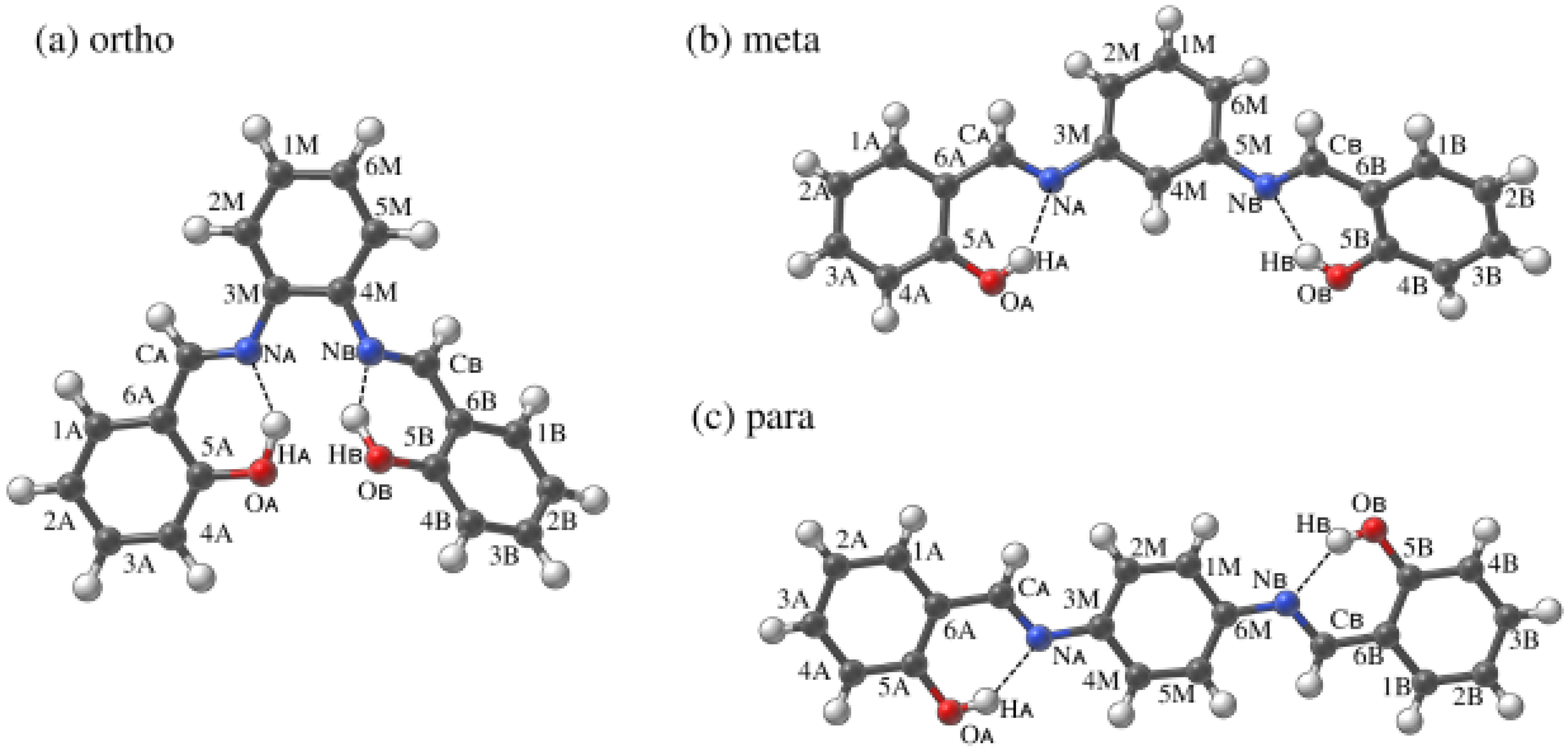
In this work three isomeric Schiff bases from salen analogues group (see Figure 1) are presented. They are the ortho, meta and para isomers of the N,N’-bis-(salicylidene)-X-phenylenediamine. These compounds contain a benzene-(1,2 or 1,3 or 1,4)-diamine bridge, which replaces more conformationally flexible ethylenediamine characteristic for salens. The middle fragment (M) is substituted by two outer phenyl groups bonded to hydroxyl groups (A and B) that come from salicylic aldehyde [24]. The hydroxyl groups are in the positions enabling the formation of intramolecular hydrogen bonds (HB) with imine nitrogen atoms (Figure 1).
Hydrogen bond is classified as a non-covalent interaction occurring between a hydrogen atom (bonded to an electronegative atom X, called ’donor atom’) and a Y atom, which has a lone electron pair (called ’acceptor atom’). This forms a three-component X-H⋯Y system, sometimes called a hydrogen bridge. Due to the type of the X atom (most commonly oxygen or nitrogen), electrons in the X-H bond are strongly shifted towards the former and the partial charges are formed: negative on X atom and positive on the hydrogen atom. This dipole interacts with a Y atom through a hydrogen atom and the HB is formed [25,26]. The influence of this interaction is strictly connected both with the character of HB (inter- or intramolecular [27]), strength [28,29], symmetry [30]) and other molecular properties (e.g., functional groups and their position [31]). In some conditions, proton transfers phenomenon between the donor and acceptor atoms can be observed [32,33], resulting in X⋯H-Y formation (called proton-transferred form). HB properties can be modulated by many factors, such as changes in the conformation of compounds or the electron density of aromatic rings, which can result from, e.g., introduction of substituents (steric and inductive effects) [34]. Analysis of these changes can be crucial for understanding the action of salen analogues as ligands [24]. It is also worth noting that the symmetry of ligands or complexes (which are susceptible to change through mechanisms such as those described above) is of great importance in modern chemistry. Symmetry influences catalytic reaction paths [35], can modulate bioavailability of drugs [36,37] or can even be of high importance in developing quantum bits [38] used in quantum computers. Schiff bases are excellent systems for basic studies and applications of externally induced symmetry changes. For example, they undergo not only cis-trans photoisomerisation, but also preferential molecular reorientation in a polarised light, which can be demonstrated, e.g., on Au nanoparticles and in protein binding pockets [39,40].
The main aim of the current study is dedicated to the intra- and inter-molecular non-covalent interactions investigations in the isomeric forms of selected Schiff bases. We analysed the crystal structures of the compounds, as well as we employed Density Functional Theory (DFT) method [41,42] for energy minimisation in vacuo and with implicit solvation model (IEF-PCM formulation) [43]. In particular, our attention was focused on: (i) changes in metric parameters associated with different states of aggregation; (ii) non-covalent interactions in molecular crystals; (iii) aromaticity changes in the studied compounds; (iv) topology and electronic structure changes and (v) energy decomposition in the crystal structure of the selected dimers. We present the transition from single molecules through dimers to molecular crystals by analysing the various factors responsible for self-organisation. Changes in the symmetry of the molecules associated with the presence of HBs in the molecules studied were also taken into account.
2. Materials and Methods
2.1. Density Functional Theory (DFT)
The initial geometry for the DFT [41,42] simulations of the monomers was taken from the crystallographic data deposited in the Cambridge Crystallographic Data Centre (CCDC) [44]. The CCDC codes are:
The quantum-chemical simulations were carried out at the B97XD/6-311+G(2d,2p) level of theory [48,49,50]. The energy minimisation was performed in vacuo and using implicit solvation model (IEF-PCM) and water as a solvent [43]. Harmonic frequencies then were computed to confirm that the obtained geometries correspond with the minima on the Potential Energy Surface (PES). In the next step, the wavefunction files were generated for further analyses using Qunatum Theory of Atoms in Molecules (QTAIM) [51], Non-Covalent Interactions (NCI) index [52], Electron Localisation Function (ELF) [53] and Core-Valence Bifurcation (CVB) [54] index. The DFT simulations were performed using the Gaussian 16 Rev. C.01. [55] suite of programs. The molecular structures of the investigated Schiff bases were visualised using the Samson R1 2022 program [56].
2.2. Harmonic Oscillator Model of Aromaticity (HOMA)
The HOMA aromaticity index [57] was computed in accordance with the generalised equation:
where is the empirical constant, chosen to give HOMA = 1 for the system with all bonds equal to optimal bond length, n is the amount of bonds, R is the bond length for C-C in benzene and R is the length of each individual bond [58]. In the HOMA index computations, the R = 1.388 Å was used [58] for the DFT data while for the molecular crystals R = 1.378 Å obtained for benzene in −3 °C [59] was applied.
2.3. Hirshfeld Surface and Independent Gradient Model (IGM)
In order to study intermolecular local contacts in the solid state, Hirshfeld surfaces [60] were obtained based on the crystal structures of the studied compounds. First, the supercells were built according to the geometry of the crystal lattice. Then, for the central molecule the surfaces were computed and the fingerprint analysis was performed [61]. To visually investigate interfragment interactions in various dimers from crystal structures, an Independent Gradient Model (IGM) based on promolecular density [62] was used. The scatter graphs as well as 3D isosurfaces were prepared and analysed. The dimers were extracted from the crystal structures of the studied Schiff bases using the Mercury 2020.1 program [63]. The data analysis was performed with assistance of the Multiwfn 3.8 [64] program and the custom scripts prepared by K.M.K. The visualisations of the obtained results were prepared with the VMD 1.9.3 [65] and Gnuplot 5.4.4 [66] programs.
2.4. Quantum Theory of Atoms in Molecules (QTAIM) and Non-Covalent Interactions (NCI) Index
The topological analysis of the structures obtained in quantum mechanical simulations (in vacuo and with application of the PCM) was performed on the basis of Quantum Theory of Atoms in Molecules (QTAIM) [51] and Non-Covalent Interactions (NCI) index [52]. The combination of these methods allowed us to fully investigate weak and strong inter- and intra-molecular interactions. On the basis of wavefunction obtained from DFT simulations, Bond and Ring Critical Points (BCPs and RCPs) and bond paths were obtained, and the Reduced Density Gradient (RDG) function was calculated. The calculations were carried out with the Multiwfn 3.8 [64] program and the custom scripts prepared by K.M.K. The collected data were analysed, combined and visualised on the scatter graphs, topological maps and 3D isosurfaces, using the VMD 1.9.3 [65] and Gnuplot 5.4.4 [66] programs.
2.5. Electron Localisation Function (ELF) and Core-Valence Bifurcation (CVB) Index
To study the strength of intramolecular hydrogen bonding in the DFT-based structures, the Core-Valence Bifurcation (CVB) index [54] obtained by topological analysis of the Electron Localisation Function (ELF) [53,67] was employed. The ELF basins were generated, and the bifurcation points between them were located and characterised. On the basis of the analysis, CVB indexes are defined as:
ELF(C-V, D) is the core-valence bifurcation point value of the donor atom. ELF(DH-A) represents the bifurcation point value between the valence basin formed by the donor atom and its bonding H atom and valence bath of the acceptor atom. The computations and visualisations were performed using the Multiwfn 3.8 program [64].
2.6. Symmetry-Adapted Perturbation Theory (SAPT)
Decomposition of the interaction energies was carried out for selected dimeric structures taken directly from the crystal structures of the investigated Schiff bases in order to preserve geometries and reproduce intermolecular forces. Two dimers were extracted for each of the studied Schiff bases using the Mercury 2020.1 program [63]. The symmetry elements used to obtain each of the dimers from the CIF files [45,46,47] are given in Table 1. The energy decomposition was carried out on the basis of Symmetry-Adapted Perturbation Theory (SAPT) [68]. The density-fitting formulation [69] and diffuse correlation-consistent jun-cc-pVDZ basis set [70,71] were employed to expand the molecular orbitals. The SAPT2 level of perturbational treatment [68,72] was selected to provide a good accuracy for the weak non-covalent interactions [73]. The basis set superposition error (BSSE) correction [74] was included in the simulations of the dimers (the homodimers were divided into “monomers” in order to fulfil the requirements of the Boys–Bernardi method). The SAPT analysis was carried out with the PSI4 1.6.1 program [75].
3. Results and Discussion
3.1. Selected Metric Parameters Analysis of the Studied Schiff Bases
The metric parameters analysis was performed for the structures obtained as a result of DFT simulations in vacuo and with the solvent reaction field (with water as a solvent) as well as for the molecular crystals (the data for crystals of the Schiff bases was taken from the literature [45,46,47]). The computed geometric parameters with respect to experimental data are presented in Table 2 and Table 3.
The quasi-ring bond lengths analysis showed that the B97XD/6-311+G(2d,2p) level of theory was able to reproduce correctly the selected metric parameters. The crystallographic data were taken as a reference. When discussing the bond lengths within the hydrogen bridge, the differences of around 5% are present across all of the studied compounds. This difference may be due to the fact that experimental data indicate an O-H bond length of less than 1 Å in two of the studied compounds. However, the largest difference between the crystal structure and the computed one is ca. 0.15 Å and it is associated with the metric parameters of the hydrogen bridge. The discrepancies between the crystal and computational data are mostly related to the positions of hydrogen atoms. This is not unexpected due to the well-known problems with location of hydrogen atoms in the electron density maps in the process of structure refinement. The deviations regarding heavy atom positions are much smaller.
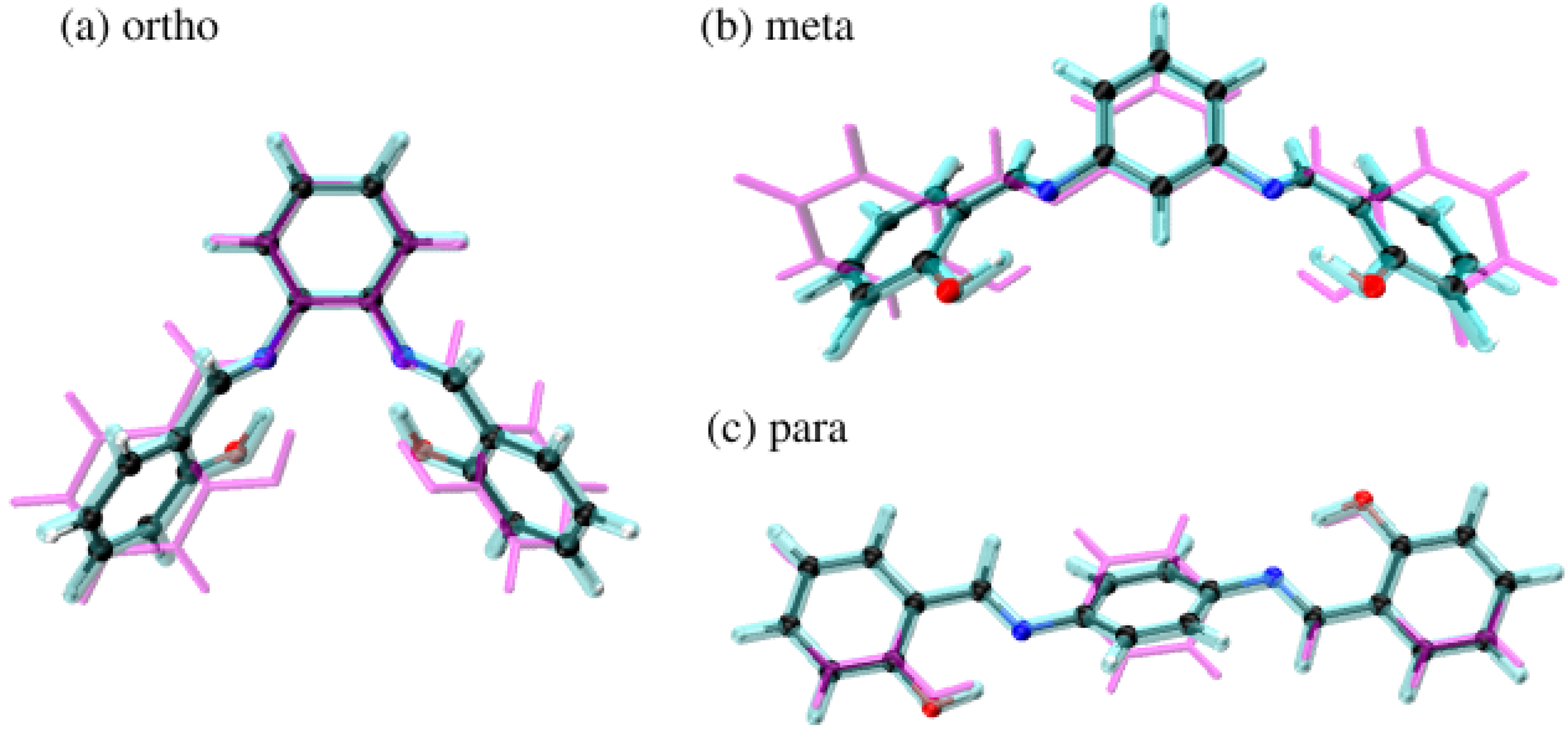
The analysis of valence and dihedral angles enabled us to discuss the symmetry changes of the studied Schiff bases when crystal data, DFT-based results in vacuo and with the application of the PCM were compared. The first significant symmetry breaking can be found in the valence angles of ortho and meta compounds, especially in the former. It is evident that the side rings of the compounds are not equal, with one experiencing more open angles (especially in OA-5A-6A to OB-5B-6B comparison to the ortho compound). This suggests that the structures are not ideally comparable, and they can be subjected to some altered interactions. Similarly, the dihedral angles within the quasi-rings are different when comparing crystal data and DFT results for ortho and meta Schiff bases derivatives. This is also, and to the largest extent, visible in the dihedral angle describing the side-group planar rotation.
All of the inequalities between side groups are not at all present in the para compound, where the molecule is almost entirely planar in the crystal. The only major difference between crystal data and the DFT results is the dihedral angle denoting the planar rotation of the side rings—in DFT they are rotated out of plane, while in crystal the molecule is almost ideally planar. The entirety of the observations are reflected in Figure 2.
To further accentuate the observations, the Root Mean Square Deviation (RMSD) analysis was performed for the investigated Schiff base derivatives. The obtained values are presented in Table 4.
Both ortho and meta compounds exhibit major symmetry breaks in the crystal, while in DFT both of these compounds are symmetrical. The situation is different for the para compound—it remains symmetrical in all studied cases, even though some of the geometrical features are different. This results in the GP-to-crystal RMSD values of 1.640, 1.405 and 0.456 for ortho, meta and para, respectively. The interactions within the molecules describing these changes, as well as intermolecular ones, is discussed in later sections.
As a final step in the geometrical analysis, the HOMA index for the side rings was calculated and is presented in Table 5. The R for the structures from DFT calculations was assumed as 1.388 Å [58], and for the solid state as 1.378 Å, consistent with the C-C bond length in benzene measured at −3 C [59].
The HOMA index values confirm that the side rings in the DFT calculations retain equal aromaticity. However, the aromaticity differences were noted in the crystal structures. The rings in the planar fragments of studied molecules showed higher aromaticity.
3.2. Intermolecular Interactions Analysis in the Crystal Structures of the Studied Schiff Base Derivatives—Hirshfeld Surface and Independent Gradient Model (IGM)
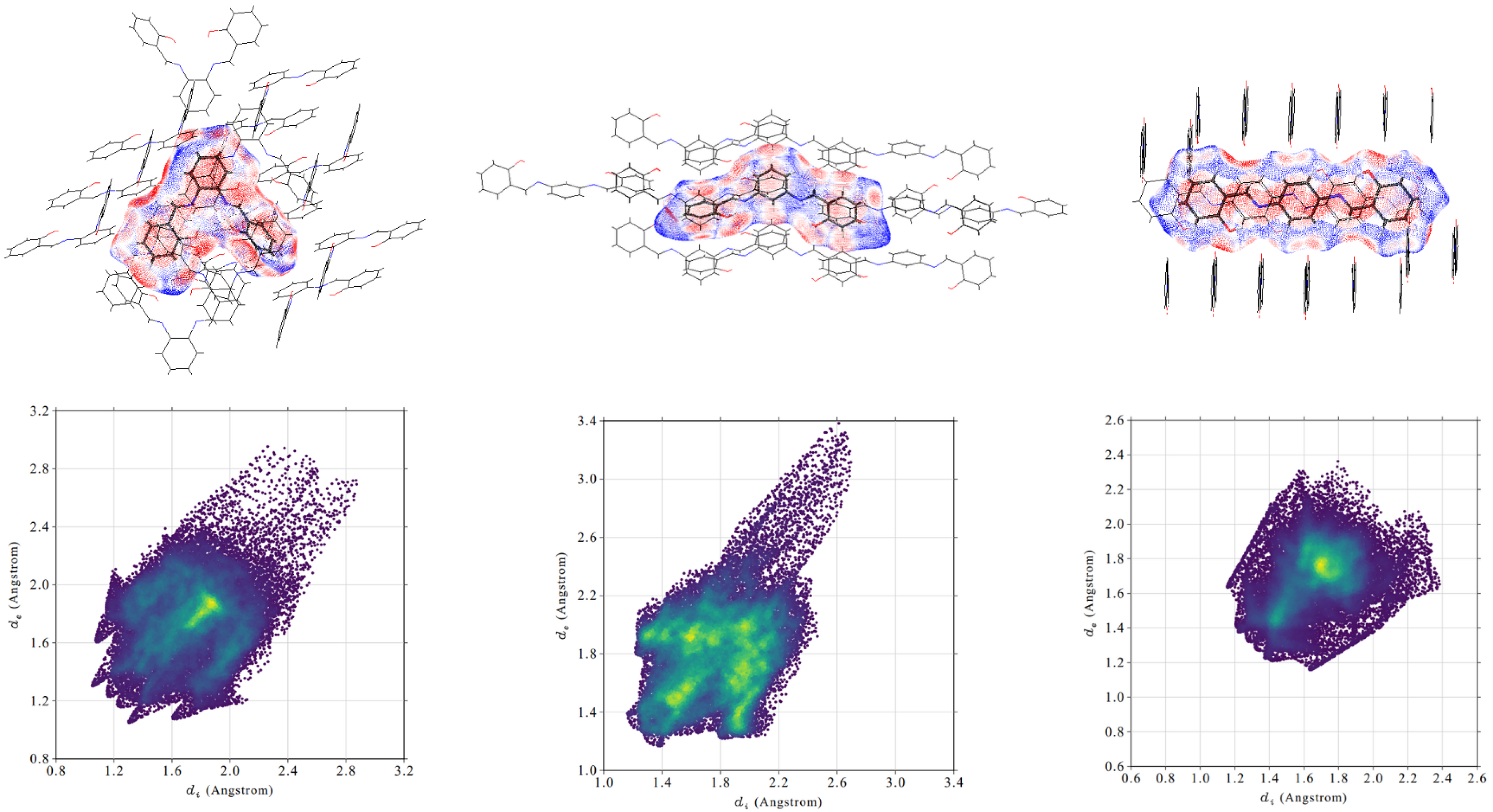
Intermolecular contacts were investigated with Hirshfeld surface analysis [60]. The surface is composed of the points where a central molecule has an equal electron density contribution to the other molecules in the supercell (based on promolecular density). The isosurface components are: d—distance from the closest external atomic nucleus to the surface and d—distance from the closest internal nucleus to the surface and van der Waals radii. They form the basis of normalised contact distance (d), which is presented using red–white–blue colour pattern on the Hirshfeld maps in Figure 3. White dots represent Van der Waals contact distances. Red dots represent d negative values, which correspond to distances shorter than vdW interactions. Blue dots mark positive values of d and contacts longer than vdW interactions. In Figure 3 it is clear that values of d are mostly negative. They indicate close intermolecular contacts and imply evident interactions. This is a result of the tight packing of molecules in the crystal unit cell. In the para compound, the values of d are the most extreme among the studied compounds. It is the effect of the highly ordered arrangement of the crystal structure. Meta and ortho compounds are not planar, and this results in less tightly packed crystal structure. However, due to the presence of the planar fragment in the ortho molecule and stacking interactions, the structure is more organised and more short-distance contacts are indicated.
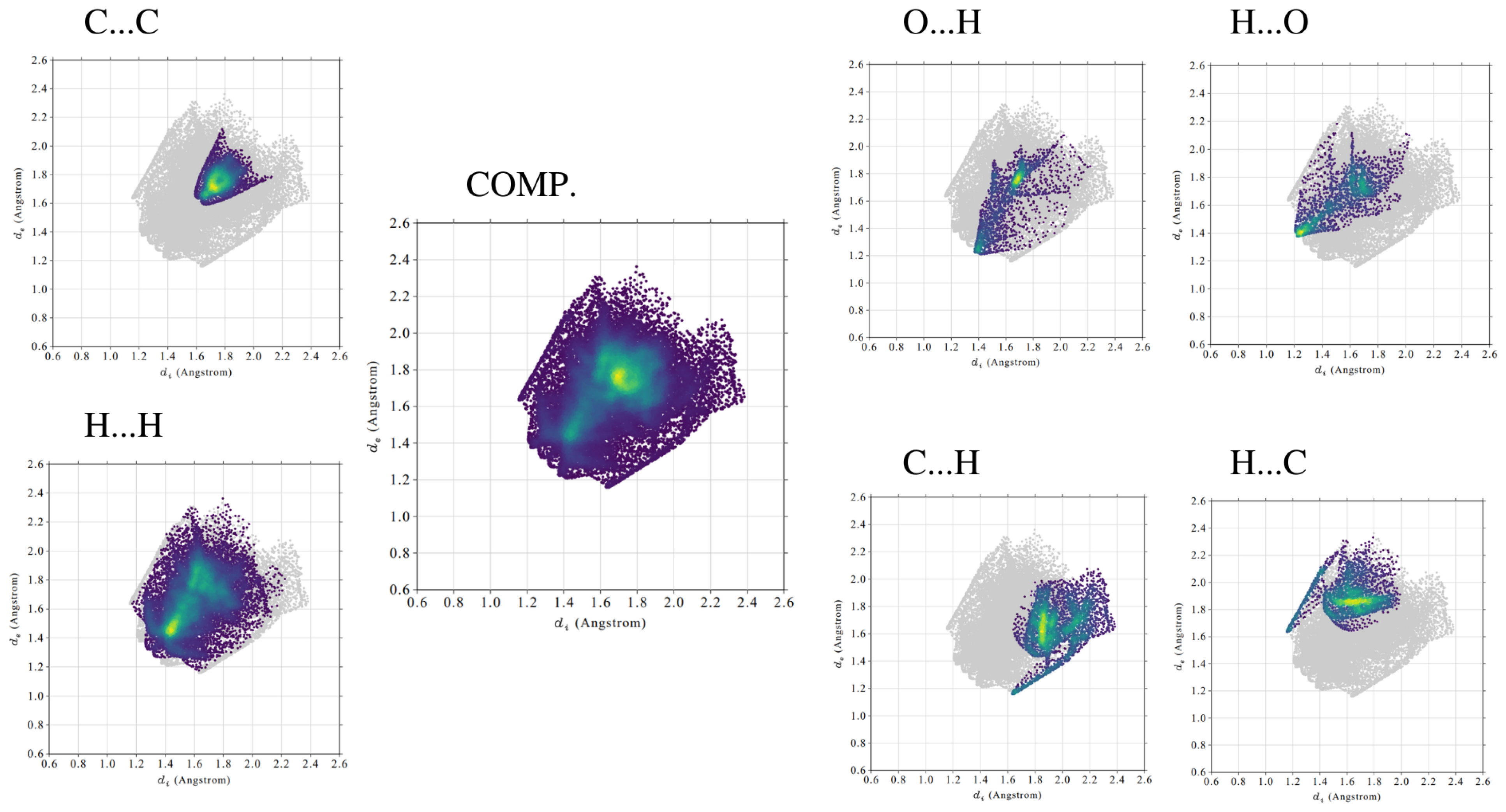
Contribution of different types of interactions (H⋯H, C⋯C, O⋯H/H⋯O, C⋯H/H⋯C) in the total contact Hirshfeld surface was studied via the fingerprint analysis [61]. The results are presented in Table 6, Figure 4 and in Supplementary Material: Figures S1–S2. Fingerprint plots are created by binning d and d pairs in the intervals of 0.01 Å and colouring each bin of the resulting 2D histogram as a function of the fraction of surface points in that bin. This ranges from purple (few points), through green, to yellow (many points). Due to their implementation, it was possible to recognise and characterise crucial intermolecular contacts in the studied compounds.
In the crystal structure of the para compound short-distance interactions originate mostly from strong stacking interactions of the aromatic rings between planar layers. This corresponds well with a significantly higher share of the C⋯C contacts in para when compared to the meta and ortho. On the fingerprint plots in Figure 4 it is also evident that a large number of C⋯C interactions are located within a relatively small space. Different effects are present in the meta isomer crystal. The molecules are not planar and the molecule organisation pattern is based on C⋯H/H⋯C contacts. The corresponding fingerprint areas are spreading and intense and their contribution to total contact surface is high. The ortho compound crystal structure is the most complex. The fingerprint analysis revealed that the C⋯C and C⋯H/H⋯C interactions have almost equal contributions to the stabilisation. It is possible due to the presence within ortho molecules both planar and rotated side rings. The former create stacking interactions, similar to the para isomer, and the latter are mostly involved in C⋯H/H⋯C contacts. It is worth noting that the O⋯H/H⋯O interactions are relevant in all of the studied crystal structures. Their involvement in stabilising the structure is comparable among different isomers and is tabulated in Table 6.
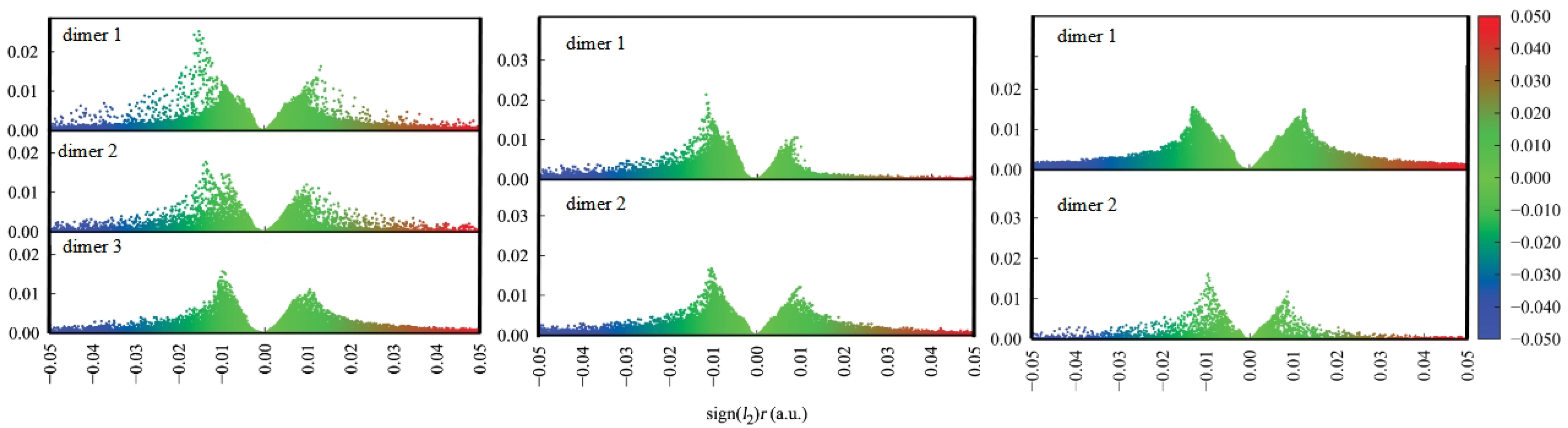
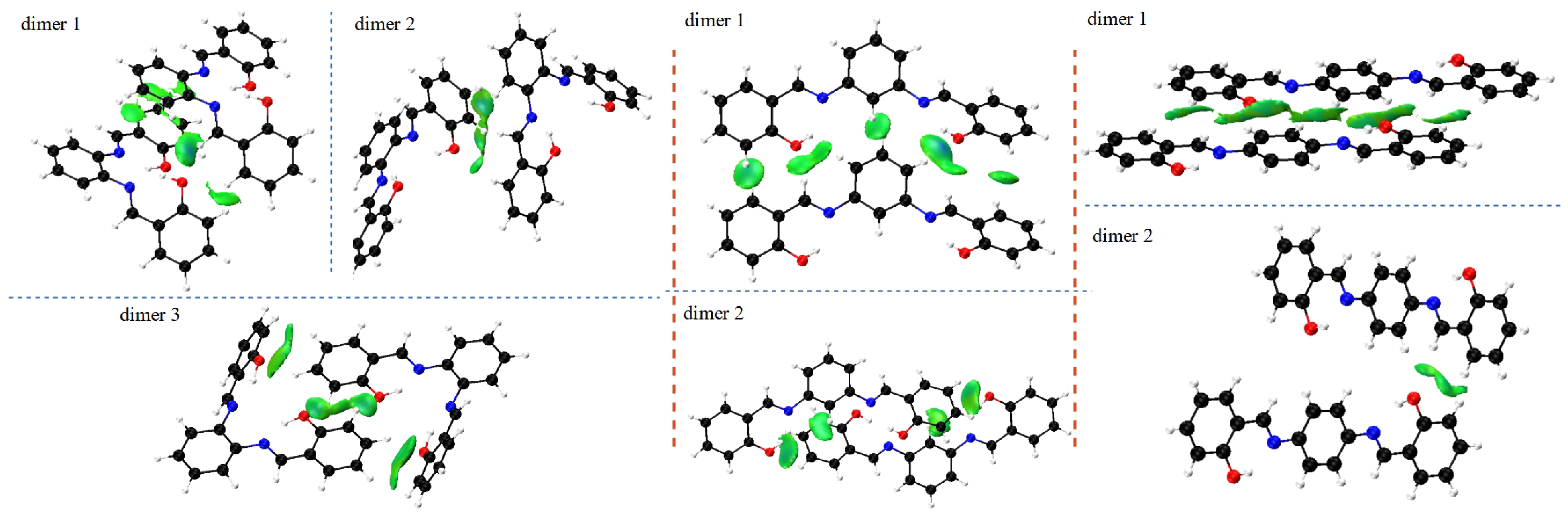
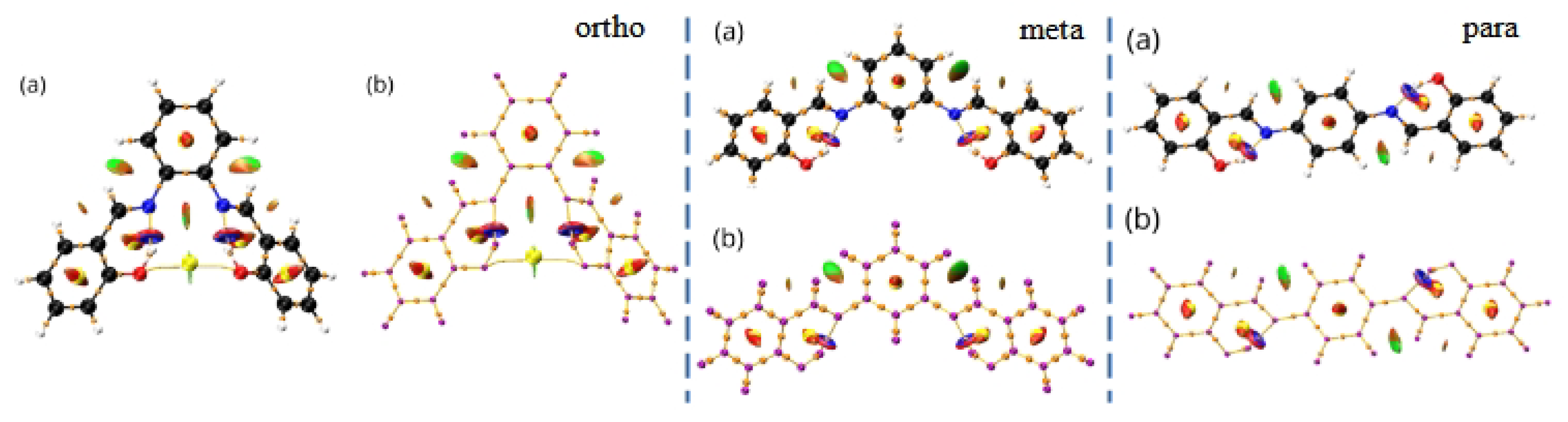
The characterisation of the contacts stabilising the crystal structure is further complemented by the use of the Independent Gradient Model (IGM) to study intermolecular interactions in all types of ortho, meta and para dimers from crystals of the Schiff base derivatives [62]. The IGM analysis allows for investigation of non-covalent interactions on the basis of the sum of spherically averaged neutral atomic densities. The core of the method is a 3D real space function, called g, which presents a unique definition of the interaction region. It can be expanded as the difference between the sum over the absolute value of the electron density gradient of each atom in the analysed fragment () and the summarised electron density gradient of each investigated atom (g). When g related to intermolecular interactions is combined with the electron density multiplied by the sign of the second Hessian eigenvalue (sign()), both: regions of the weak interaction occurrence, as well as their nature and strength, can be characterised. The interactions in the studied dimers are depicted in Figure 5 as the scatter graphs and in Figure 6 as the 3D gradient isosurfaces, where the blue, green and red regions represent attraction, Van der Waals interaction and repulsion, respectively.
Figure 6 presents the structures of the studied dimers with the IGM gradient isosurfaces represented in green.
Intermolecular stability in crystals shows significant differences when comparing the isomer to isomer. The relatively simplest is the para isomer—most of the stabilisation comes from the stacking of multiple molecular fragments between layers, with additional term brought from longer-range O⋯H and H⋯H interaction between the orthogonal planes. When analysing the scatter graphs, it is evident that the stacking interactions provide stronger stabilisation due to their accumulation and proximity of the molecules. This further confirms observations from the Hirshfeld surfaces, and is strongly related to the planar nature of the molecule. The more intricate effects can be found in the meta isomer. The meta compound does not have a typical ’layered’ dimer. Stabilisation is ensured via C(Ph)⋯H interactions and a special H(1)⋯O(2)⋯H(1) triangular system, where the number in brackets denotes the molecule within the dimer the atom is from. This ternary intermolecular entity acts as a counterpart of the stacking interactions—the meta molecules in the crystal are not planar, as in para. Examination of the scatter graphs indicates that the H⋯O⋯H interactions acquire a significantly stronger attractive character on the side of the rotated side ring in dimer 1. This is the result of cooperation of the H⋯O⋯H and parallel-displaced stacking interactions. The ortho isomer is most elaborate as it has three types of dimers. Dimer 1 is showing stacking interactions between the planar parts of the molecules, with some additional O⋯H and C-H⋯H-C contributions. Dimer 2 is stabilised by the (C)Ph⋯H interaction and the ternary H(1)⋯O(2)⋯H(1) system. Dimer 3 is the most interesting of the three—the two molecules have the planar parts in plane but with opposite turns, and the rotated side rings are out of plane, in close proximity to the planar side rings. The latter are stabilised via the H(1)⋯O(2)⋯H(1) triangles, while the former form even more elaborate, quaternary system O(1)⋯H(2)⋯H(1)⋯O(2). Similarly to meta, ortho molecules in the crystal are not planar, yet retain some planar stacking in para. Scatter graphs suggest the strongest attractive character of the H⋯O⋯H interactions appear in dimer 1. It may be concluded that it is due to the strong cooperation of interactions, as is the case of the meta isomer. This phenomenon does not occur in dimer 3 and only Van der Waals interaction are observed. Results of IGM analysis are consistent with the ones obtained from the Hirshfeld surfaces and together form a comprehensive description of intermolecular interactions between the molecules.
3.3. Intramolecular Interactions Analysis on the Basis of DFT Results—Quantum Theory of Atoms in Molecules (QTAIM) and Non-Covalent Interactions (NCI) Index
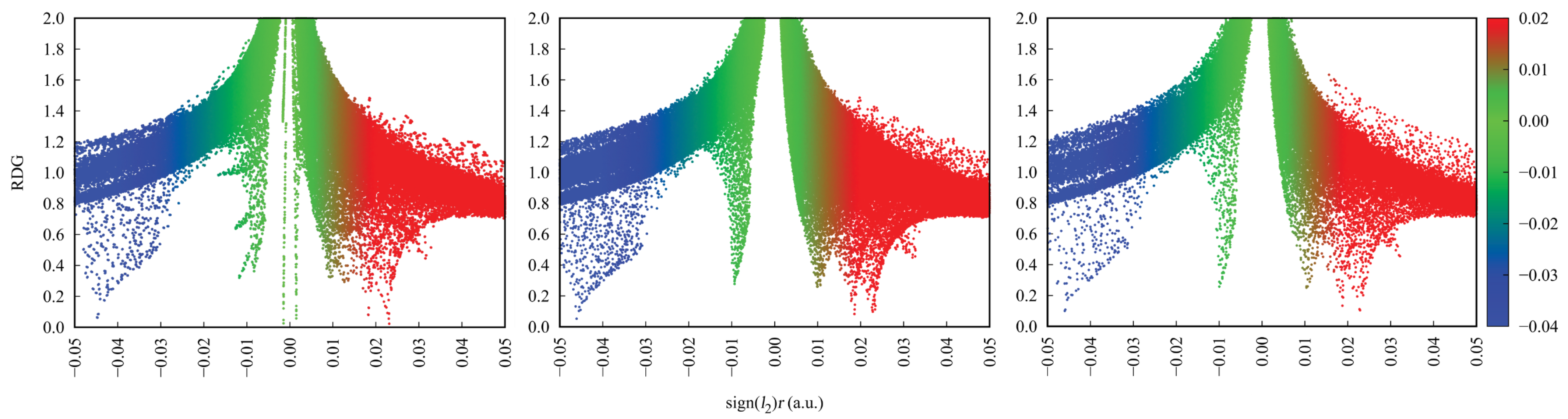
Intramolecular interactions in DFT-based structures in vacuo and in the PCM were studied using Quantum Theory of Atoms in Molecules (QTAIM) combined with Non-Covalent Interactions (NCI) index [51,52]. QTAIM theory describes bonding in a molecule by characterising stationary points of the electron density field, called Bond Critical Points (BCP), Ring Critical Points (RCPs), Cage Critical Points (CCPs) and connections between them, named bond paths. For each of them, both the exact location and electronic structure properties are determined. The combination of QTAIM and NCI analyses allow not only to identify the bonding pattern in a molecule, but also to explore additional weak interactions and describe their nature and strength. The NCI index is based on electron density and employs the dependence between Reduced Density Gradient (RDG) and the electron density multiplied by the sign of the second Hessian eigenvalue (sign()). When sign() takes the value lower than 0, it implies molecular attraction. When sign() ≈ 0, Van der Waals interactions are denoted and finally the value sign() higher than 0 stands for molecular repulsion.
Figure 7 presents the results of the NCI and AIM topological analyses for the studied compounds in vacuo. The results for the PCM can be found in Supplementary Material: Figure S3. In all of the presented structures, the yellow lines indicate the intramolecular interaction paths, while the yellow and orange dots mark the Ring Critical Points (RCPs) and Bond Critical Points (BCPs), respectively. Red regions—intramolecular repulsion, blue—attraction and green—Van der Waals interactions.
As evident from Figure 7 in all of the studied compounds, the hydrogen bridges with their neighbouring atoms constitute distinct quasi-rings. This is indicated by both the isosurfaces from the NCI analysis, as well as the RCPs from the QTAIM analysis. Such insights are well established in the literature [76]. Additionally, the ortho compound has an additional, macro-ring located in between the side wings. This is indicated by the distinctive NCI pattern of repulsion and vdW interactions, as well as the presence of RCP located on the O–O axis. These specific interactions in the ortho isomer significantly affect the NCI scatter plots, presented in Figure 8 and in Supplementary Material: Figure S4. Additional green and red regions correspond to vdW and steric interactions are observed. It is also worth noting that the inclusion of PCM does not change the results significantly. The O⋯O distance is 3.523 Å in the experimental structure, 3.721 Å in the GP and 3.520 Å in the PCM model. Thus, the presence of the macrocyclic ring involving the O–O axis is expected in the crystal structure as well.
QTAIM theory provides meaningful information of intramolecular hydrogen bonding present in the studied compounds. HBs constitute the essential part of the molecular structure and geometry. Investigating electronic properties at BCPs between atoms involved in the hydrogen bridge demonstrated differences among studied isomers. The results for all of the compounds obtained in vacuo are presented in Table 7 and in Supplementary Material: Table S1 for the structures computed in PCM. For the covalent O—H bonds absolute values of the electron density, its Laplacians and the density of potential energy decrease in the following order: ortho > meta > para. The opposite effect is observed for the H⋯N in the hydrogen bond. Each of the above-mentioned absolute values at BCPs increase from ortho to para isomer. It is therefore clear that HBs in the investigated compounds have the following relations regarding HB strength:
- (i)
- HB in the ortho isomer is significantly weaker than the corresponding bonds in meta and para,
- (ii)
- in the para isomer HBs are the strongest.
This effect is related to differences in the proximity of the hydrogen bridges in all of the studied compounds. In the ortho, bridges are located in close vicinity to each other and show mutual competition, which results in lowering their strength. In the meta, hydrogen bridges compete with each other; however, they have more spatial freedom and the binding energy lowering effect is weaker. In the para, HBs are the strongest due to their specific, alternate arrangement and planarity of the molecule. As such, HBs do not exhibit as strong of a competition.
3.4. Intramolecular Hydrogen Bonding Examination—Electron Localisation Function and Core-Valence Bifurcation Index (CVB)
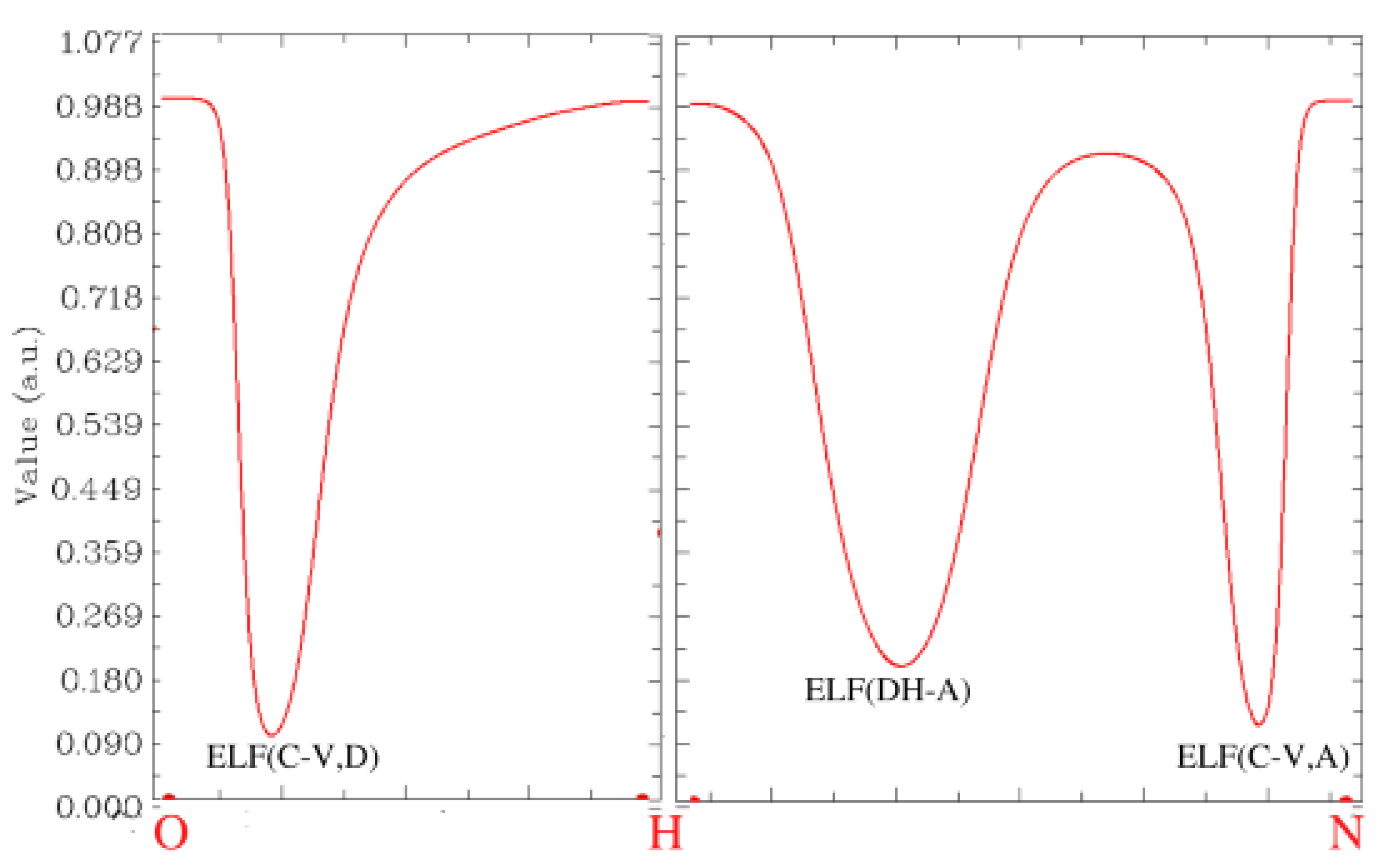
Conclusions from the QTAIM analysis regarding intramolecular HBs strength were confirmed by the Core-Valence Bifurcation index (CVB) based on topological analysis of the Electron Localisation Function (ELF) [46,54,77]. Note that the stronger the HB, the more negative the CVB index. This is happening due to a shortening of the distance between the acceptor and hydrogen atom and the enhancing covalent character of the HB. Therefore, the bifurcation point value between valence basin formed by the donor atom and its bonding hydrogen atom and valence bath of the acceptor atom increases, resulting in decreasing the value of CVB index. The ELF plots can be found in Figure 9 and in Supplementary Material: Figures S5–S7.
The CVB indexes for all of the investigated compounds, presented in Table 8 and in Supplementary Material: Table S2, are negative. This indicates the presence of hydrogen bonding between analysed atoms. As shown in the QTAIM BCPs analysis, the intramolecular HB in the para isomer is the strongest (CVB is the most negative) and in the ortho it is the weakest. Applying the PCM in DFT calculations does not affect molecular topology significantly; consequently, the results are almost analogous to those obtained in vacuo.
Considering the competition between intramolecular HBs in the ortho and meta isomers and geometrical analysis, it can be concluded that the symmetry breaking phenomenon in the crystal structure appears due to the attempt to arrange the system and maintain the strongest HBs, which stabilise single molecules and also have their share in stabilising the whole crystal structure by cooperation with H⋯O⋯H intermolecular interactions. Through rotating one of the side rings, the distance between two hydrogen bridges increases and the competition affects HBs less significantly. The rotation is strongest in the ortho isomer, where strongest competitive contacts between the bridges occur. Moreover, due to the appearance of the rotation phenomenon, it is possible to achieve stronger stabilisation of the crystal structure by creating stacking or (C)Ph⋯H interactions with some fragments of the molecules. The gas phase structures are not affected by additional intermolecular interactions; therefore, they exhibit the tendency of aromatic systems to maintain planarity as much as allowed by repulsion between the substituents. Due to this, the GP-optimised structures are less prone to symmetry breaking. The issue of symmetry of molecular adducts in the gas phase is currently considered a challenging topic because of diverse time scales involved from the experimental side, as well as intricate competition between non-covalent interactions responsible for shaping and stabilising the adducts [78].
3.5. Interaction Energy Decomposition—Symmetry-Adapted Perturbation Theory (SAPT)
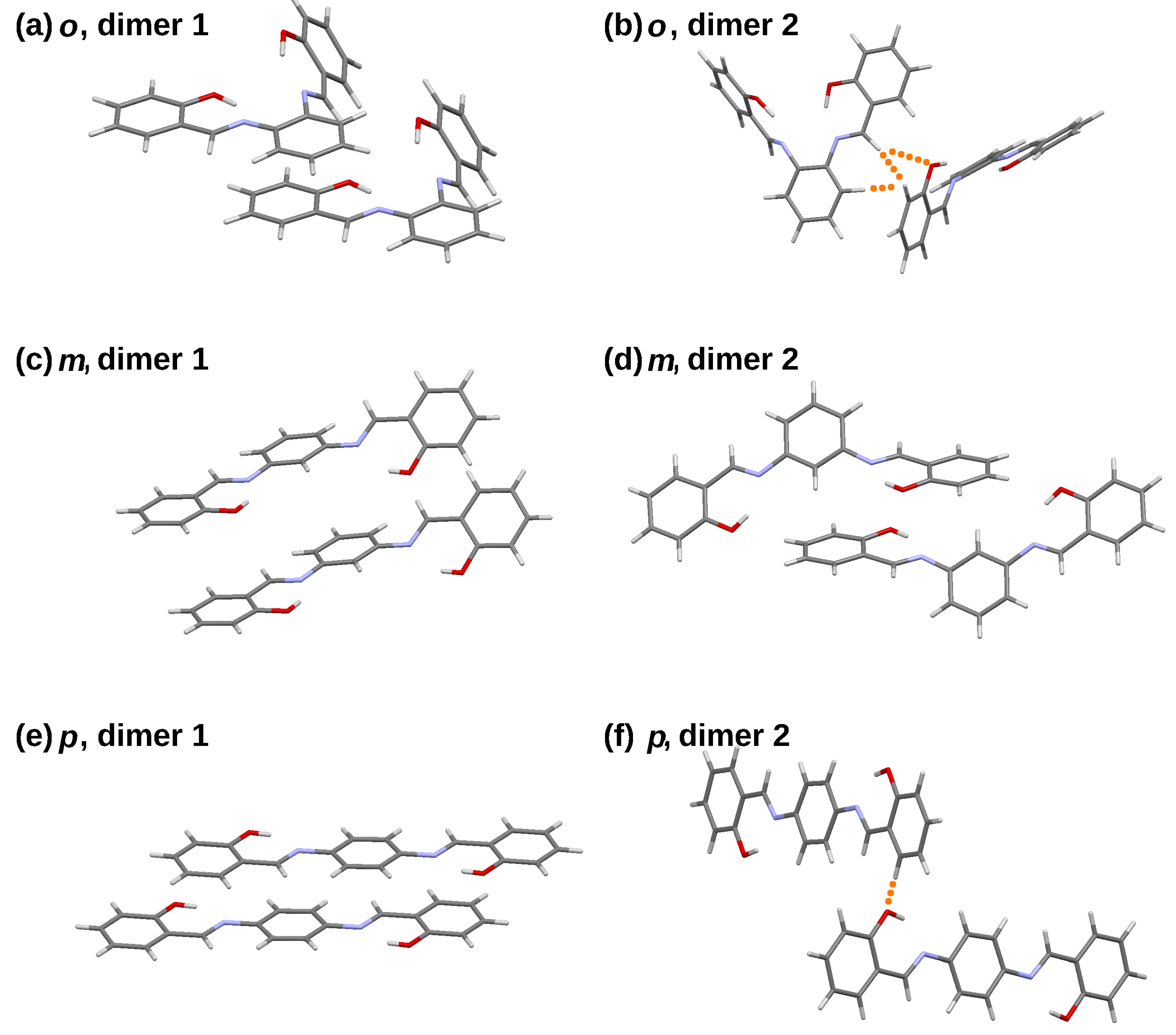
The QTAIM, NCI or ELF approaches allow insight into the details of specific interactions. On the other hand, the Symmetry-Adapted Perturbation Theory (SAPT) decomposition scheme provides a more general picture, at the price of losing the ability to pinpoint specific interaction. SAPT divides the interaction energy into several groups of terms: electrostatic, exchange, induction and dispersion, which is possible by the use of proper imposition of the wavefunction permutational symmetry [68]. The electrostatic term describes Coulombic interaction between two parts constituting the dimer, but with the electron densities taken from “isolated molecule” scenario: the two parts interact, but their densities are kept frozen. The same holds for the exchange term, describing the Pauli repulsion. The induction term introduces relaxation of the electron densities of the monomers—polarisation by the presence of the other partner, and associated changes in the Coulombic and Pauli energies. Finally, the correlation between fluctuations of electron densities between the monomers gives rise to dispersion forces. All these terms can be estimated to a given perturbational order, yielding particular levels of SAPT theory [68,72]. The most popular are SAPT0 (without inclusion of intra-monomer electron correlation effects) and SAPT2, while recent proposals include sSAPT0 (scaled SAPT0) deemed to be potentially very accurate at the fraction of the computational cost of SAPT2 [73]. These three levels, SAPT0, sSAPT0 and SAPT2, were evaluated for the dimers of the studied compounds taken from the crystal structures (see Table 1 and Figure 10), and the results are presented in Table 9.
The detailed division of interaction energy terms in Table 9 is presented for the highest SAPT2 level, so that the electrostatics, exchange, induction and dispersion values sum up to the SAPT2 energy. The interaction is rendered ca. 1–2 kcal mol stronger at the SAPT0 level; we have already observed such overbinding for similar systems [33]. The scaled SAPT0 level performs in a very similar way to the SAPT0.
The dimers extracted from the ortho and para crystal structures share a common motif: they can be described as “stacked” (dimer 1) and “held by C-H⋯O bonds” (dimer 2). Moreover, the planes formed by the corresponding rings of the constituent monomers are parallel in dimer 1 and almost perpendicular in dimer 2 (see Figure 10). This leads to large differences in the respective terms of the SAPT decomposition. In particular, the exchange contributions are very much different between the dimer 1 and dimer 2 series, which is the consequence of the mutual orientation of the monomers. The stacking arrangement relies on an overlap of large, relatively flat areas, which results in increased total Pauli repulsion. On the other hand, in dimer 2, the intermolecular contacts are scarce (see Figure 10 where they are denoted with orange dots) and the exchange repulsion is small in absolute value. Electrostatics, resulting from permanent charge distribution of the original monomers, is also larger for the dimer 1 series. This is easier to understand for the para- isomers, where dimer 2 is composed of two almost perpendicular molecules, yielding most unfavourable arrangement for dipoles, although good for quadrupole moments. However, the difference in the contact area between the dimer 1 and 2 series is best visible in the dispersion term. For the ortho compound, the dispersion term is three times smaller for dimer 2, whereas in the para series, the difference is ten-fold. Seemingly, dispersion is the most important binding component for the stacked dimers (dimer 1 series).
The meta isomer behaves differently. Both dimers extracted from the crystal structure can be considered “stacked”, and dispersion is crucial for their stability; the difference between its magnitude for the meta dimers 1 and 2 is less than twofold. The same is true for the exchange term. Speculatively, the difference between the ortho, meta and para dimers might be attributed to the combination of their (non-)planarity and spatial crowding. Although the planar para compound with the highest symmetry among the studied molecules is able to form strong stacking interactions, this is less rewarding for the ortho and meta isomers. Further, the meta isomer molecule is less crowded sterically, which is clearly visible upon comparison of the NCI gradient isosurfaces of the ortho and meta dimers—see earlier sections and Figure 7. This enables the formation of two different stacked dimer types in the crystal structure of the ortho compound.
The final remark of the SAPT study is curiously small role of the induction term—its values, ranging from −0.80 to −4.45 kcal mol, cannot be considered as decisive factors for the stability of the dimers. This stability arises from electrostatics and dispersion, while the exchange component determines the intermolecular separation.
4. Conclusions
The three studied N,N’-bis(salicylidene)-phenylenediamines are conformationally diversified. Only the para isomer is found to be very close to planarity in the crystal structure. Nevertheless, our study shows that the arrangement of the molecules in the respective crystals gives rise to numerous stacking, C-H⋯O and C-H⋯C contacts. Spatially resolved non-covalent interaction analysis schemes (such as Hirshfeld surface, QTAIM, NCI, ELF) show that apparent steric crowding in the ortho isomer makes it possible to form a large quasi-macrocyclic ring in addition to the quasi-aromatic rings formed by the intramolecular hydrogen bonds. Interestingly, the more sterically crowded ortho and meta isomers exhibit major symmetry breaks in the solid state, while DFT calculations render both of these compounds as symmetrical. The para compound remains essentially symmetrical in all the studied cases. The aromatic rings retain however their aromaticity in the molecular structures. Despite having potentially easily modulated intramolecular hydrogen bonds, the studied molecules were not affected strongly by the implicit PCM solvation model, and also the polarisation term of the SAPT decomposition does not play any significant role in the formation of dimeric structures. Thus, the studied salen analogues are excellent examples of structural diversity in a seemingly closely related set of compounds. Summarising, our study has revealed:
- (i)
- The computational approaches have indicated the presence of diverse types of non-covalent interactions shaping the conformational landscape of the crystals;
- (ii)
- The employed QTAIM and NCI analyses have illustrated distribution of non-covalent interactions in the optimised structures;
- (iii)
- Application of QTAIM enabled the quantitative description of the electron density distribution in the hydrogen bonds;
- (iv)
- Application of the symmetry-adapted perturbation theory for the dimers derived from crystallographic data provided new information on the energy components in the studied dimers; the important role of dispersion was noted, especially in comparison with rather small induction terms.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/sym15020424/s1, Table of contents: I. Figure S1. Examples of two-dimensional (2D) Hirshfeld (fingerprint) diagrams of various intermolecular interactions in the crystal structure for the meta compound. II. Figure S2. Examples of two-dimensional (2D) Hirshfeld (fingerprint) diagrams of various intermolecular interactions in the crystal structure for the para compound. III. Figure S3. Combined QTAIM and NCI results for all of the studied Schiff bases in PCM. IV. Figure S4. NCI scatter graphs for all of the compounds in PCM. V. Table S1. QTAIM Bond Critical Points (BCPs) properties for hydrogen bond in all of the compounds in PCM. VI. Figure S5. ELF for the intramolecular hydrogen bond in the ortho compound in vacuo and in PCM. VII. Figure S6. ELF for the intramolecular hydrogen bond in the meta compound in vacuo and in PCM. VIII. Figure S7. ELF for the intramolecular hydrogen bond in the para compound in vacuo and in PCM. IX. Table S2. Core-valence bifurcation point values and CVB indexes for all of the compounds in PCM.
Author Contributions
Conceptualisation, K.M.K. and A.J.; methodology, K.M.K., J.J.P. and A.J.; validation, K.M.K.; formal analysis, K.M.K., S.B., J.J.P. and A.J.; investigation, K.M.K., S.B., M.P. and J.J.P.; data curation, K.M.K. and J.J.P.; writing—original draft preparation, K.M.K., S.B., M.P., J.J.P. and A.J.; writing—review and editing, K.M.K., M.P., J.J.P. and A.J.; visualisation, K.M.K. and M.P.; supervision, A.J.; project administration, K.M.K. and A.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank Aleksander Filarowski for a fruitful discussion. Additionally, the authors gratefully acknowledge the Wrocław Centre for Networking and Supercomputing (WCSS), Academic Computer Centre ACK Cyfronet in Kraków (ARES supercomputer) and the Poznań Supercomputing and Networking Center (PSNC) for generous grants of computer time and facilities.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| BCP | Bond Critical Point |
| BSSE | Basis Set Superposition Error |
| CCP | Cage Critical Point |
| CVB | Core-Valence Bifurcation index |
| DFT | Density Functional Theory |
| ELF | Electron Localisation Function |
| GP | Gas Phase |
| HB | Hydrogen Bond |
| HOMA | Harmonic Oscillator Model of Aromaticity |
| IEF | Integral Equation Formalism |
| IGM | Independent Gradient Model |
| NCI | Non-Covalent Interactions index |
| PES | Potential Energy Surface |
| PCM | Polarisable Continuum Model |
| RCP | Ring Critical Point |
| RDG | Reduced Density Gradient |
| RMSD | Root Mean Square Deviation |
| QTAIM | Quantum Theory of Atoms in Molecules |
| SAPT | Symmetry-Adapted Perturbation Theory |
| vdW | Van der Waals interactions |
References
- Schiff, H. Mittheilungen aus dem Universitätslaboratorium in Pisa: Eine neue Reihe organischer Basen. Liebigs Ann. 1864, 131, 118–119. [Google Scholar] [CrossRef]
- Moss, G.P.; Smith, P.A.S.; Tavernier, D. Glossary of class names of organic compounds and reactivity intermediates based on structure (IUPAC Recommendations 1995). Pure Appl. Chem. 1995, 67, 1307–1375. [Google Scholar] [CrossRef]
- Tidwell, T. Hugo (Ugo) Schiff, Schiff Bases, and a Century of β-Lactam Synthesis. Angew. Chem. Int. Ed. Engl. 2008, 47, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.J. The Enantioselective Synthesis of α-Amino Acids by Phase-Transfer Catalysis with Achiral Schiff Base Esters. Acc. Chem. Res. 2004, 37, 506–517. [Google Scholar] [CrossRef]
- Filarowski, A.; Lopatkova, M.; Lipkowski, P.; der Auweraer, M.V.; Leen, V.; Dehaen, W. Solvatochromism of BODIPY-Schiff Dye. J. Phys. Chem. B 2014, 119, 2576–2584. [Google Scholar] [CrossRef]
- Jia, Y.; Li, J. Molecular Assembly of Schiff Base Interactions: Construction and Application. Chem. Rev. 2014, 115, 1597–1621. [Google Scholar] [CrossRef]
- Wang, J.; Meng, Q.; Yang, Y.; Zhong, S.; Zhang, R.; Fang, Y.; Gao, Y.; Cui, X. Schiff Base Aggregation-Induced Emission Luminogens for Sensing Applications: A Review. ACS Sens. 2022, 7, 2521–2536. [Google Scholar] [CrossRef]
- Chen, J.; Peng, Q.; Peng, X.; Zhang, H.; Zeng, H. Probing and Manipulating Noncovalent Interactions in Functional Polymeric Systems. Chem. Rev. 2022, 122, 14594–14678. [Google Scholar] [CrossRef]
- Kumari, S.; Chauhan, G.S. New Cellulose–Lysine Schiff-Base-Based Sensor–Adsorbent for Mercury Ions. ACS Appl. Mater. Interf. 2014, 6, 5908–5917. [Google Scholar] [CrossRef]
- Raczuk, E.; Dmochowska, B.; Samaszko-Fiertek, J.; Madaj, J. Different Schiff Bases—Structure, Importance and Classification. Molecules 2022, 27, 787. [Google Scholar] [CrossRef]
- Catalano, A.; Sinicropi, M.S.; Iacopetta, D.; Ceramella, J.; Mariconda, A.; Rosano, C.; Scali, E.; Saturnino, C.; Longo, P. A Review on the Advancements in the Field of Metal Complexes with Schiff Bases as Antiproliferative Agents. Appl. Sci. 2021, 11, 6027. [Google Scholar] [CrossRef]
- Curtis, N.F. The advent of macrocyclic chemistry. Supramol. Chem. 2012, 24, 439–447. [Google Scholar] [CrossRef]
- Gregoliński, J.; Ślepokura, K. Lanthanide(III) and yttrium(III) coordination compounds of diastereomeric (2+2) macrocyclic imines derived from 2, 6-diformylpyridine and trans-1, 2-diaminocyclopentane. Polyhedron 2018, 147, 15–25. [Google Scholar] [CrossRef]
- Guerriero, P.; Tarnburini, S.; Vigato, P. From mononuclear to polynuclear macrocyclic or macroacyclic complexes. Coord. Chem. Rev. 1995, 139, 17–243. [Google Scholar] [CrossRef]
- Cozzi, P.G. Metal–Salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Irie, R.; Noda, K.; Ito, Y.; Matsumoto, N.; Katsuki, T. Catalytic asymmetric epoxidation of unfunctionalized olefins using chiral (salen)manganese(III) complexes. Tetrahedron Asymmetry 1991, 2, 481–494. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Zhang, W.; Muci, A.R.; Ecker, J.R.; Deng, L. Highly enantioselective epoxidation catalysts derived from 1, 2-diaminocyclohexane. J. Am. Chem. Soc. 1991, 113, 7063–7064. [Google Scholar] [CrossRef]
- Gruzdev, M.; Chervonova, U.; Vorobeva, V. The Branched Schiff Base Cationic Complexes of Iron(III) with Different Counter-Ions. Symmetry 2022, 14. [Google Scholar] [CrossRef]
- Lagerspets, E.; Lagerblom, K.; Heliövaara, E.; Hiltunen, O.M.; Moslova, K.; Nieger, M.; Repo, T. Schiff base Cu(I) catalyst for aerobic oxidation of primary alcohols. Mol. Catal. 2019, 468, 75–79. [Google Scholar] [CrossRef]
- Nichols, A.W.; Chatterjee, S.; Sabat, M.; Machan, C.W. Electrocatalytic Reduction of CO2 to Formate by an Iron Schiff Base Complex. Inorg. Chem. 2018, 57, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Englinger, B.; Pirker, C.; Heffeter, P.; Terenzi, A.; Kowol, C.R.; Keppler, B.K.; Berger, W. Metal Drugs and the Anticancer Immune Response. Chem. Rev. 2018, 119, 1519–1624. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, I.P.; Colbon, P.J.J. The Growing Importance of Chirality in 3D Chemical Space Exploration and Modern Drug Discovery Approaches for Hit-ID. ACS Med. Chem. Lett. 2021, 12, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Cort, A.D.; Bernardin, P.D.; Forte, G.; Mihan, F.Y. Metal–salophen-based receptors for anions. Chem. Soc. Rev. 2010, 39, 3863. [Google Scholar] [CrossRef]
- Latimer, W.M.; Rodebush, W.H. Polarity and ionization from the standpoint of the Lewis theory of valence. J. Am. Chem. Soc. 1920, 42, 1419–1433. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1939. [Google Scholar]
- Jaffé, H.H. Inter- and Intramolecular Hydrogen Bonds1. J. Am. Chem. Soc. 1957, 79, 2373–2375. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces; Academic Press: London, UK, 1992. [Google Scholar]
- Szalewicz, K. Hydrogen Bond, Encyclopedia of Physical Science and Technology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2001; pp. 505–538. [Google Scholar]
- Clements, R.; Dean, R.L.; Singh, T.R.; Wood, J.L. Proton double minimum potentials in symmetric hydrogen bonds. J. Chem. Soc. D 1971, 18, 1125–1126. [Google Scholar] [CrossRef]
- Krupka, K.M.; Pocheć, M.; Panek, J.J.; Jezierska, A. Comprehensive Empirical Model of Substitution—Influence on Hydrogen Bonding in Aromatic Schiff Bases. Int. J. Mol. Sci. 2022, 23, 12439. [Google Scholar] [CrossRef]
- Martyniak, A.; Majerz, I.; Filarowski, A. Peculiarities of quasi-aromatic hydrogen bonding. RSC Adv. 2012, 2, 8135. [Google Scholar] [CrossRef]
- Kułacz, K.; Pocheć, M.; Jezierska, A.; Panek, J.J. Naphthazarin Derivatives in the Light of Intra- and Intermolecular Forces. Molecules 2021, 26, 5642. [Google Scholar] [CrossRef]
- Singh, A.; Barman, P.; Gogoi, H.P. Influence of Steric and Electronic Effects in Structure-Activity Relationships of Schiff Base Ligands: Green Synthesis, Characterization, DFT/TD-DFT Calculations, Molecular Docking and Biological Studies. ChemistrySelect 2022, 7. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Bredikhina, Z.A. Stereoselective Crystallization as a Basis for Single-Enantiomer Drug Production. Chem. Eng. Technol. 2017, 40, 1211–1220. [Google Scholar] [CrossRef]
- Gal, J. The Discovery of Stereoselectivity at Biological Receptors: Arnaldo Piutti and the Taste of the Asparagine Enantiomers-History and Analysis on the 125th Anniversary. Chirality 2012, 24, 959–976. [Google Scholar] [CrossRef]
- Gal, J. Molecular Chirality in Chemistry and Biology: Historical Milestones. Helv. Chim. Acta 2013, 96, 1617–1657. [Google Scholar] [CrossRef]
- Kazmierczak, N.P.; Mirzoyan, R.; Hadt, R.G. The Impact of Ligand Field Symmetry on Molecular Qubit Coherence. J. Am. Chem. Soc. 2021, 143, 17305–17315. [Google Scholar] [CrossRef]
- Sunaga, N.; Haraguchi, T.; Akitsu, T. Orientation of Chiral Schiff Base Metal Complexes Involving Azo-Groups for Induced CD on Gold Nanoparticles by Polarized UV Light Irradiation. Symmetry 2019, 11, 1094. [Google Scholar] [CrossRef]
- Kajiwara, K.; Pradhan, S.; Haraguchi, T.; Sinha, C.; Parida, R.; Giri, S.; Roymahaptra, G.; Akitsu, T. Photo-Tunable Azobenzene-Anthraquinone Schiff Base Copper Complexes as Mediators for Laccase in Biofuel Cell Cathode. Symmetry 2020, 12, 797. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- CCDC Structural Database. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 10 June 2022).
- Pahor, N.B.; Calligaris, M.; Delise, P.; Dodic, G.; Nardin, G.; Randaccio, L. Structural effects of the co-ordination of quadridentate Schiff bases to transition-metal atoms. Structure of NN′-(o-phenylene)bis(salicylideneamine) and of its cobalt(II) complex. J. Chem. Soc. Dalton Trans. 1976, 23, 2478–2483. [Google Scholar] [CrossRef]
- Safin, D.A.; Robeyns, K.; Garcia, Y. Solid-state thermo- and photochromism in N, N′-bis(5-X-salicylidene)diamines (X = H, Br). RSC Adv. 2012, 2, 11379. [Google Scholar] [CrossRef]
- Niu, C.; Zhao, L.; Fang, T.; Deng, X.; Ma, H.; Zhang, J.; Na, N.; Han, J.; Ouyang, J. Color- and Morphology-Controlled Self-Assembly of New Electron-Donor-Substituted Aggregation-Induced Emission Compounds. Langmuir 2014, 30, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Bader, R. Atoms in Molecules: A Quantum Theory; International Series of Monographs on Chemistry; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Fuster, F.; Silvi, B. Does the topological approach characterize the hydrogen bond? Theor. Chem. Acc. 2000, 104, 13–21. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- OneAngstrom. SAMSON 2022 R1 Platform for Molecular Design. Available online: https://www.samson-connect.net/ (accessed on 15 December 2022).
- Kruszewski, J.; Krygowski, T. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972, 13, 3839–3842. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Cyrański, M.K. Structural Aspects of Aromaticity. Chem. Rev. 2001, 101, 1385–1420. [Google Scholar] [CrossRef]
- COX, E.G.; SMITH, J.A.S. Crystal Structure of Benzene at −3 °C. Nature 1954, 173, 75. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Mercury—Crystal Structure Visualisation. Available online: http://www.ccdc.cam.ac.uk/Solutions/CSDSystem/Pages/Mercury.aspx (accessed on 15 December 2022).
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD – Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C. Gnuplot 5.4.4: An Interactive Plotting Program. Available online: https://sourceforge.net/projects/gnuplot/files/gnuplot/5.4.4/ (accessed on 20 December 2022).
- Savin, A.; Jepsen, O.; Flad, J.; Andersen, O.K.; Preuss, H.; von Schnering, H.G. Electron Localization in Solid-State Structures of the Elements: The Diamond Structure. Angew. Chem. Int. Ed. Engl. 1992, 31, 187–188. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, E.G.; Sherrill, C.D. Wavefunction methods for noncovalent interactions. WIREs Comput. Mol. Sci. 2012, 2, 304–326. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Remigio, R.D.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Bankiewicz, B.; Czarnocki, Z.; Palusiak, M. Quasi-aromaticity—what does it mean? Tetrahedron 2015, 71, 4895–4908. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Actual Symmetry of Symmetric Molecular Adducts in the Gas Phase, Solution and in the Solid State. Symmetry 2021, 13, 756. [Google Scholar] [CrossRef]
Figure 1.
Molecular structure of N,N’-bis-(salicylidene)-o-phenylenediamine (a) and its two isomers: meta (b) and para (c). The dotted line indicates the presence of intramolecular hydrogen bond. Atoms numbering scheme is presented as well. Atoms colour coding is as follows: black—carbon, blue—nitrogen, red—oxygen and white—hydrogen.
Figure 1.
Molecular structure of N,N’-bis-(salicylidene)-o-phenylenediamine (a) and its two isomers: meta (b) and para (c). The dotted line indicates the presence of intramolecular hydrogen bond. Atoms numbering scheme is presented as well. Atoms colour coding is as follows: black—carbon, blue—nitrogen, red—oxygen and white—hydrogen.

Figure 2.
Superpositions of the molecular structures obtained from the X-ray diffraction experiment (magenta) [45,46,47] DFT calculations with PCM (cyan) and in the gas phase (balls and sticks). Atoms colour coding: black—carbon, blue—nitrogen, red—oxygen and white—hydrogen.

Figure 3.
Hirshfeld isosurfaces (top) and two-dimensional (2D) fingerprint plots (bottom) of the intermolecular interactions in the crystal structure of the ortho (left), meta (middle) and para (right) studied Schiff bases.
Figure 3.
Hirshfeld isosurfaces (top) and two-dimensional (2D) fingerprint plots (bottom) of the intermolecular interactions in the crystal structure of the ortho (left), meta (middle) and para (right) studied Schiff bases.

Figure 4.
Examples of two-dimensional (2D) Hirshfeld (fingerprint) diagrams of various intermolecular interactions in the crystal structure for the para compound. The number of surface points in the d and d bin varies from purple (few points), through green, to yellow (many points).
Figure 4.
Examples of two-dimensional (2D) Hirshfeld (fingerprint) diagrams of various intermolecular interactions in the crystal structure for the para compound. The number of surface points in the d and d bin varies from purple (few points), through green, to yellow (many points).

Figure 5.
IGM scatter graphs for dimers of the ortho (left), meta (middle) and para (right) structures. The figure was prepared based on literature crystallographic data [45,46,47].

Figure 6.
IGM gradient isosurfaces for dimers of the ortho (left), meta (middle) and para (right) structures. The figure was prepared based on the literature crystallographic data [45,46,47].

Figure 7.
Combined QTAIM and NCI results for all of the studied Schiff bases in vacuo: molecular structure with ball and stick visualisation (a) and only stick visualisation for clarity of BCPs and RCPs as well as other intramolecular interactions identified by NCI index (b).
Figure 7.
Combined QTAIM and NCI results for all of the studied Schiff bases in vacuo: molecular structure with ball and stick visualisation (a) and only stick visualisation for clarity of BCPs and RCPs as well as other intramolecular interactions identified by NCI index (b).

Figure 8.
NCI scatter graphs for the studied Schiff base derivatives in vacuo: ortho (left), meta (middle) and para (right).
Figure 8.
NCI scatter graphs for the studied Schiff base derivatives in vacuo: ortho (left), meta (middle) and para (right).

Figure 9.
ELF plot for the intramolecular hydrogen bond in the ortho compound in vacuo.

Figure 10.
Dimers of the studied compounds taken from their crystal structures and used in the SAPT interaction energy analysis. Orange dots represent hydrogen-bonded intermolecular contacts. The figure was prepared based on the literature crystallographic data [45,46,47].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Symmetry operations used to generate dimers based on available crystal data of the Schiff bases [45,46,47].
| Structure | CCDC Deposit No. | Symmetry Code |
|---|---|---|
| o, dimer 1 | 1226150 | −1+x, y, z |
| o, dimer 2 | 1226150 | x, −½−y, −½+z |
| m, dimer 1 | 828843 | x, −1+y, z |
| m, dimer 2 | 828843 | 1−x, 2−y, −z |
| p, dimer 1 | 962961 | x, −1+y, z |
| p, dimer 2 | 962961 | x, 1.5−y, ½+z |
Table 2.
Quasi-ring bond lengths in [Å] of the studied Schiff bases. DFT simulations were performed in the gas phase and with PCM at the B97XD/6-311+G(2d,2p) level of theory.
Table 2.
Quasi-ring bond lengths in [Å] of the studied Schiff bases. DFT simulations were performed in the gas phase and with PCM at the B97XD/6-311+G(2d,2p) level of theory.
| Bonds | Crystal Data | Gas Phase (GP) | Diff (Crystal, GP) | PCM | Diff (Crystal Data, PCM) |
|---|---|---|---|---|---|
| Ortho 1 | |||||
| HA...NA | 1.709 | 1.766 | −0.057 | 1.728 | −0.019 |
| OA-HA | 1.074 | 0.933 | 0.141 | 0.988 | 0.086 |
| OA-5A | 1.346 | 1.333 | 0.013 | 1.340 | 0.006 |
| 5A-6A | 1.425 | 1.410 | 0.015 | 1.410 | 0.015 |
| 6A-CA | 1.442 | 1.450 | −0.008 | 1.453 | −0.011 |
| CA-NA | 1.292 | 1.278 | 0.014 | 1.278 | 0.014 |
| HB...NB | 1.670 | 1.767 | −0.097 | 1.727 | −0.057 |
| OB-HB | 1.023 | 0.933 | 0.090 | 0.988 | 0.035 |
| OB-5B | 1.345 | 1.333 | 0.012 | 1.340 | 0.005 |
| 5B-6B | 1.416 | 1.410 | 0.006 | 1.410 | 0.006 |
| 6B-CB | 1.456 | 1.450 | 0.006 | 1.452 | 0.004 |
| CB-NB | 1.284 | 1.278 | 0.006 | 1.279 | 0.005 |
| Meta 2 | |||||
| HA...NA | 1.897 | 1.758 | 0.139 | 1.729 | 0.168 |
| OA-HA | 0.820 | 0.984 | −0.164 | 0.988 | −0.168 |
| OA-5A | 1.357 | 1.334 | 0.023 | 1.341 | 0.016 |
| 5A-6A | 1.416 | 1.410 | 0.006 | 1.410 | 0.006 |
| 6A-CA | 1.449 | 1.450 | −0.001 | 1.452 | −0.003 |
| CA-NA | 1.228 | 1.278 | 0.050 | 1.279 | −0.051 |
| HB...NB | 1.902 | 1.758 | 0.144 | 1.729 | 0.173 |
| OB-HB | 0.821 | 0.984 | −0.163 | 0.988 | −0.167 |
| OB-5B | 1.357 | 1.334 | 0.023 | 1.341 | 0.016 |
| 5B-6B | 1.417 | 1.411 | 0.006 | 1.410 | 0.007 |
| 6B-CB | 1.450 | 1.450 | 0.000 | 1.452 | −0.002 |
| CB-NB | 1.279 | 1.278 | 0.001 | 1.279 | 0.000 |
| Para 3,4 | |||||
| H...N | 1.857 | 1.755 | 0.102 | 1.725 | 0.132 |
| O-H | 0.840 | 0.984 | −0.144 | 0.989 | −0.149 |
| O-5 | 1.350 | 1.334 | 0.016 | 1.341 | 0.009 |
| 5-6 | 1.412 | 1.411 | 0.001 | 1.410 | 0.002 |
| 6-C | 1.452 | 1.450 | 0.002 | 1.452 | 0.000 |
| C-N | 1.288 | 1.278 | 0.010 | 1.279 | 0.009 |
Table 3.
Selected valence (V) and dihedral (D) angle values in [o] of the investigated Schiff bases. DFT simulations were performed in the gas phase and with PCM at the B97XD/6-311 + G(2d, 2p) level of theory.
Table 3.
Selected valence (V) and dihedral (D) angle values in [o] of the investigated Schiff bases. DFT simulations were performed in the gas phase and with PCM at the B97XD/6-311 + G(2d, 2p) level of theory.
| Atoms | Crystal Data | Gas Phase (GP) | Diff (Crystal, GP) | PCM | Diff (Crystal, PCM) |
|---|---|---|---|---|---|
| Ortho 1 | |||||
| NA-HA-OA (V) | 140.3 | 145.2 | −4.9 | 146.3 | −6.0 |
| HA-OA-5A (V) | 109.8 | 108.3 | 1.5 | 107.9 | 1.9 |
| OA-5A-6A (V) | 121.6 | 122.5 | −0.9 | 122.0 | −0.4 |
| CA-NA-HA (V) | 103.1 | 99.0 | 4.1 | 98.9 | 4.2 |
| NA-HA-OA-5A (D) | 18.0 | 3.1 | 14.9 | 2.0 | 16.0 |
| OA-5A-6A-CA (D) | −1.9 | −0.1 | −1.8 | −0.1 | −1.8 |
| CA-NA-HA-OA (D) | −15.7 | −2.7 | −13.0 | −1.7 | −14.0 |
| CA-NA-3M-4M (D) | −132.2 | −133.9 | 1.7 | −135.5 | 3.3 |
| NB-HB-OB (V) | 144.1 | 139.3 | 4.8 | 137.1 | 7.0 |
| HB-OB-5B (V) | 109.8 | 103.9 | 5.9 | 103.8 | 6.0 |
| OB-5B-6B (V) | 120.1 | 122.1 | −2.0 | 122.1 | −2.0 |
| CB-NB-HB (V) | 101.9 | 112.0 | −10.1 | 113.1 | −11.2 |
| NB-HB-OB-5B (D) | −15.0 | −3.1 | −11.9 | −1.9 | −13.1 |
| OB-5B-6B-CB (D) | 1.6 | 0.1 | 1.5 | 0.1 | 1.5 |
| CB-NB-HB-OB (D) | 18.6 | 2.8 | 15.8 | 2.4 | 16.2 |
| CB-NB-4M-3M (D) | 178.0 | 133.9 | 44.1 | 137.3 | 40.7 |
| Meta 2 | |||||
| NA-HA-OA (V) | 147.8 | 146.7 | 1.1 | 147.7 | 0.1 |
| HA-OA-5A (V) | 109.5 | 107.8 | 1.7 | 107.3 | 2.2 |
| OA-5A-6A (V) | 121.0 | 122.1 | −1.1 | 121.7 | −0.7 |
| CA-NA-HA (V) | 96.9 | 99.3 | −2.4 | 99.8 | −2.9 |
| NA-HA-OA-5A (D) | −6.3 | 0.9 | −7.2 | 1.3 | −7.6 |
| OA-5A-6A-CA (D) | −7.7 | −0.1 | −7.6 | −0.1 | −7.6 |
| CA-NA-HA-OA (D) | 4.3 | −0.6 | 4.9 | −1.4 | 5.7 |
| CA-NA-3M-4M (D) | −146.1 | −138.7 | −7.4 | −141.8 | −4.3 |
| NB-HB-OB (V) | 147.1 | 146.7 | 0.4 | 147.7 | −0.6 |
| HB-OB-5B (V) | 109.6 | 107.8 | 1.8 | 107.3 | 2.3 |
| OB-5B-6B (V) | 121.2 | 122.1 | −0.9 | 121.7 | −0.5 |
| CB-NB-HB (V) | 97.8 | 99.3 | −1.5 | 99.8 | −2.0 |
| NB-HB-OB-5B (D) | 2.4 | −0.9 | 3.3 | −1.3 | 3.7 |
| OB-5B-6B-CB (D) | −4.2 | 0.1 | −4.3 | 0.1 | −4.3 |
| CB-NB-HB-OB (D) | −2.5 | 0.6 | −3.1 | 1.4 | −3.9 |
| CB-NB-5M-4M (D) | 172.4 | 138.8 | 33.6 | 141.8 | 30.6 |
| Para 3,4 | |||||
| N-H-O (V) | 147.4 | 147.0 | 0.4 | 147.9 | −0.5 |
| H-O-5 (V) | 109.5 | 107.7 | 1.8 | 107.2 | 2.3 |
| O-5-6 (V) | 121.5 | 122.1 | −0.6 | 121.7 | −0.2 |
| C-N-H (V) | 98.4 | 99.3 | −0.9 | 99.8 | −1.4 |
| N-H-O-5 (D) | 1.6 | 0.6 | 1.0 | 1.3 | 0.3 |
| O-5-6-C (D) | 1.8 | −0.2 | 2.0 | −0.2 | 2.0 |
| C-N-H-O (D) | 1.3 | −0.4 | 1.7 | −1.3 | 2.6 |
| C-N-3M-4M (D) | −176.2 | −140.2 | −36.0 | −142.6 | −33.6 |
Table 4.
RMSD values of the Schiff base derivatives.
| RMSD—All Atoms | RMSD—without H Atoms | |
|---|---|---|
| Ortho | ||
| GP vs. PCM | 0.087 | 0.067 |
| GP vs. crystal | 1.640 | 1.232 |
| PCM vs. crystal | 1.590 | 1.194 |
| Meta | ||
| GP vs. PCM | 0.094 | 0.074 |
| GP vs. crystal | 1.405 | 1.108 |
| PCM vs. crystal | 1.34 | 1.052 |
| Para | ||
| GP vs. PCM | 0.040 | 0.028 |
| GP vs. crystal | 0.456 | 0.306 |
| PCM vs. crystal | 0.423 | 0.282 |
Table 5.
HOMA index values for side rings of the studied Schiff base derivatives. The ring atoms labelling is consistent with that presented Figure 1.
Table 5.
HOMA index values for side rings of the studied Schiff base derivatives. The ring atoms labelling is consistent with that presented Figure 1.
| Side Aromatic Ring | HOMA Index—Ring A | HOMA Index—Ring B |
|---|---|---|
| Ortho (DFT) | 0.960 | 0.960 |
| Ortho (Crystal) | 0.811 | 0.873 |
| Para (DFT) | 0.961 | 0.961 |
| Para (Crystal) | 0.892 | 0.892 |
| Meta (DFT) | 0.961 | 0.960 |
| Meta (Crystal) | 0.882 | 0.899 |
Table 6.
Hirshfeld local contact surfaces and their percentage share in the total contact surface for crystal structures of the studied Schiff base derivatives.
Table 6.
Hirshfeld local contact surfaces and their percentage share in the total contact surface for crystal structures of the studied Schiff base derivatives.
| Ortho | Meta | Para | |
|---|---|---|---|
| Total Contact Surface [Å] | 366.773 | 371.716 | 353.213 |
| C⋯C | |||
| Local contact surface [Å] | 20.685 | 2.873 | 37.204 |
| Percentage share [%] | 5.64 | 0.77 | 10.53 |
| H⋯H | |||
| Local contact surface [Å] | 161.387 | 159.475 | 180.149 |
| Percentage share [%] | 44.00 | 42.90 | 51.00 |
| O⋯H | |||
| Local contact surface [Å] | 23.653 | 18.298 | 22.212 |
| Percentage share [%] | 6.45 | 4.92 | 6.29 |
| H⋯O | |||
| Local contact surface [Å] | 26.507 | 18.403 | 21.373 |
| Percentage share [%] | 7.23 | 4.95 | 6.05 |
| C⋯H | |||
| Local contact surface [Å] | 66.361 | 78.738 | 38.979 |
| Percentage share [%] | 18.09 | 21.18 | 11.04 |
| H⋯C | |||
| Local contact surface [Å] | 52.824 | 65.738 | 28.944 |
| Percentage share [%] | 14.40 | 17.63 | 8.19 |
Table 7.
Electron density, , is given in atomic units and its Laplacian , is in units at the BCPs. stands for BCP potential energy density. They are properties derived from QTAIM. The results were obtained using B97XD/6-311 + G(2d,2p) level of theory in vacuo.
Table 7.
Electron density, , is given in atomic units and its Laplacian , is in units at the BCPs. stands for BCP potential energy density. They are properties derived from QTAIM. The results were obtained using B97XD/6-311 + G(2d,2p) level of theory in vacuo.
| Ortho | Meta | Para | |
|---|---|---|---|
| O–H | |||
| BCP | 0.3465 | 0.3455 | 0.3451 |
| ∇BCP | −2.6382 | −2.6264 | −2.6206 |
| VBCP | −0.8018 | −0.7996 | −0.7985 |
| H...N | |||
| BCP | 0.0450 | 0.0461 | 0.0465 |
| ∇BCP | 0.1081 | 0.1082 | 0.1082 |
| VBCP | −0.0382 | −0.0393 | −0.0397 |
Table 8.
Core-valence bifurcation point values and CVB indexes for all of the compounds in the gas phase.
Table 8.
Core-valence bifurcation point values and CVB indexes for all of the compounds in the gas phase.
| Ortho | Meta | Para | |
|---|---|---|---|
| ELF(C-V,D) | 0.1021 | 0.1024 | 0.1024 |
| ELF(C-V,A) | 0.1173 | 0.1171 | 0.1172 |
| ELF(DH-A) | 0.2005 | 0.2066 | 0.2093 |
| CVB index | −0.0984 | −0.1043 | −0.1068 |
Table 9.
SAPT2 interaction energy partitioning for the dimeric structures taken from the crystal structures [45,46,47]—all terms in kcal mol−1. See the main text for the description of particular terms.
| o, Dimer 1 | o, Dimer 2 | m, Dimer 1 | m, Dimer 2 | p, Dimer 1 | p, Dimer 2 | |
|---|---|---|---|---|---|---|
| Electrostatics | −9.99 | −4.56 | −4.68 | −6.67 | −15.92 | −3.12 |
| Exchange | 18.74 | 6.71 | 6.22 | 10.96 | 37.33 | 3.50 |
| Induction | −2.95 | −1.55 | −1.05 | −1.27 | −4.45 | −0.80 |
| Dispersion | −22.16 | −6.96 | −8.33 | −15.28 | −39.17 | −3.64 |
| SAPT0 | −18.43 | −7.68 | −8.97 | −13.31 | −23.06 | −4.91 |
| sSAPT0 | −18.38 | −7.66 | −8.96 | −13.28 | −22.94 | −4.91 |
| SAPT2 | −16.36 | −6.37 | −7.85 | −12.25 | −22.21 | −4.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Krupka, K.M.; Banach, S.; Pocheć, M.; Panek, J.J.; Jezierska, A. Making and Breaking—Insight into the Symmetry of Salen Analogues. Symmetry 2023, 15, 424. https://doi.org/10.3390/sym15020424
AMA Style
Krupka KM, Banach S, Pocheć M, Panek JJ, Jezierska A. Making and Breaking—Insight into the Symmetry of Salen Analogues. Symmetry. 2023; 15(2):424. https://doi.org/10.3390/sym15020424
Chicago/Turabian StyleKrupka, Katarzyna M., Sylwia Banach, Michał Pocheć, Jarosław J. Panek, and Aneta Jezierska. 2023. "Making and Breaking—Insight into the Symmetry of Salen Analogues" Symmetry 15, no. 2: 424. https://doi.org/10.3390/sym15020424
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.