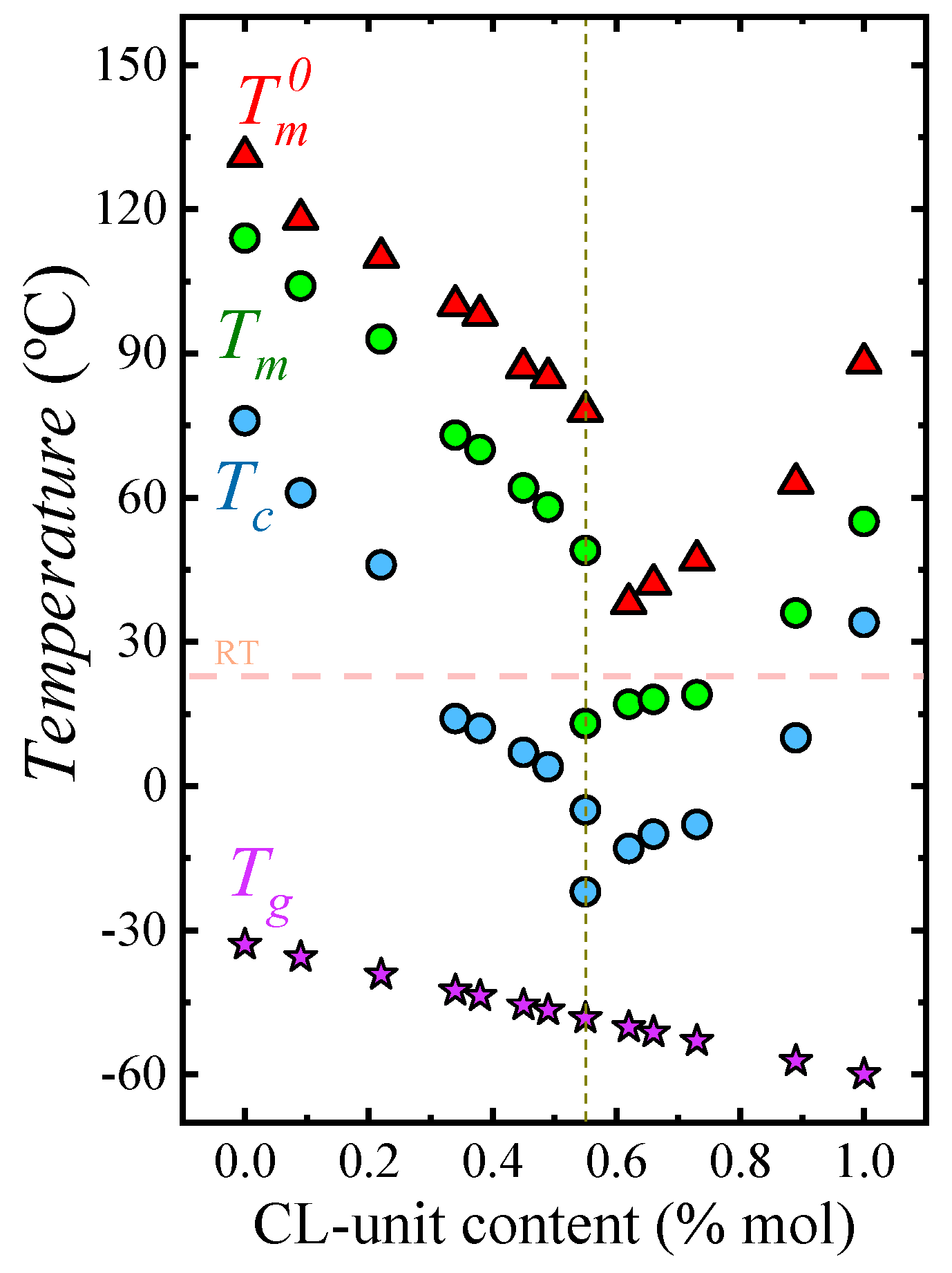
Figure 1 presents a phase diagram for the PBS-ran-PCL system. These random copolymers exhibit a single-phase melt and a single glass transition temperature, as expected for random copolymers. Upon cooling from the melt, the materials are capable of crystallizing in the entire composition range, in spite of being random, as demonstrated by NMR studies [
17]. The copolymers display a pseudo-eutectic point at the composition BS
45CL
55. This BS
45CL
55 copolymer is the only one in the series that can form two crystalline phases upon cooling from the melt, i.e., a PBS-rich phase and a PCL-rich phase (as evidenced earlier by WAXS and DSC [
17]), hence the two melting point values reported in
Figure 1 for this composition. To each side of the pseudo-eutectic point, a single crystalline phase is formed, either a PBS-rich phase (i.e., left-hand side of the eutectic) or a PCL-rich phase (i.e., right-hand side of the eutectic), with crystalline unit cells resembling those of PBS and PCL respectively.
The phase diagram shown in
Figure 1 illustrates the versatility of isodimorphic copolymers. It is well known that the optimal mechanical properties in terms of ductility and toughness of thermoplastic semicrystalline materials are generally observed at temperatures in between
Tg and
Tm. Thanks to random copolymerization, the copolymers exhibit a single
Tg value that is independent of the melting point of the phase (or phases in the case of the composition at the pseudo-eutectic point) that is able to crystallize. This remarkable behavior provides a separate control of
Tg and
Tm which cannot be obtained in homopolymers. Additionally, as
Figure 1 shows, depending on composition, the samples can be molten at room temperature or they can be semicrystalline. Such wide range of thermal properties can lead to fine tuning mechanical properties and crystallinities to tailor applications.
3.1. Nucleation Kinetics Studied by PLOM
Counting the number of spherulites in PLOM experiments is the usual way of obtaining nucleation data by assuming that each spherulite grows from one heterogeneous nucleus. In this work, we studied the nucleation kinetics by determining the nucleation density as a function of time by PLOM, from which nucleation rates can be calculated.
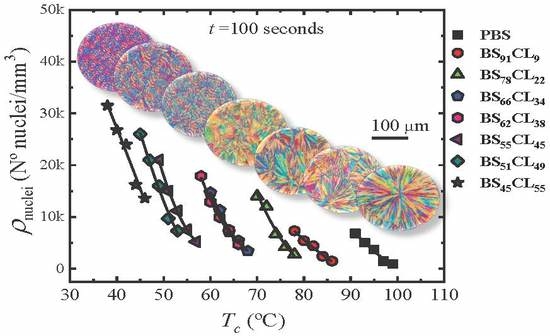
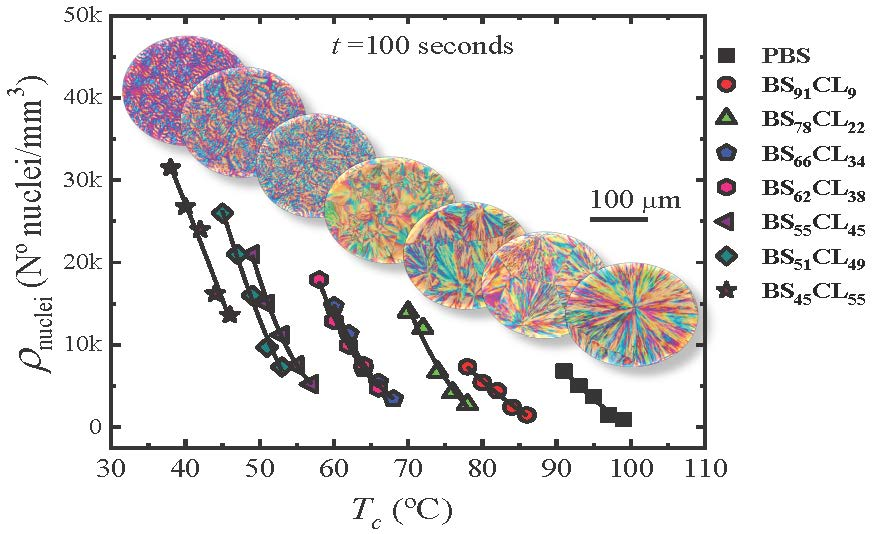
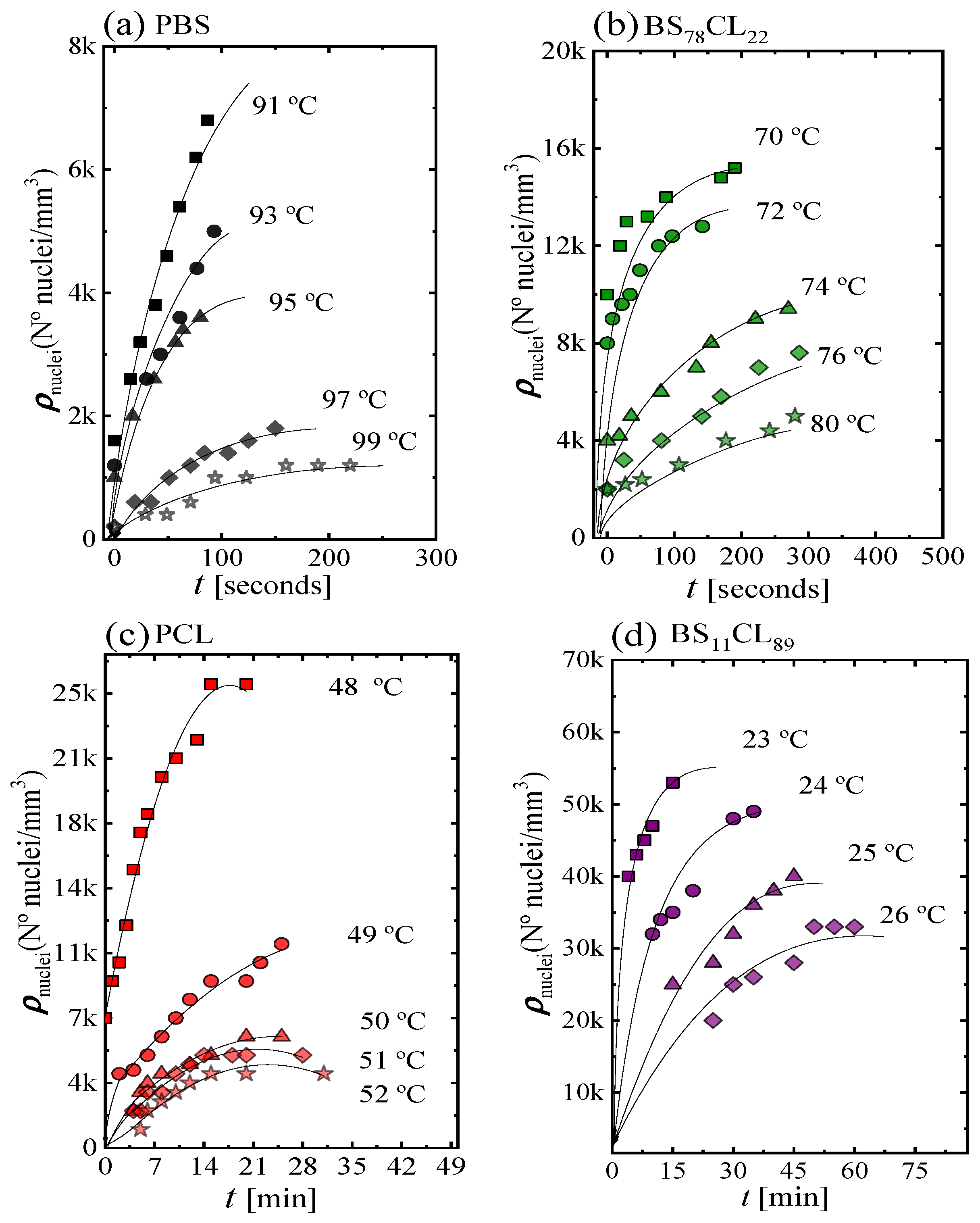
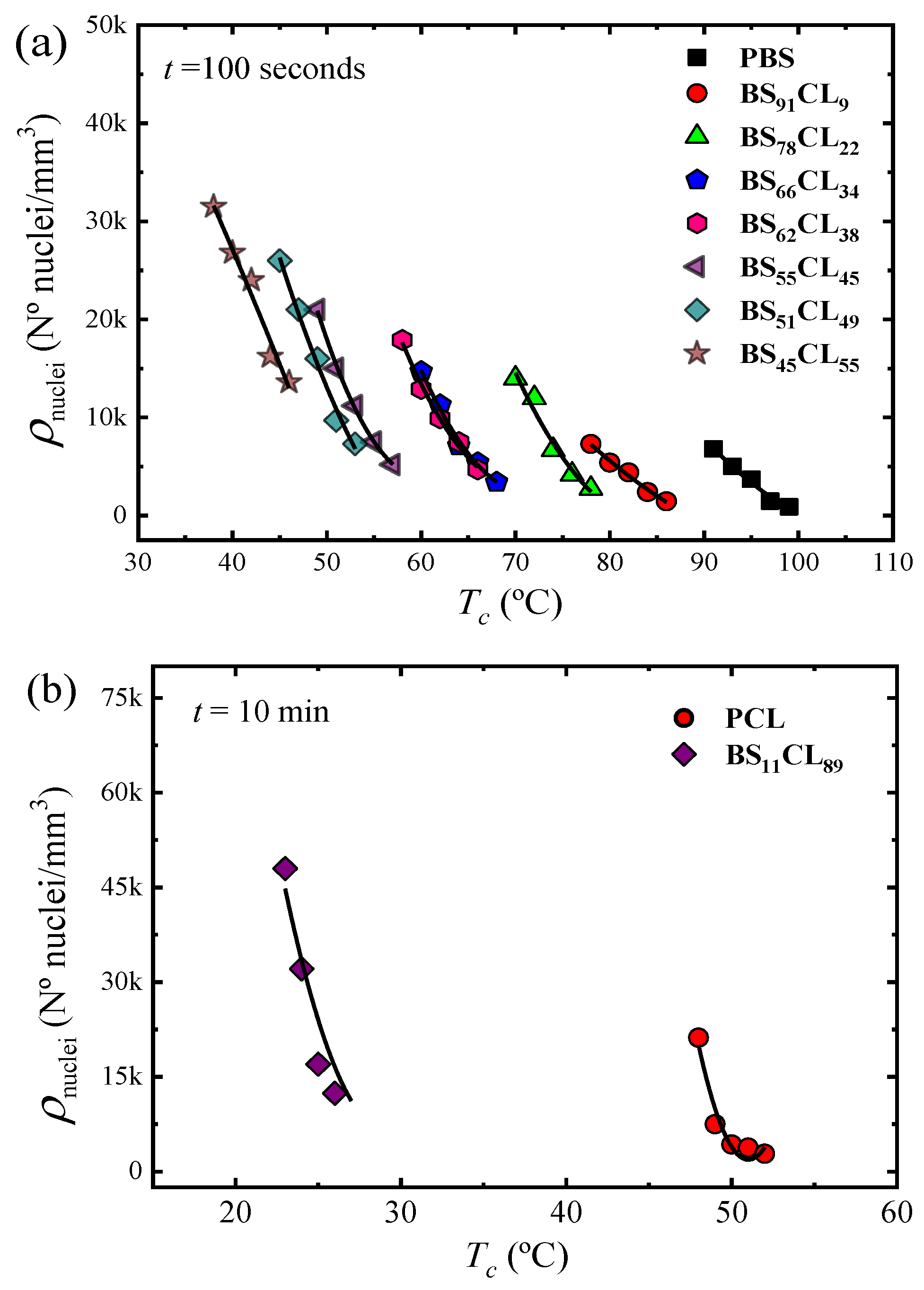
Figure 2 shows four examples of plots of the nucleation density ρ
nuclei (nuclei/mm
3) as a function of time for neat PBS, neat PCL, and two sample copolymers. The rest of the data can be found in the
Supplementary Information (Figure SI-1). The nucleation density increases almost linearly with time at short times, then it tends to saturate. The number of heterogeneous nuclei that are activated at longer times increases as nucleation temperature decreases, a typical behavior of polymer nucleation [
20]. As expected, the nucleation density at any given time increases as
Tc decreases, because the thermodynamic driving force for primary nucleation increases with supercooling [
21].
Figure 3 shows plots of nucleation density versus temperature taken at a constant nucleation time of 100 s for neat PBS and PBS-rich copolymers (
Figure 3a) and 10 min in the case of PCL and BS
11CL
89 copolymer (
Figure 3b).
PBS exhibits the lowest nucleation density of all samples, therefore, the largest spherulites (see Figure 5 below). As the amount of CL comonomer increases in the PBS-rich copolymers (
Figure 3a), the nucleation density increases, as well as the supercooling needed for nucleation. In the case of PBS-rich copolymers,
Figure 3a (and
Figure SI-1 in the SI) shows nucleation data for seven different copolymers, with compositions ranging from 91% to 45% PBS.
The dependence of nucleation density on supercooling, can be observed in
Figure SI-2. The data presented in
Figure 3a can be reduced to a supercooling range between 32 and 40 °C, i.e., only 8 °C. This means that a large part of the horizontal shift in the curves of
Figure 3a (spanning nearly 60 °C in crystallization temperature) is due to changes in supercooling. These changes are caused by the variations in equilibrium melting temperatures with composition (see
Figure 1).
PCL has a higher nucleation density than PBS when compared at equal supercoolings (see
Figure SI-2b). When a small amount of BS comonomer is incorporated, as in random copolymer BS
11CL
89, the nucleation density increases significantly (
Figure 3b). Due to the very high nucleation density of the other PCL-rich composition copolymers (with higher amounts of PBS), it was impossible to determine their nucleation kinetics. Examples of the microspherulitic morphologies obtained for such PCL-rich copolymers can be observed in Figure 5 below.
It is interesting to note than in both sides of the pseudo-eutectic point (i.e., the PBS-rich side represented in
Figure 3a and the PCL-rich side represented in
Figure 3b, see also
Figure 1), the copolymers exhibit higher nucleation density than their corresponding homopolymers. This behavior could be somewhat analogous to what has been observed in long-chain branched polylactides (PLLAs) [
22] or long-chain branched polypropylenes (PPs) with respect to linear analogs [
23]. The interruption of crystallizable linear sequences with defects has been reported to increase nucleation density although the reasons are not clear. In the present case, the linear crystallizable sequence of PBS, for instance, is being changed by the introduction of randomly placed PCL repeat units. Even though the random copolyesters can form a single phase in the melt, there may be at the segmental level, some preference for PBS-PBS local chain segmental contacts in comparison to less favorable PBS-PCL contacts. We speculate that this may drive the enhancement of nucleation, but more in-depth studies would be needed to ascertain the exact reason for this behavior.
The Fisher–Turnbull nucleation theory [
24] can be used to quantify the activation free energy of primary nucleation. This theory gives the steady-state rate of primary nucleation per unit volume and time,
I = dN/dt, for a heterogeneous nucleation process on a preexisting flat surface (or heterogeneous nucleus) as:
where
I0 is related to the diffusion of polymeric segments from the melt to the nucleation site, Δ
F* is a parameter proportional to the primary nucleation free energy, and
σ and
σe are the lateral and fold surface free energies, respectively. Δ
T is the supercooling defined as Δ
T =
−
and
is the equilibrium melting point. Δ
σ is the interfacial free energy difference, given by:
in which
σs/c is the crystal-substrate interfacial energy and
σs/m is the melt-substrate interfacial energy. Therefore, Δ
σ can be considered proportional to the surface tension properties of the substrate, polymer crystal and polymer melt. The interfacial free energy difference is a convenient way to express the nucleating ability of the substrate towards the polymer melt.
In this work, the values of
(listed in
Table SI-1 and plotted in
Figure 1) were obtained by isothermal crystallization DSC experiments followed by Hoffman–Weeks extrapolations (see
Figure SI-3). Δ
Hv is the volumetric melting enthalpy (J/m
3) and it was estimated by
, so that
ρ = 1.26 g/cm
3 and
= 213 J/g for neat PBS [
25] and
ρ = 1.14 g/cm
3 and
= 139.5 J/g for neat PCL [
26].
In this work, we employed the value of
= 213 J/g for neat PBS and PBS-rich phase composition that has been recently obtained by some of us [
25]. This value was determined employing a combined DSC and X-ray diffraction method using isothermal crystallization data. This experimentally extrapolated value is higher than that of
= 110 J/g, estimated empirically by the group contribution method [
27], but very close to the value of 210 J/g reported by Papageorgiou et al. [
28].
The values of the nucleation rate I were calculated from the initial slope (i.e., at short measurement times, where linear trends were obtained) of the plots shown in
Figure 2 and
Figure SI-1.
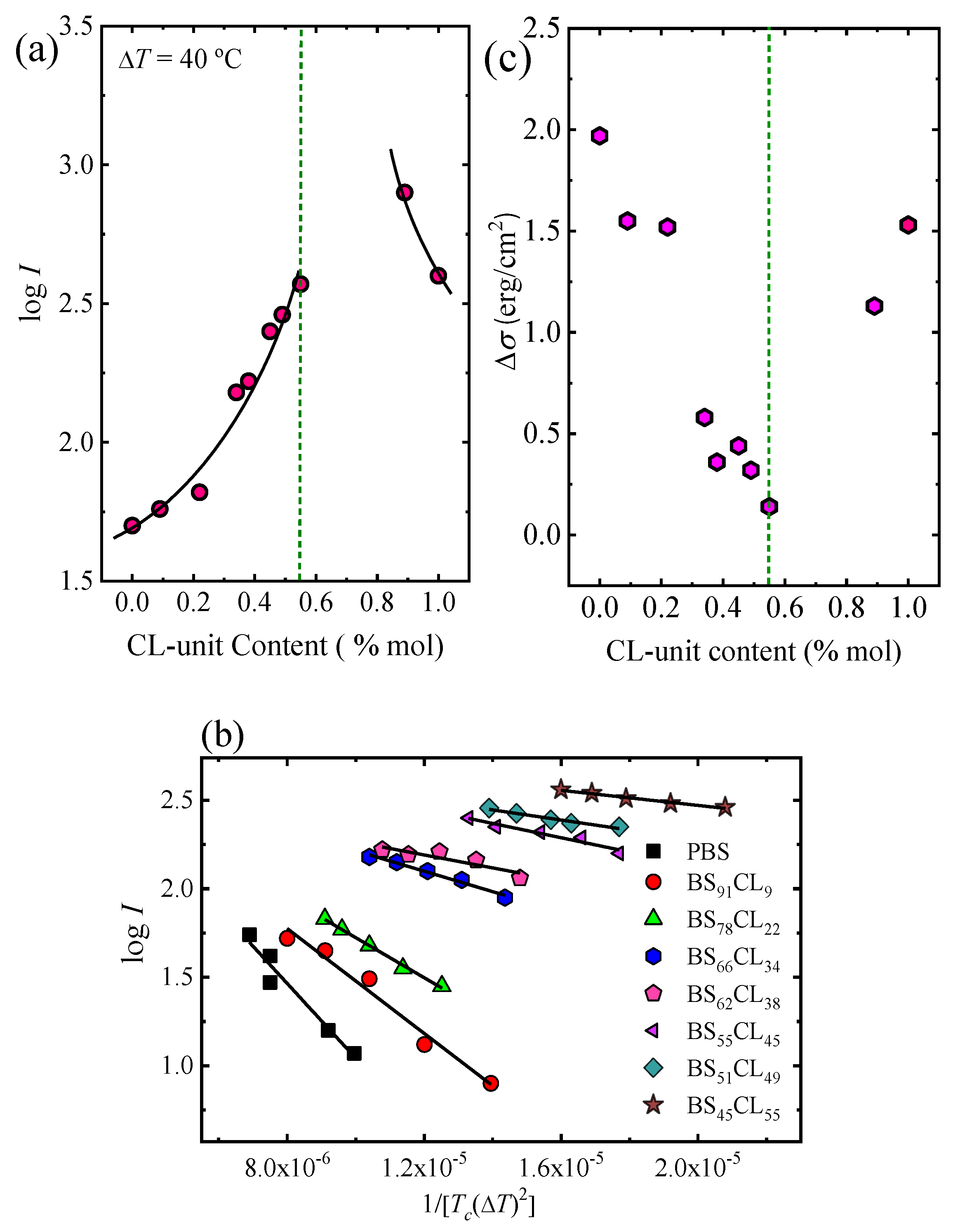
Figure 4a shows log I as a function of CL-unit molar fraction for a constant supercooling of Δ
T = 40 °C. The nucleation rate strongly depends on copolymer composition. Adding a comonomer randomly along the chain to either PBS or PCL largely increases the nucleation rate. In the PBS-rich composition side (to the left of the pseudo-eutectic point signaled by a vertical line in
Figure 4a) the nucleation rate increases up to 7.5 times with respect to neat PBS, as the amount of PCL units in the random copolymer increases. Neat PCL nucleates faster than neat PBS. In the PCL-rich composition side, only one copolymer was measured (whose nucleation rate increased two-fold with respect to neat PCL), as increasing PBS content towards the pseudo-eutectic point increased nucleation rate so much that measurements were no longer possible.
Figure 4b shows the Turnbull–Fisher plots for PBS and PBS-rich compositions based on Equation (1). Turnbull–Fisher plots for PCL and BS
11CL
89 copolymer are presented in the
supplementary information (Figure SI-4). The nucleation data can be successfully fitted with the linearized version of Equation (1). From the slope, a value of the interfacial free energy difference (Δ
σ) can be obtained.
Small values of Δ
σ are indicative of good nucleation efficiency since a lower amount of interfacial energy is required to form the crystal–substrate interface.
Table 2 reports a value of Δ
σ for PBS equal to 1.97 erg/cm
2. As seen in
Figure 4c (and
Table 2), this interfacial free energy difference progressively decreases in the copolymers as the amount of CL comonomer increases, indicating that the primary nucleation process is facilitated by copolymerization with PCL until the pseudo-eutectic point is reached. On the right-hand side of the pseudo-eutetic point in
Figure 4c, PCL has a Δ
σ value of 1.53 erg/cm
2, which is, as expected, smaller than that of PBS, as PCL has a larger nucleation density at equivalent supercoolings than PBS. The copolymer B
11CL
89 shows an even smaller value of Δ
σ, as the incorporation of PBS in the copolymer increases its nucleation capacity.
3.2. Kinetics of Superstructural Growth (Secondary Nucleation) by PLOM
PBS, PCL, and all the random copolymers prepared in this work exhibited spherulitic superstructural morphologies. Examples of the spherulites obtained at a constant supercooling value of 40 °C can be observed in
Figure 5. Both PBS and PCL exhibited well-developed spherulites without banding. PBS-rich copolymers that contain more than 34% PCL exhibit clear banding. This is consistent with previous works indicating that the addition of diluents (for PBS-rich compositions, crystallization occurs while PCL chains are in the liquid state) to several polyesters induces banding [
29,
30].
Isothermal crystallization experiments were performed to follow the growth of spherulites as a function of time using PLOM. The growth rate was calculated from the slope of spherulite radius versus time plots, which were always observed to be highly linear [
18,
31].
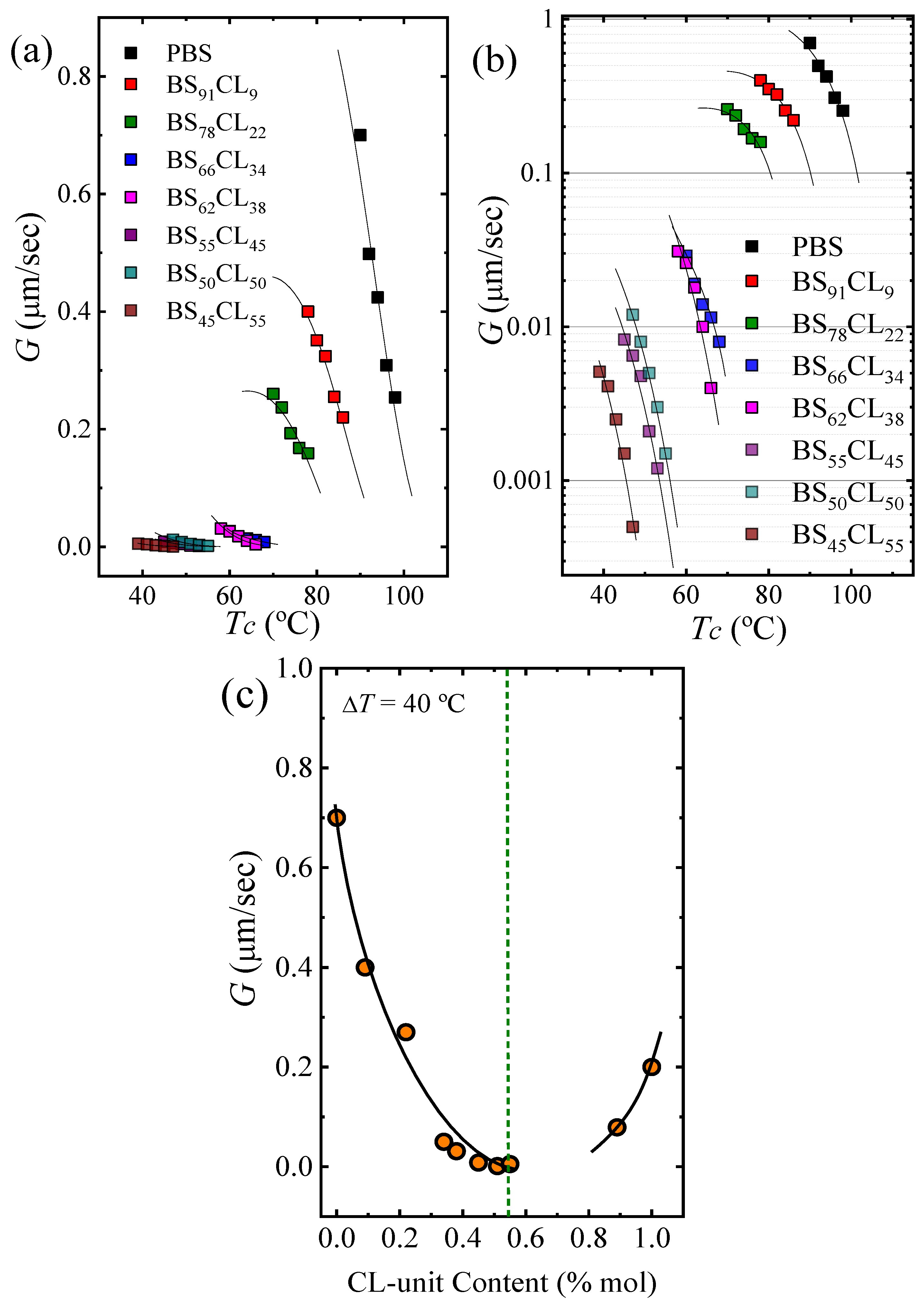
The experimental growth rates are plotted as a function of the isothermal crystallization temperatures employed in
Figure 6a with a linear scale and in
Figure 6b with a log scale, so that differences in G values for PBS-rich samples with PCL contents larger than 22% are observed. The incorporation of PCL repeat units in the random copolymers have a dramatic influence on the growth rate of the PBS-rich phase spherulites, as G decreases up to 3.5 orders of magnitude (
Figure 6b). The decrease in G values with comonomer incorporation for the PBS rich copolymers is due to two reasons. Firstly, as in any isodimorphic copolymer, there is a competition between inclusion and exclusion of repeat units within the PBS crystal lattice, where exclusion typically predominates. Secondly, incorporation of PCL repeat units in the copolymer chains reduces
Tg values (as shown in
Figure 1), thereby causing a plasticization effect on the PBS-rich phase. In the case of the PCL-rich compositions, the spherulitic growth rate was determined for only one copolymer (i.e., BS
11CL
89), as in the other cases, as pointed out above, the nucleation rate and nucleation density were so high, that it was impossible to measure the extremely fast growth of very small spherulites. For this copolymer, the growth rate decreased in relative terms (see
Figure 6c) by a factor of approximately 2.5 at a supercooling of 40 °C.
The data presented in
Figure 6a are plotted as a function of supercooling in the
Supplementary Information (Figure SI-5). The PBS-rich growth rate data is shifted horizontally and but there is no overlap in the y axis values. If we were dealing with a simple solvent effect, the growth rate curves at different compositions should completely overlap in a master curve when plotted as a function of supercooling. The lack of superposition is due to the fact that PCL repeat units are randomly incorporated and covalently bonded with the PBS repeat units. The interruption of crystallizable PBS repeat units (by the majority of PCL repeat units that are excluded from the crystals) makes more difficult the secondary nucleation process.
Figure 6c shows how the growth rate depends on composition at a constant supercooling of 40 °C. The trend is the opposite as that obtained for primary nucleation (compare
Figure 6c with
Figure 4a). In order to quantify the restrictions imposed by the comonomer on the crystallization of the major component, we employed the Lauritzen and Hoffman theory, as it allows the calculation of energetic terms related to the secondary nucleation process (i.e., growth process).
The Lauritzen and Hoffman (LH) nucleation and growth theory [
32] was used to fit the spherulitic growth rate data as a function of isothermal crystallization temperature, according to the following equation:
where
G0 is the growth rate constant that includes all the terms that are temperature-insensitive,
U* is the transport activation energy which characterizes molecular diffusion across the interfacial boundary between melt and crystals (in this work, we employ a constant value of 1500 cal/mol).
Tc is the crystallization temperature and
T0 is a hypothetical temperature at which all chain movements freeze (taken as
T0 =
Tg − 30 °C);
is the equilibrium melting temperature and
f is a temperature correction factor given by the following expression:
f = 2
Tc/(
Tc +
).
The equilibrium melting temperatures
were estimated by the Hoffman–Weeks linear extrapolation (
Figure SI-3 and Table SI-1). The parameter
is proportional to the energy barrier for secondary nucleation or spherulitic growth and is given by:
where
j is assumed to be equal to 2 for crystallization in the so-called Regime II, a regime where both secondary nucleation at the growth front and the rate of spread along the growing crystal face are comparable [
26]. The other terms in the equation are the width of the chain
bo, the lateral surface free energy
σ, the fold surface free energy
σe, the Boltzman constant k, and the equilibrium latent heat of fusion,
.
Plotting
versus 1/
Tc(Δ
T)
f (i.e., the Lauritzen and Hoffman plots) gives a straight line and its slope and intercept are equal to
and
G0 respectively. Examples of LH plots can be found in the
Supplementary Information, Figure SI-6. Having the value of
, the magnitude of
σσe can be calculated from Equation (5). In order to calculate separately the values of
σ and
σe, the following expression can be used [
33]:
where
a0b0 is the cross sectional area of the chain. To obtain the parameters of the LH theory, the following values were used for neat PBS and BS-rich compositions [
34,
35]:
a0 = 5.25 Å and
b0 = 4.04 Å, and for neat PCL and CL-rich compositions [
36]:
a0 = 4.52 Å and
b0 = 4.12 Å.
Finally,
q, the work done by the macromolecule to form a fold is given by [
33]:
The solid lines in
Figure 6a,b correspond to fittings to Equation (3).
Table 2 shows that
values (which are proportional to the energy barrier for spherulitic growth) for the PBS-rich crystal phase tend to increase as PCL repeat units are incorporated in the random copolymers until a maximum value is reached at the pseudo-eutectic point. Similar trends are observed for the fold surface free energy and for the work done to form folds.
A plot of fold surface free energy versus composition can be found in the
Supplementary Information (Figure SI-7). These results quantitatively measure how comonomer incorporation makes difficult the spherulitic growth of the PBS-rich phase. A similar interpretation can be done to the mirror values presented in
Table 2 for PCL and the BS
11CL
89 copolymer with respect to the PCL phase.
The results presented in the two sections above can be summarized by comparing
Figure 4 with
Figure 6. The incorporation of comonomers at each side of the eutectic causes an increase in the nucleation density and nucleation rate but at the same time a decrease in spherulitic growth rate. These two processes, primary nucleation and growth are combined when a semicrystalline polymer is crystallized from the melt. Their simultaneous effect can be ascertained by determining overall crystallization kinetics by DSC.
3.3. Overall Crystallization Kinetics Studied by DSC
The overall isothermal crystallization kinetics considers both nucleation and growth, and can be conveniently determined by isothermal DSC experiments.
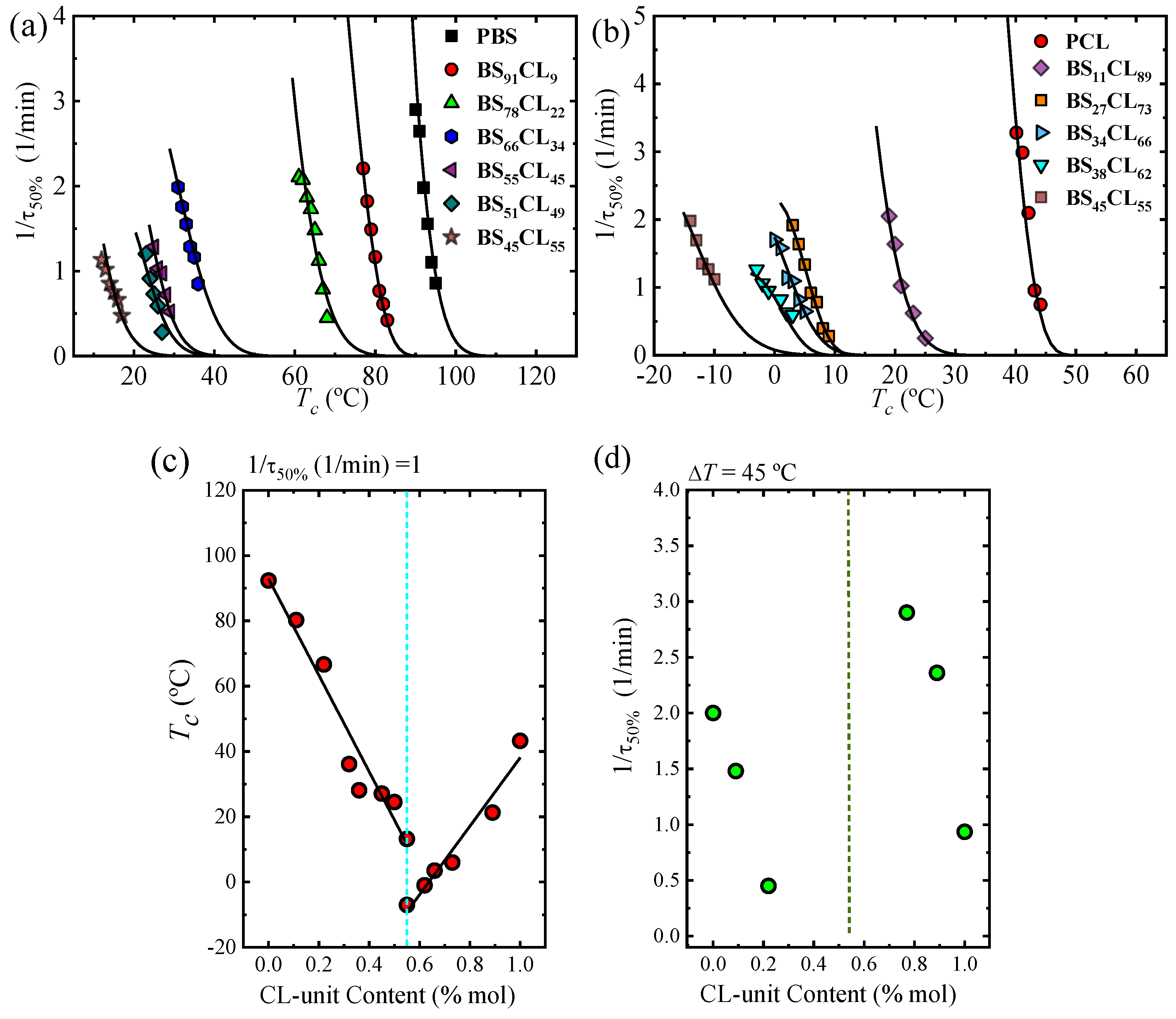
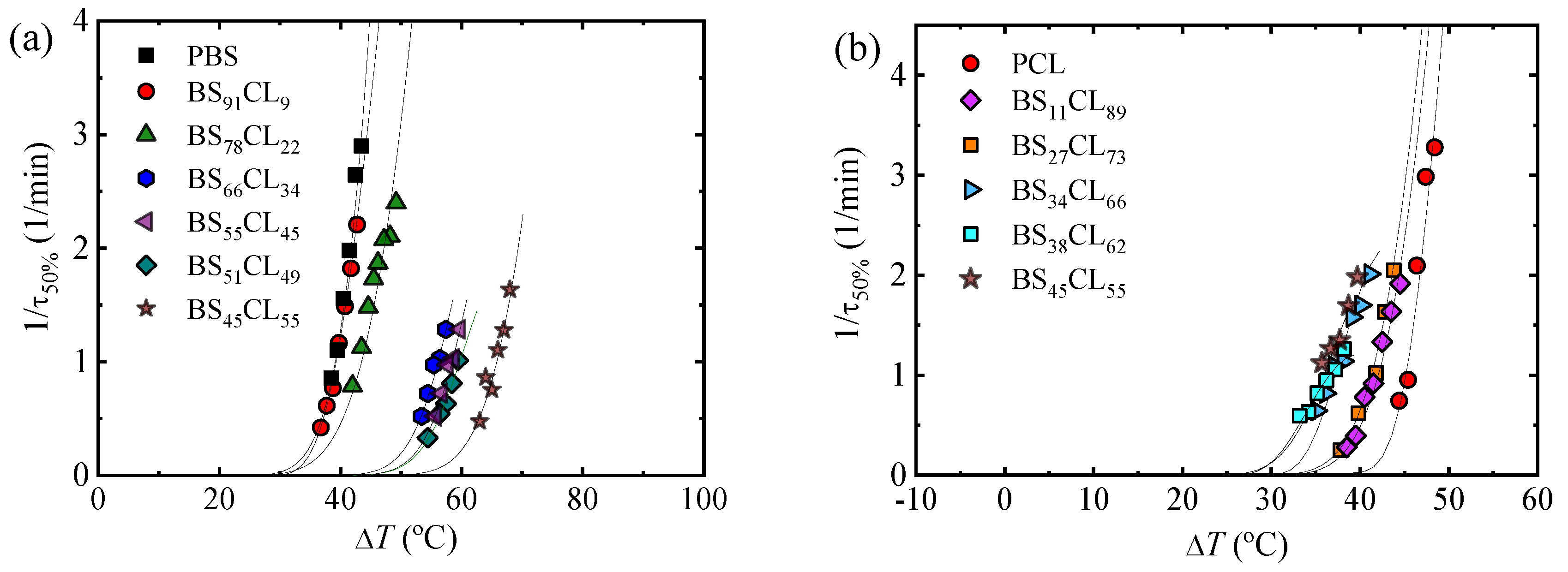
Figure 7a,b shows the experimental overall crystallization rate expressed as the inverse of the crystallization half time (
τ50%). By DSC, we were able to determine the overall isothermal crystallization kinetics for both homopolymers and all copolymers (five copolymers where only the PBS-rich phase crystallized and four copolymers where only the PCL-rich phase can crystallize).
For the special composition at the eutectic point (i.e., BS45CL55) that shows two crystalline phases, PBS-rich phase and PCL-rich phase, we performed isothermal crystallization using different protocols. For the PBS-rich phase crystallization, isothermal DSC experiments were performed at temperatures where the PCL-rich phase is in the melt and cannot crystallize, while in the PCL-rich phase, a special protocol was adopted to previously crystallize the PBS-rich phase to saturation (see experimental part).
Figure 7a,b shows the strong dependence of the overall crystallization rate and the temperature range where measurements were possible on copolymer composition. In the case of the PBS and all PBS-rich compositions, the overall crystallization proceeds from a single-phase melt. Upon increasing PCL content, the amount of the crystallizable PBS-rich phase decreases and there will be more molten PCL component causing a plasticization (“solvent effect”). In addition, the effect of PCL exclusion in the PBS-rich crystal lattice may cause some further reduction in crystallization rate.
Figure 7a shows that the temperature needed for crystallization decreases as PCL content in the copolymer increases, while the overall crystallization rate measured at the minimum
Tc value possible tends to decrease.
Figure 7c plots the crystallization temperature needed to obtain the same overall crystallization rate of 1 min
−1. These T
c values monotonically decrease with PCL content until the pseudo-eutectic region is reached. On the PCL-rich side,
Figure 7b,c shows similar results, as the crystallization temperatures needed to crystallize the PCL phase decrease as PBS repeat units are added to the copolymer.
To check if the supercooling is playing a major role upon changing composition, we plot the data contained in
Figure 7a,b as a function of Δ
T in
Figure 8a,b, respectively. Surprisingly, the trends are quite different depending on the phase under consideration, or the composition range.
Figure 8a shows that the curves of PBS-rich overall growth rate data that originally spanned a
Tc range of approximately 90 °C (in
Figure 7a) are now within 30 °C in supercooling, attesting for the thermodynamic compensation of the solvent effect, as the PCL-rich phase is in the melt. In fact, the curves of PBS and BS
91CL
9 completely overlap, while that of BS
78CL
22 is relatively close to that of neat PBS. However, beyond 22% CL incorporation in the copolymer, the samples require much larger supercooling to crystallize. It is clear that the dominant factor to the left of the eutectic point is the growth rate, as the results presented in
Figure 6a and
Figure 7a imply an overall crystallization rate reduction with PCL incorporation in the copolymer, both in terms of crystallization temperature or supercooling. In spite of the increase in nucleation density and nucleation rate (see
Figure 3 and
Figure 4) with PCL incorporation in the copolymer, it is the very large decrease in growth rate (of up to three orders of magnitude, see
Figure 6b) that dominates, leading to a decrease in overall crystallization rate (
Figure 7a and
Figure 8a).
Figure 7d represents the overall crystallization rate as a function of composition for a constant supercooling of 45 °C. In the PBS-rich side of the pseudo-eutectic region (left-hand side of
Figure 7d), only three data points are plotted, as they are the only ones that could be measured at such constant supercooling value (check
Figure 8b). The trend clearly shows a significant decrease in overall crystallization rate as CL unit content increases, as expected from
Figure 7a and
Figure 8a and the discussion above. Even though the value of supercooling is not exactly the same (with only 5 °C difference), a comparison with
Figure 4a and
Figure 6c clearly indicates that the PBS-rich phase overall crystallization is dominated by growth rate.
Figure 8b shows remarkable results for the PCL-rich phase overall crystallization. The 1/
τ50% curves versus temperature span a temperature range of 65 °C (
Figure 6b). When they are plot as a function of supercooling, they only span 15 °C. However, they do not overlap, as would be expected for a simple solvent effect. In fact, the supercooling needed for crystallization of the PCL-rich phase remarkably decreases as PBS repeat units are included in the copolymer. The results indicate an acceleration of the overall crystallization rate (at constant supercooling) that can only be explained by the increase in both nucleation density and nucleation rate. We were only able to measure the increase in nucleation density and nucleation rate in
Figure 3 and
Figure 4 for neat PCL and BS
11CL
89, as further incorporation of BS units increased the nucleation density so much that measurements by polarized optical microscopy of nucleation rate became impossible. Hence, we are convinced that primary nucleation enhancement upon PBS repeat unit incorporation in the copolymers is the reason behind the acceleration of the overall crystallization kinetics, when this is considered in terms of supercooling. In the right-hand side of the pseudo-eutectic point in
Figure 7d, the increase in overall crystallization rate at a constant supercooling of 45 °C can be appreciated.
The Lauritzen and Hoffman theory can also be applied to fit the overall crystallization data presented above. Equation (3) has to be modified to employ, as a characteristic rate, the inverse of the half-crystallization time determined by DSC, as follows [
37]:
where all the terms have been defined above, except for
, which now represents a parameter proportional to the energy barrier for both primary nucleation and spherulitic growth. The superscript τ is used to indicate its origin (coming from DSC data, and hence from fitting 1/τ
50% versus crystallization temperature). In this way, it is different from
, defined in Equation (3), derived from growth rate data and therefore proportional just to the free energy barrier for secondary nucleation or growth. The solid lines in
Figure 7a,b and
Figure 8a,b represent the fits to the Lauritzen and Hoffman theory.
Table 3 on the other hand reports all the relevant parameters.
There are no results in the literature regarding the isothermal crystallization of PBS-ran-PCL copolymers, therefore, we cannot compare the parameters reported in
Table 2 and
Table 3 with literature values. In the case of the homopolymers, Wu et al. [
38] and Papageorgiou et al. [
39] reported
values for neat PBS equal to 1.157 × 10
5 and 2.64 × 10
5 K
2, respectively, which are close to the value obtained in this work, i.e., 2.04 × 10
5 K
2. A value for
for neat PBS reported [
39] equal to 1.88 × 10
5 has been reported, that is somewhat higher than that obtained in this work, i.e., 0.87 × 10
5 K
2. For neat PCL, the energetic parameters previously reported based on fits of the Lauritzen and Hoffman theories [
40] are in the same order of magnitude as those reported here [
33].
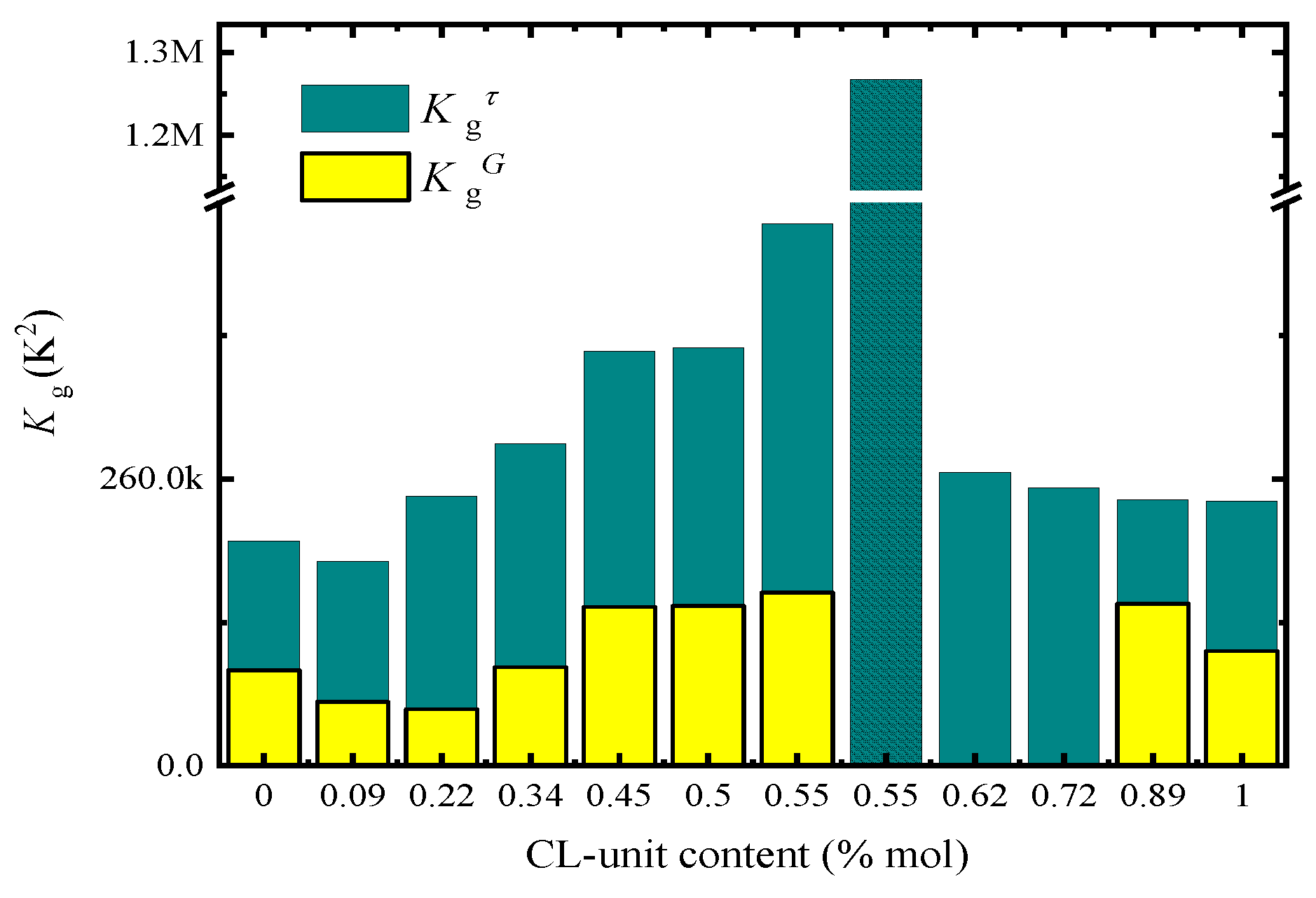
Figure 9 plots both K
g values, obtained by PLOM (
) and DSC (
) as a function of CL-unit molar content. As expected, all
values are larger than
values, as DSC measurements take into account both nucleation and growth, while PLOM measurements considered only growth (see more details in [
37]).
In the case of
values, the trends observed are expected in view of the results obtained in
Figure 6. The energy barrier for crystal growth increased with CL-unit molar content, since growth rate decreased as comonomer incorporation increased. On the other hand, when we analyzed the results obtained for
in
Figure 9, we noticed that there is a clear asymmetry depending on which side of the pseudo-eutectic region the material is. On the PBS-rich side (left-hand side of
Figure 9),
values rapidly increased upon CL units addition. This is expected from the results presented in
Figure 7d, where a large decrease in overall crystallization rate for the PBS-rich side of the composition range can be observed.
In the case of the PCL-rich composition range, we would have expected a decrease in
values with PCL content increases according to
Figure 7d. Instead, we observe in
Figure 9 that the energy barrier for both nucleation and growth does not significantly change with composition (see right-hand side of
Figure 9). We have to remember that for the PCL-rich copolymers the situation is particularly complicated as the nucleation density and nucleation rate increase with CL-unit content but the growth rate decreases. Hence, even though according to
Figure 7d the overall crystallization rate at constant supercooling seems to be dominated by primary nucleation, the values of
are obtained from the slope of the Lauritzen and Hoffman plots that take into account the full range of supercoolings where the measurements were taken. Therefore, it seems that when the overall energy barrier is considered, there is a balance between nucleation and growth which keeps the
values constant with composition.
3.4. Double Crystallization at the Pseudo-Eutectic Point
For the copolymer whose composition is within the pseudo-eutectic point, i.e., BS
45CL
55, we performed isothermal crystallization in a wide range of crystallization temperatures
Tc, to find the temperature region where only one of the phases, PBS or PCL, is able to crystallize.
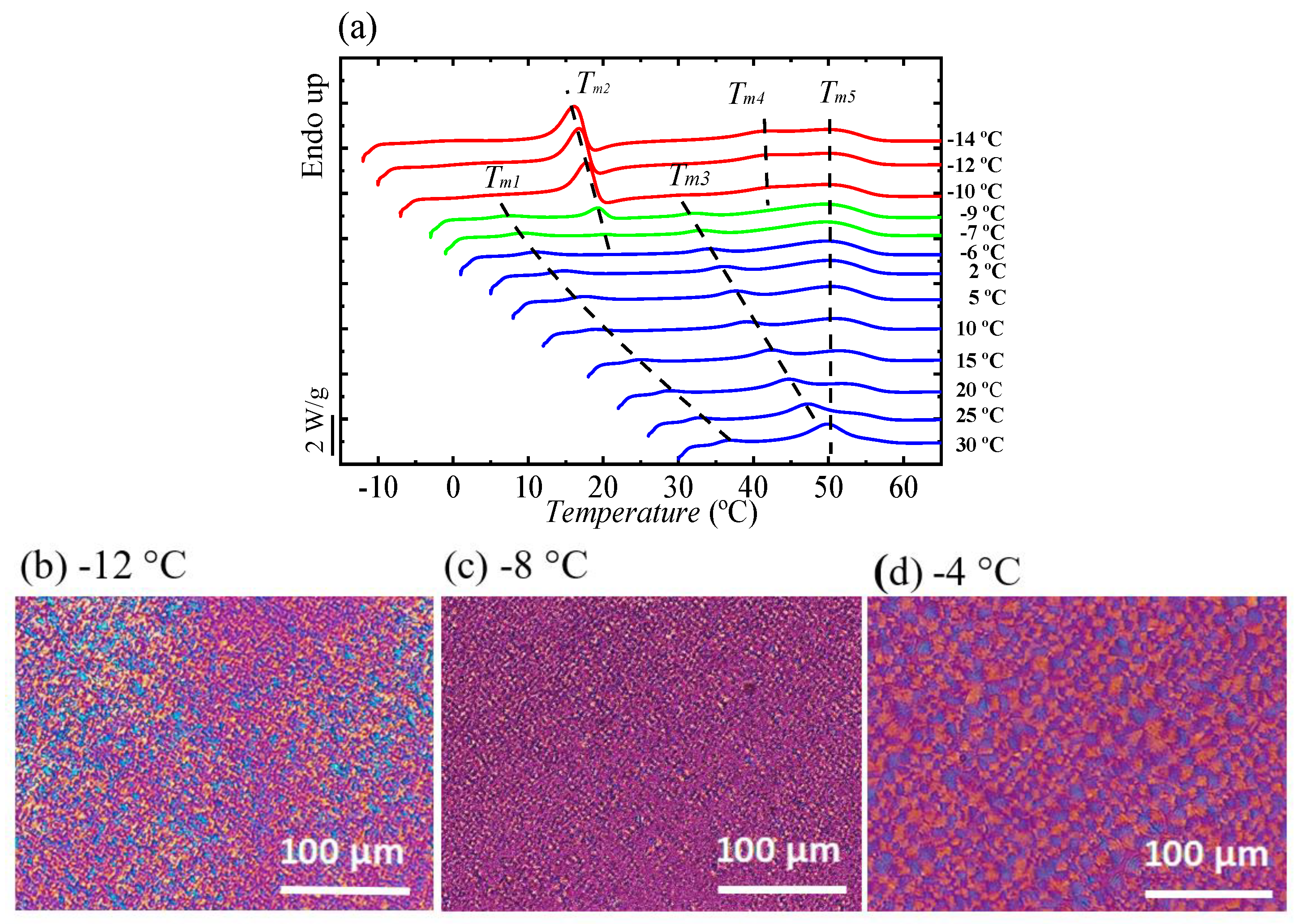
Figure 10 shows the heating DSC scan recorded at 10 °C/min for BS
45CL
55 sample after it was isothermally crystallized at the indicated
Tc values.
At least five different endotherms can be found upon close examination of
Figure 10 and we indicated with dashed lines how these endotherms approximately shift depending on the
Tc values employed before heating the samples. The first melting peak
Tm1 is present in all melting curves and its location is almost at 7 °C higher than the crystallization temperature. This peak has been traditionally regarded as the melting of thin crystals formed during the secondary crystallization process [
41]. The second peak, labeled
Tm2, appeared at
Tc values lower than −6 °C and corresponds to the melting of PCL-rich crystals. The third peak or
Tm3 labeled peak in
Figure 10 highly depends on the isothermal crystallization temperature and corresponds to the melting of the PBS-rich crystals, which were formed during the isothermal crystallization.
In addition, a melting peak (
Tm5) at around 50 °C and another one just below it (
Tm4) were observed. The melting peak labeled
Tm5 corresponds to the melting of PBS crystals that have reorganized during the heating scan, and have a melting point which is almost constant at around 50 °C, regardless of the crystallization temperature [
42,
43]. The
Tm2 and
Tm3 peaks increase almost linearly with increasing
Tc. As shown in
Figure 10,
Tm3 disappeared in the DSC heating curves where the crystallization temperature is less than −9 °C.
The morphologies obtained after isothermal crystallization at three selected temperatures can be observed in
Figure 10b–d. As it will be shown below, WAXS experiments confirmed that at very low
Tc values including −12 °C, only PCL-rich crystals can be formed.
Figure 10d shows small spherulites that were formed at
Tc = −4 °C with spherulites size around 10 µm. At
Tc = −8 °C, where both PCL and PBS crystals can form, there are two crystals sizes, one with 4 µm radii (PBS crystals) and another with around 1.5 µm size (PCL crystals), see
Figure 10d.
Figure 10b shows only PCL crystals with small spherulites size (less than 1 µm) at
Tc = −12 °C.
We performed in situ synchrotron WAXS experiments for the sample at the pseudo-eutectic point, to clarify the temperature range of crystallization of the PBS-rich and the PCL-rich phases and corroborate the assignment of the thermal transitions in
Figure 10. These experiments were performed during isothermal crystallization (for 20 min) at three different
Tc values chosen from three different crystallization regions in
Figure 10.
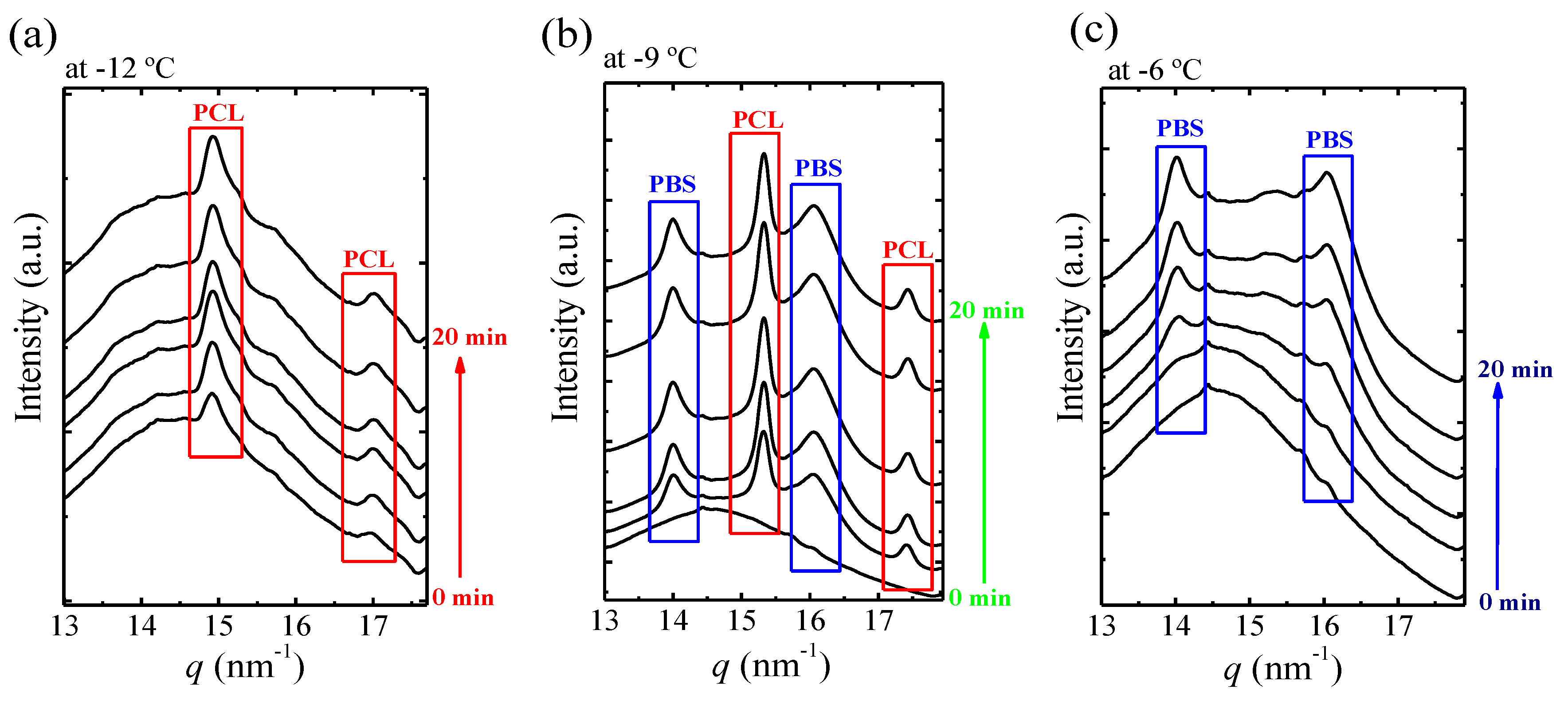
Figure 11a–c shows selected real-time WAXS diffractograms for BS
45CL
55 (i.e., the sample at the pseudo-eutectic point) measured during isothermal crystallization at −12, −9, and −6 °C. If the sample shows characteristic reflections at q = 13.9 and 16.1 nm
−1, they correspond to the PBS (020) and (110) crystallographic planes. If the sample exhibits reflections at q = 15.3 and 17.4 nm
−1, they belong to the PCL (110) and (200) planes [
10].
Changes in the crystallization temperature strongly affect the diffraction pattern at the pseudo-eutectic point. As can be seen in
Figure 11, at −12 °C only the PCL-rich phase is able to crystalize (
Figure 11a) while at −6 °C (
Figure 11c) only the PBS-rich phase crystallizes. On the other hand, at the intermediate
Tc value of −9 °C, both PBS-rich and PCL-rich phases can crystallize. If the DSC curves of
Figure 10 are considered again, the WAXS assignments are consistent with the heating runs after crystallization for all samples crystallized at −9 °C and higher. In the case of low crystallization temperatures, i.e., below −9 °C, it should be noted that WAXS indicate that only the PCL-rich phase can crystallize. The DSC heating runs shown in
Figure 10 also show melting transitions corresponding to the melting of PBS-rich phase. These PBS-rich phase crystals must be formed by cold-crystallization during the heating scan for the samples crystallized at −10, −12, and −14 °C in
Figure 10. In fact, upon close examination of
Figure 10, the end of a cold crystallization process can be observed just after the melting peak of the PCL-rich phase crystals.
Taking into account the WAXS and DSC results presented in
Figure 10 and
Figure 11, the DSC curves in
Figure 10 were plotted with a color code to indicate which phases can crystallize during isothermal crystallization depending on the
Tc values employed. If the
Tc values are −10 °C or lower, only the PCL-rich phase can crystallize, and the curves were arbitrarily plotted in red in
Figure 10a. If the
Tc values are between −9 and −7 °C (including these two temperatures), both the PCL-rich and the PBS-rich phases can crystallize (green curves in
Figure 10). Finally, if the
Tc temperatures are −6 °C and above, only the PBS-rich phase can crystallize (blue curves in
Figure 10).
The pseudo-eutectic sample, BS
45CL
55, exhibits a very interesting phase behavior, as depending on the crystallization conditions, one or both phases can be formed. We have studied previously the nonisothermal crystallization of the same copolymers employed here [
17]. It is interesting to note that under nonisothermal conditions, the cooling rate employed determines which phase can crystallize and also if one or two phases are formed. In this work, on the other hand, we show that one or two phases can be formed depending on the isothermal crystallization temperature chosen. Therefore, the properties of this isodimorphic copolyester with pseudo-eutectic composition can be tailored by varying both nonisothermal or isothermal crystallization conditions, a remarkable and novel behavior, as far as the authors are aware.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}