
A High-Performance Fluorescence Immunoassay Based on the Relaxation of Quenching, Exemplified by Detection of Cardiac Troponin I

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Protein Fragmentation-Based Quenching Relaxation
2.2.1. Conjugation of Antibody or SA with Red BSA
2.2.2. Protease-Mediated Red BSA Fragmentation
2.2.3. Optimization of Immunoassay Performance
2.3. DNA Dehybridization-Based Quenching Relaxation
2.3.1. Preparation of ssDNA-Grafted Gold Nanoparticles
2.3.2. Hybridization of Dye-Labeled Probe Strands
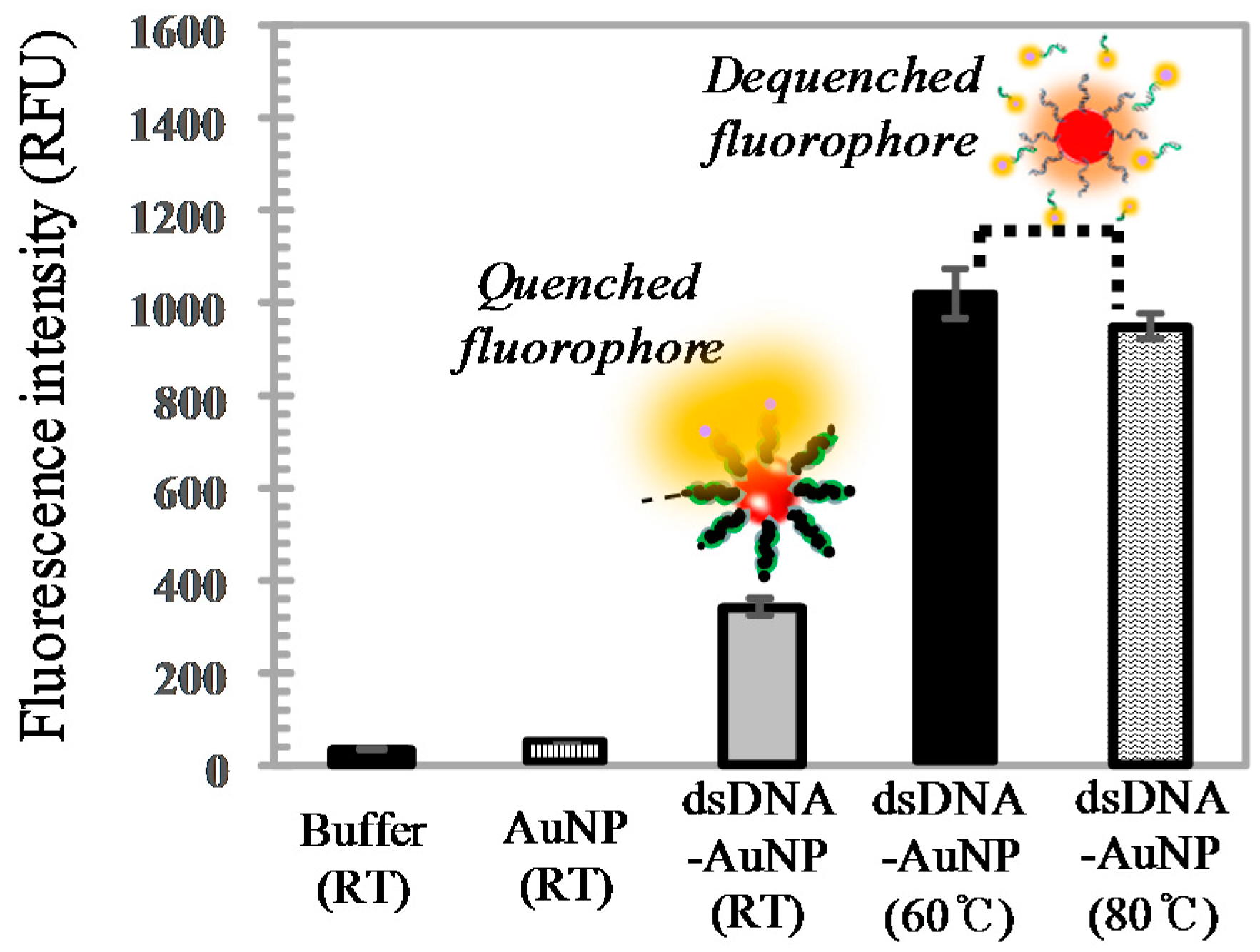
2.3.3. Temperature-Controlled Dehybridization
2.4. Conventional Fluorescent Immunoassay
2.4.1. Conjugation of Antibody to Fluorescent Dye
2.4.2. Analytical Procedure
3. Results and Discussion
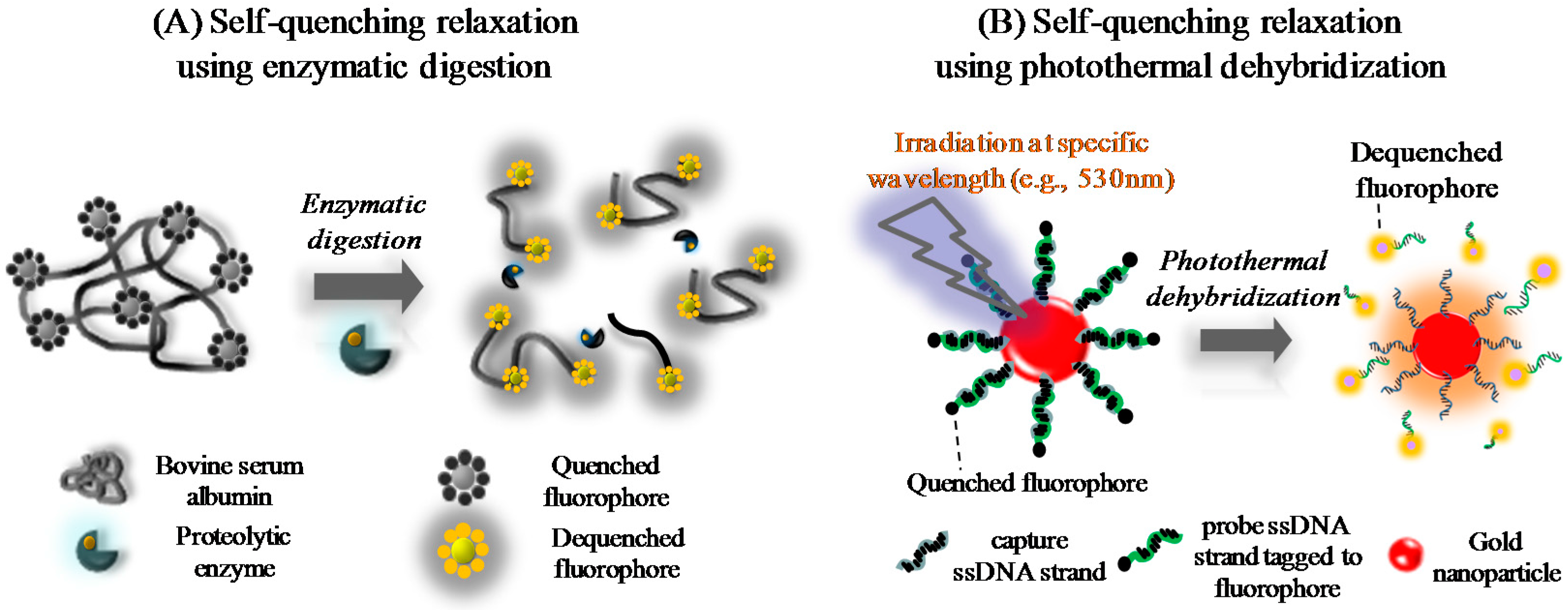
3.1. Analytical Concepts
Definitions of Terms
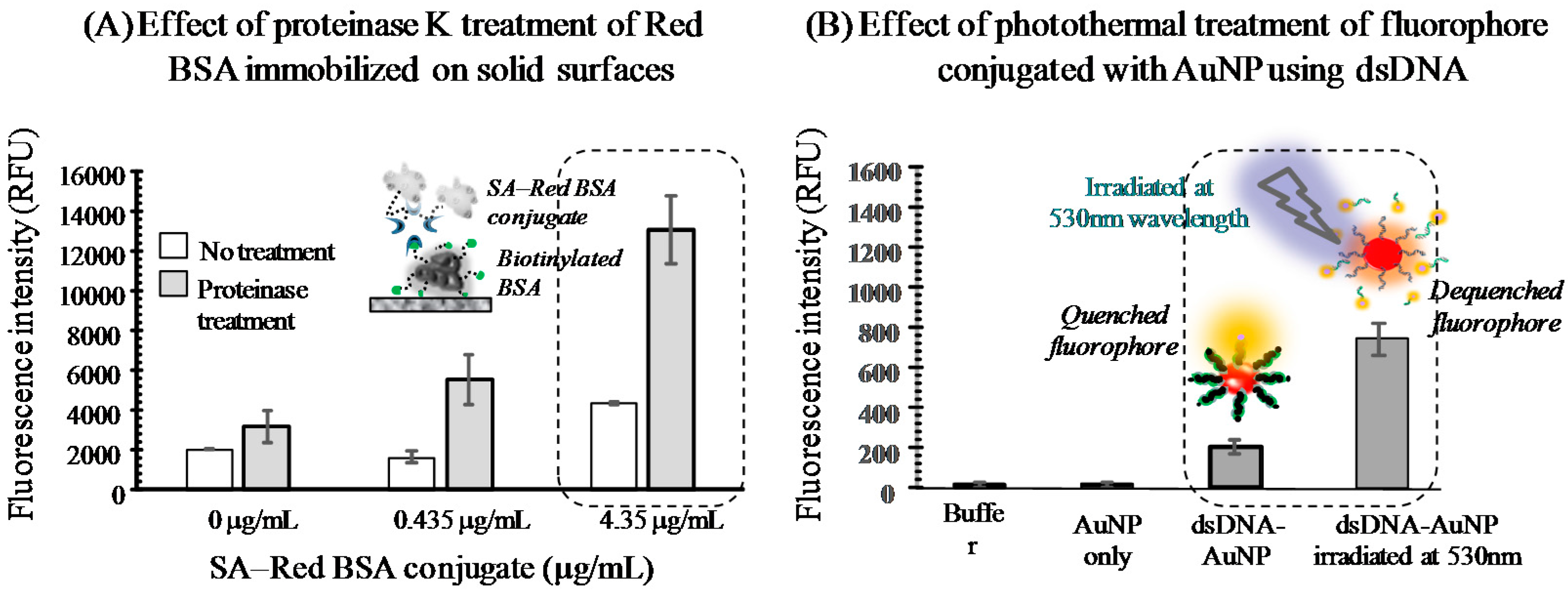
3.2. Preliminary Experimental Models
3.2.1. Protease-Assisted Protein Fragmentation Model
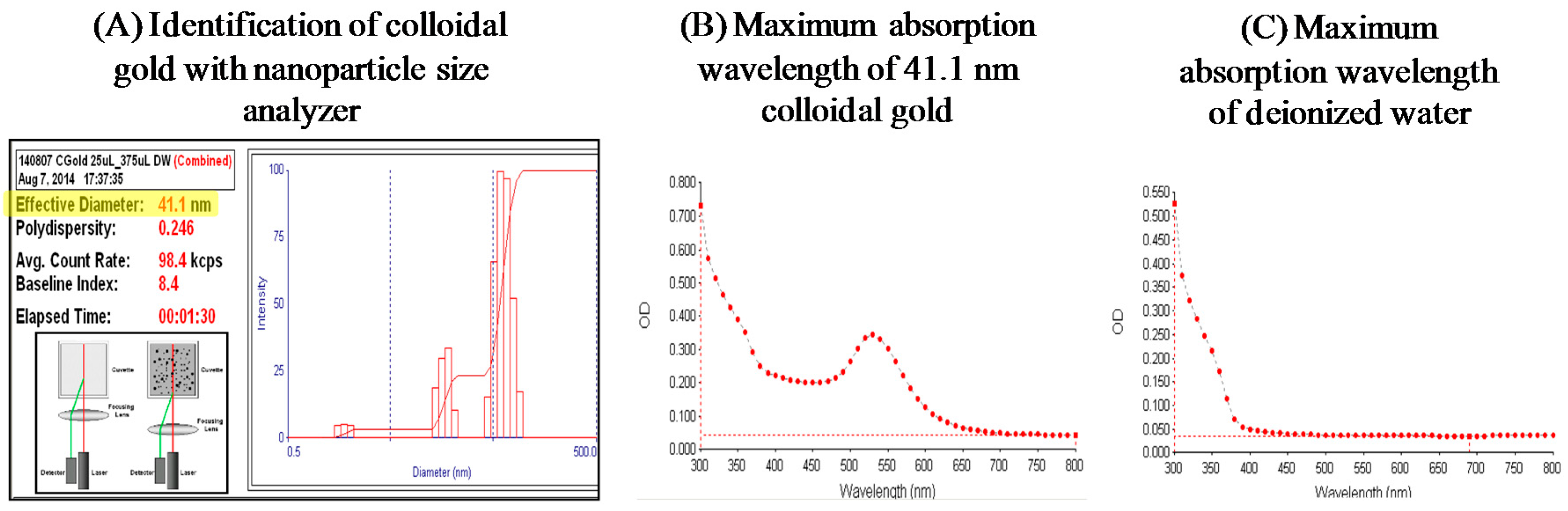
3.2.2. Photothermal Energy-Induced Dehybridization Model
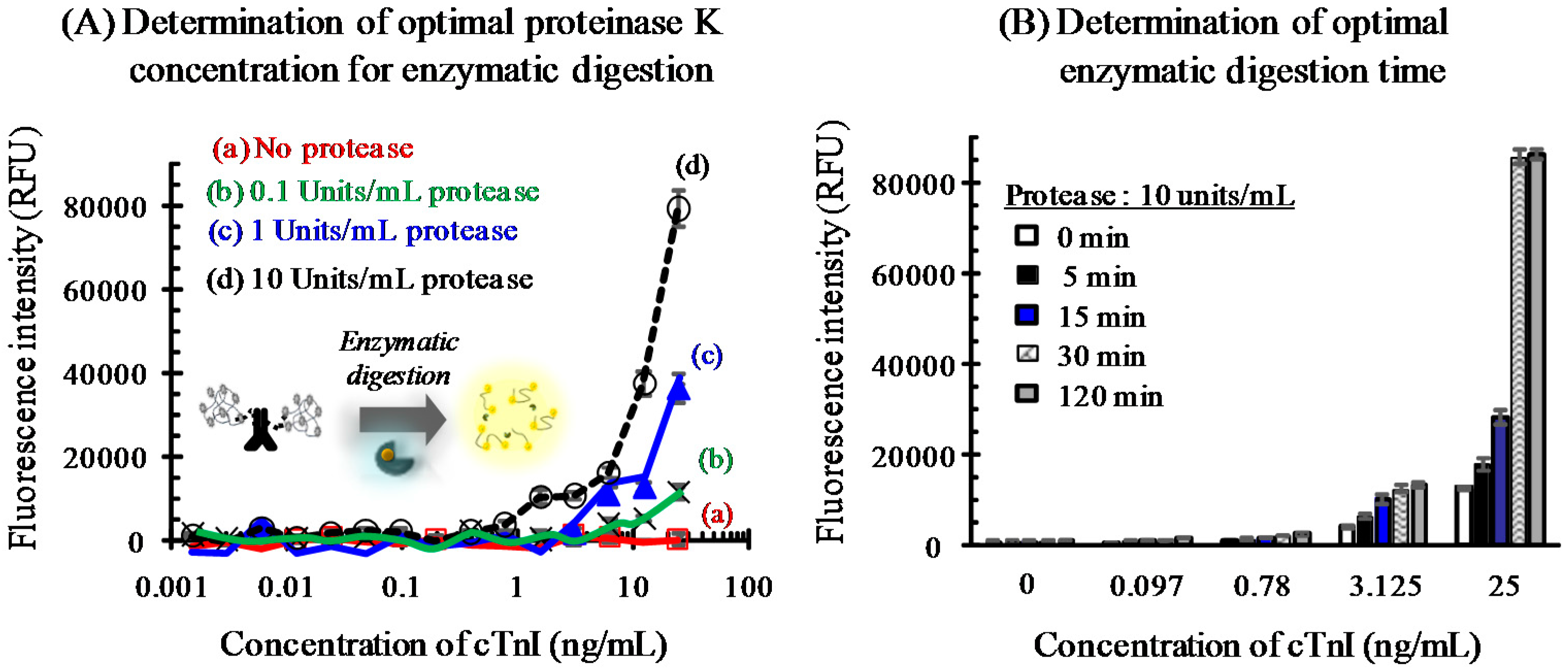
3.3. Immunoassay Employing Protease-Assisted Signal Enhancement
3.3.1. Optimization for Fluorescent Signal Generation
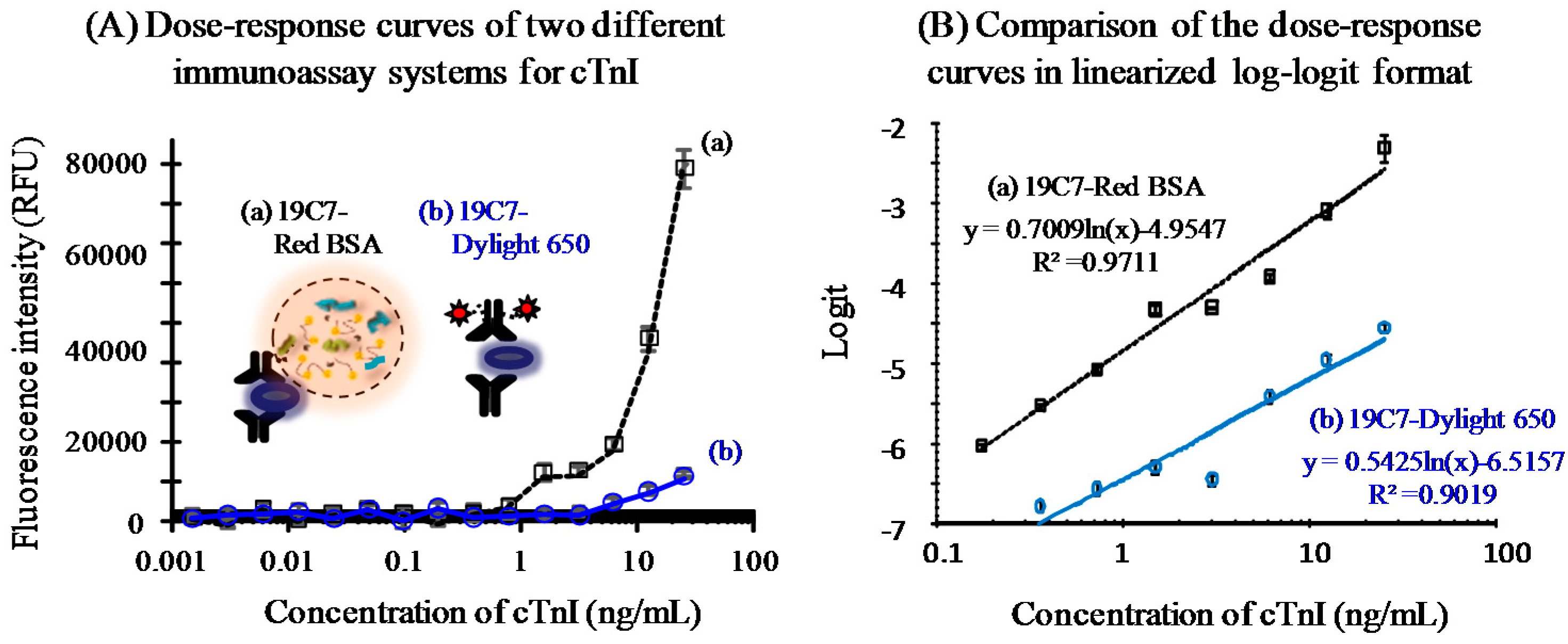
3.3.2. Immunoassay Performance
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Ds-DNA | Double strand-deoxyribonucleic acid |
| AuNPs | Gold nanoparticles |
| cTnI | Cardiac troponin I |
| FRET | Fluorescence resonance energy transfer |
| BSA | Bovine serum albumin |
| PBS | Phosphate buffered saline |
| DTT | Dithiothreitol |
| RT | Room temperature |
Appendix A


References
- Morrow, D.A.; Cannon, C.P.; Rifai, N.; Frey, M.J.; Vicari, R.; Lakkis, N.; Robertson, D.H.; Hille, D.A.; DeLucca, P.T.; DiBattiste, P.M.; et al. Ability of minor elevations of troponins I and T to predict benefit from an early invasive strategy in patients with unstable angina and non-ST elevation myocardial infarction: Results from a randomized trial. J. Am. Med. Assoc. 2001, 286, 2405–2412. [Google Scholar] [CrossRef]
- Roberts, I.M.; Jones, S.L.; Premier, R.R.; Cox, J.C. A comparison of the sensitivity and specificity of enzyme immunoassays and time-resolved fluoroimmunoassay. J. Immunol. Methods 1991, 143, 49–56. [Google Scholar] [CrossRef]
- Luppa, P.B.; Sokoll, L.J.; Chan, D.W. Immunosensors—principles and applications to clinical chemistry. Clin. Chim. Acta 2001, 314, 1–26. [Google Scholar] [CrossRef]
- Sukhanova, A.; Devy, J.; Venteo, L.; Kaplan, H.; Artemyev, M.; Oleinikov, V.; Klinov, D.; Pluot, M.; Cohen, J.H.; Nabiev, I. Biocompatible fluorescent nanocrystals for immunolabeling of membrane proteins and cells. Anal. Biochem. 2004, 324, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Decher, G.; Nerambourg, N.; Praho, R.; Werts, M.H.; Blanchard-Desce, M. Distance-dependent fluorescence quenching on gold nanoparticles ensheathed with layer-by-layer assembled polyelectrolytes. Nano Lett. 2006, 6, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Ao, L.; Gao, F.; Pan, B.; He, R.; Cui, D. Fluoroimmunoassay for antigen based on fluorescence quenching signal of gold nanoparticles. Anal. Chem. 2006, 78, 1104–1106. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Holmes, K.L.; Lantz, L.M. Protein labeling with fluorescent probes. Methods cell biol. 2001, 63, 185–204. [Google Scholar] [PubMed]
- Bhargava, S.K.; Booth, J.M.; Agrawal, S.; Coloe, P.; Kar, G. Gold nanoparticle formation during bromoaurate reduction by amino acids. Langmuir 2005, 21, 5949–5956. [Google Scholar] [CrossRef] [PubMed]
- Bodor, G.S.; Porter, S.; Landt, Y.; Ladenson, J.H. Development of monoclonal antibodies for an assay of cardiac troponin-I and preliminary results in suspected cases of myocardial infarction. Clin. Chem. 1992, 38, 2203–2214. [Google Scholar] [PubMed]
- Casari, G.; Sander, C.; Valencia, A. A method to predict functional residues in proteins. Nature struct. Biol. 1995, 2, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.F.; Knutson, J.R. Mechanism of fluorescence concentration quenching of carboxyfluorescein in liposomes: Energy transfer to nonfluorescent dimers. Anal. Biochem. 1988, 172, 61–77. [Google Scholar] [CrossRef]
- Ebeling, W.; Hennrich, N.; Klockow, M.; Metz, H.; Orth, H.D.; Lang, H. Proteinase K from Tritirachium album Limber. Eur. J. Biochem. 1974, 47, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Munechika, K.; Ginger, D.S. Dependence of fluorescence intensity on the spectral overlap between fluorophores and plasmon resonant single silver nanoparticles. Nano lett. 2007, 7, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Mitsudome, T.; Noujima, A.; Mikami, Y.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Supported gold and silver nanoparticles for catalytic deoxygenation of epoxides into alkenes. Angew. Chem. 2010, 49, 5545–5548. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dai, Q.; Austin, L.; Coutts, J.; Knowles, G.; Zou, J.; Chen, H.; Huo, Q. A one-step homogeneous immunoassay for cancer biomarker detection using gold nanoparticle probes coupled with dynamic light scattering. J. Am. Chem. Soc. 2008, 130, 2780–2782. [Google Scholar] [CrossRef] [PubMed]
- Arjmandi, N.; Van Roy, W.; Lagae, L.; Borghs, G. Measuring the electric charge and zeta potential of nanometer-sized objects using pyramidal-shaped nanopores. Anal. Chem. 2012, 84, 8490–8496. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Cho, J.H.; Cha, G.S.; Lee, C.W.; Kim, H.B.; Paek, S.H. Conductimetric membrane strip immunosensor with polyaniline-bound gold colloids as signal generator. Biosens. Bioelectron. 2000, 14, 907–915. [Google Scholar] [CrossRef]
- Reichlin, T.; Hochholzer, W.; Bassetti, S.; Steuer, S.; Stelzig, C.; Hartwiger, S.; Biedert, S.; Schaub, N.; Buerge, C.; Potocki, M.; et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N. Engl. J. Med. 2009, 361, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Kim, M.H.; Mok, R.S.; Jeon, J.W.; Lim, G.S.; Chai, C.Y.; Paek, S.H. Two-dimensional paper chromatography-based fluorescent immunosensor for detecting acute myocardial infarction markers. J. Chromatogr. B. 2014, 967, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Kohl, T.; Heinze, K.G.; Kuhlemann, R.; Koltermann, A.; Schwille, P. A protease assay for two-photon crosscorrelation and FRET analysis based solely on fluorescent proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 12161–12166. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Ohashi, H.; Iijima, I.; Ihara, M.; Takagi, H.; Hohsaka, T.; Ueda, H. “Quenchbodies”: Quench-based antibody probes that show antigen-dependent fluorescence. J. Am. Chem. Soc. 2011, 133, 17386–17394. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, L.; Calza, P.; Sordello, F.; Giovannoli, C.; Di Nardo, F.; Passini, C.; Cerruti, M.; Goryacheva, I.Y.; Speranskaya, E.S.; Baggiani, C. Multi-analyte homogenous immunoassay based on quenching of quantum dots by functionalized graphene. Anal. Bioanal. Chem. 2014, 406, 4841–4849. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-W.; Cho, I.-H.; Park, J.-N.; Seo, S.-M.; Paek, S.-H. A High-Performance Fluorescence Immunoassay Based on the Relaxation of Quenching, Exemplified by Detection of Cardiac Troponin I. Sensors 2016, 16, 669. https://doi.org/10.3390/s16050669
Kim S-W, Cho I-H, Park J-N, Seo S-M, Paek S-H. A High-Performance Fluorescence Immunoassay Based on the Relaxation of Quenching, Exemplified by Detection of Cardiac Troponin I. Sensors. 2016; 16(5):669. https://doi.org/10.3390/s16050669
Chicago/Turabian StyleKim, Seung-Wan, Il-Hoon Cho, Ji-Na Park, Sung-Min Seo, and Se-Hwan Paek. 2016. "A High-Performance Fluorescence Immunoassay Based on the Relaxation of Quenching, Exemplified by Detection of Cardiac Troponin I" Sensors 16, no. 5: 669. https://doi.org/10.3390/s16050669