Modeling of Entangled Polymer Diffusion in Melts and Nanocomposites: A Review
by
 , , , and
, , , and
Argyrios Karatrantos
1,* ,
,
Russell J. Composto
2,
Karen I. Winey
2,
Martin Kröger
3,* and
and
Nigel Clarke
4,* 1
Materials Research and Technology, Luxembourg Institute of Science and Technology, 5, Avenue des Hauts-Fourneaux, L-4362 Esch-sur-Alzette, Luxembourg
2
Department of Materials Science and Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA
3
Polymer Physics, Department of Materials, ETH Zurich, Leopold-Ruzicka-Weg 4, CH-8093 Zurich, Switzerland
4
Department of Physics and Astronomy, University of Sheffield, Sheffield S3 7RH, UK
*
Authors to whom correspondence should be addressed.
Polymers 2019, 11(5), 876; https://doi.org/10.3390/polym11050876
Submission received: 8 March 2019
/
Revised: 6 May 2019
/
Accepted: 9 May 2019
/
Published: 14 May 2019
(This article belongs to the Special Issue Theory and Simulations of Entangled Polymers)
Abstract
:This review concerns modeling studies of the fundamental problem of entangled (reptational) homopolymer diffusion in melts and nanocomposite materials in comparison to experiments. In polymer melts, the developed united atom and multibead spring models predict an exponent of the molecular weight dependence to the polymer diffusion very similar to experiments and the tube reptation model. There are rather unexplored parameters that can influence polymer diffusion such as polymer semiflexibility or polydispersity, leading to a different exponent. Models with soft potentials or slip-springs can estimate accurately the tube model predictions in polymer melts enabling us to reach larger length scales and simulate well entangled polymers. However, in polymer nanocomposites, reptational polymer diffusion is more complicated due to nanoparticle fillers size, loading, geometry and polymer-nanoparticle interactions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
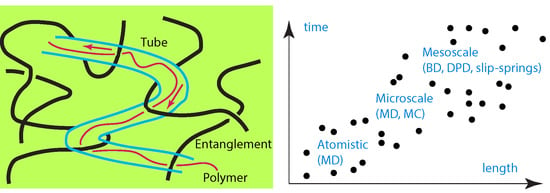
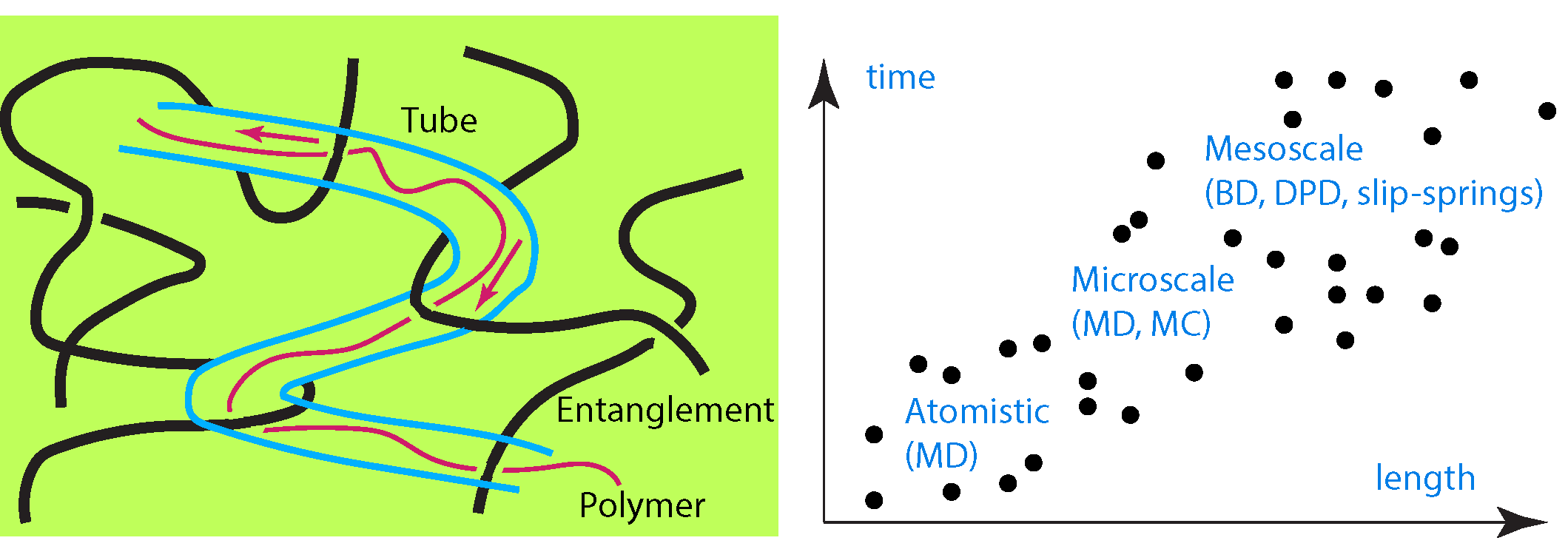
While there are numerous simulation studies investigating polymer structure [1,2,3,4,5,6,7], reinforcement [8,9,10], entanglements [11,12,13,14,15] and their effect on macroscopic properties, such as viscosity [16,17,18], and mechanical properties [19], on the contrary, there are much fewer studies focusing to the simulation of entangled polymer diffusion [20]. The rate of polymer diffusion of entangled polymers is important since it is a critical factor for the quality of polymer blends and nanocomposites. The tube (reptation) model [21,22,23,24] claims that the movement of a long polymer chain is confined to and moves along a tube, which is formed by the entanglements of neighboring chains. Implications of the reptation model have been worked out during the last decades. As an example, the mean square displacement of a Rouse segment within a chain with N segments, segment length b, where denotes the spatial position of the segment, is: for , ∼ for , ∼ for , and ∼ for . Here , , and are the times at which the segment begins to feel the tube constraint, the Rouse relaxation time, and the disentanglement time, respectively. In particular , , , where denotes the number of entanglements per chain, and the entanglement length, which is affected by density and semiflexibility of polymer chain. The self-diffusion coefficient of a reptating polymer chain is according to the tube reptation model, in contrast to the one for a Rouse: or Zimm chain involving the Flory exponent . Taking into account contour length fluctuations, has been revised by Doi [21] as where X is a numerical factor (). Corresponding differences between the viscoelastic properties, relaxation modulus, dynamic light scattering of entangled and unentangled polymer melts have been discussed in detail in the literature [21,25].
In polymer melts, the reptation model has been confirmed and tested by both equilibrium [11,26,27] and nonequilibrium simulations [28,29,30,31], and experiments [32,33,34]. The problem of entangled polymer diffusion [35] with obstacles (nanoparticles) has been addressed by a few studies, but in nanocomposites, there are several parameters that can affect the reptational tube, such as nanoparticle type or loading, confinement, polymer–nanoparticle type of interaction [36]. In this article we do not aim to report and discuss studies that investigate the dynamics of polymers under the presence of obstacles or confinement. Neither is the goal to report in detail, experimental efforts [37] nor the details of different molecular simulation techniques for measuring polymer diffusion. Here is a thorough review of theoretical research and methodologies that have been implemented to address and investigate the reptational linear homopolymer diffusion, in melts and nanocomposites, in comparison to experiments. This review is organized to discuss in order from atomistic to mesoscale and theoretical modeling of polymer melts and nanocomposites (including polymers confined between surfaces).
2. Experiments
2.1. Polymer Melts
Early measurements of the polymer chain self-diffusion coefficient D, by forward recoil spectrometry, in highly entangled poly(styrene) (PS) melts, observed a scaling relation [38], later refined to [39] where M is the molecular weight of a PS molecule. The tracer diffusion coefficients of deuterated PS diffusing into a poly(styrene) matrix of molecular weight P was measured by means of an ion-beam analysis method [40,41]. For matrices with molecular weight 200 kDa, the tracer diffusion coefficients values were independent of P, which is consistent with the reptation mechanism. As the molecular weight P of the matrix is decreased, tracer diffusion coefficient increases due to a constraint release mechanism [40] that arises from the diffusion of the surrounding matrix chains. It was found that the constraint release diffusion behaves as [42,43]. The constraint release is negligible in the measurement of when the matrix molecular weight P is sufficiently large. The temperature dependence of is reflected basically in the monomeric friction coefficient, and is accurately described by the Vogel equation [44]. In other studies of diffusion in PS melts, Antonietti et al. [45,46,47] also verified the Doi-Edwards chain reptation model [22]. The crossover to Rouse-like behavior for short tracer PS chains is at kDa for higher molecular weight matrices and kDa when the tracer and matrix molecular weights coincide [46].
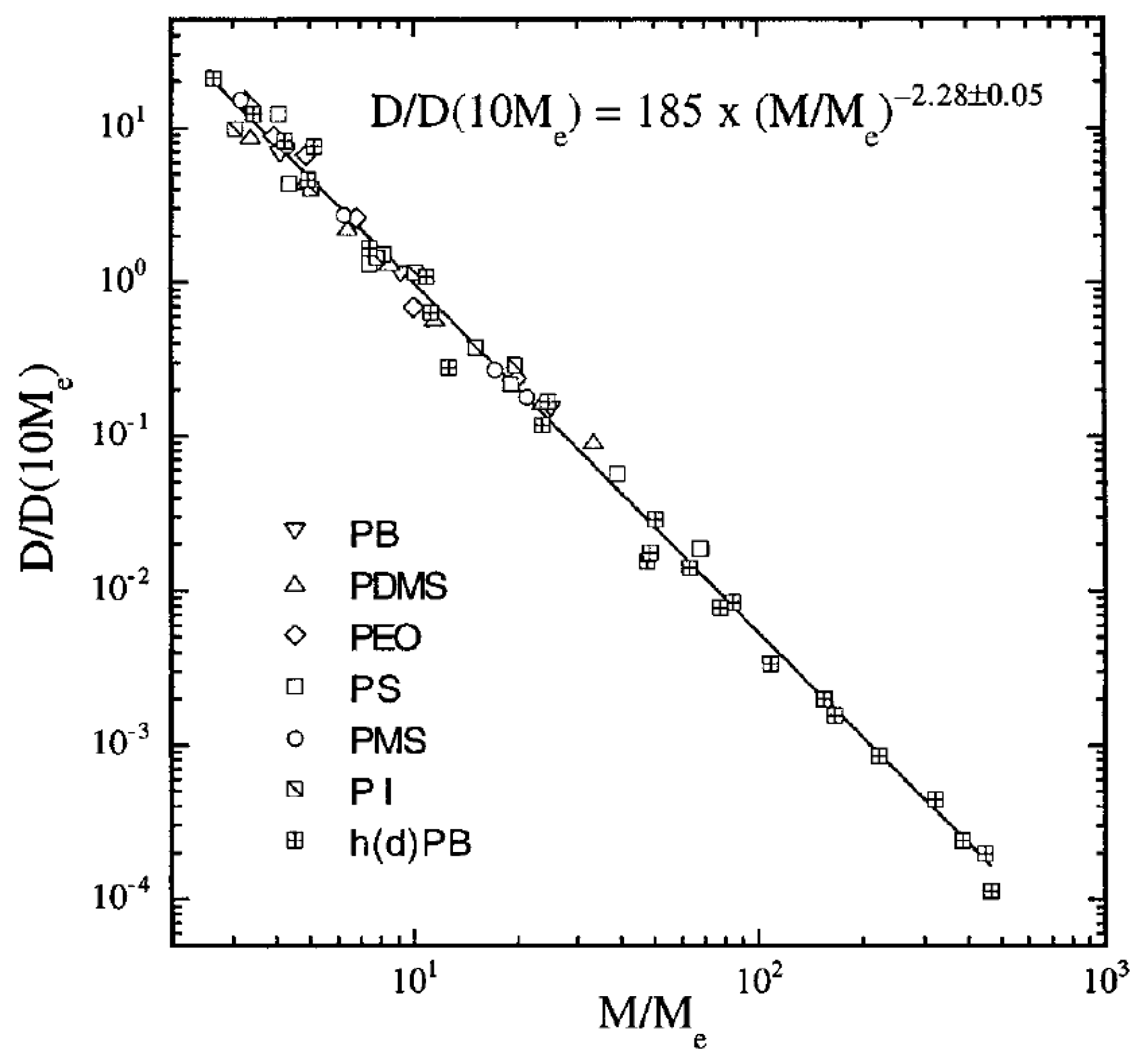
In hydrogenated poly(butadiene) (hPB) solutions and melts, it was found that (Figure 1). Although these results contradicted the widespread belief that in melts (which was observed for the PS tracer diffusion in highly entangled matrices obtained by Green et al. [42], Green and Kramer [43] and Pearson et al. [52]), it was shown that in fact the literature data for seven different polymers are consistent with scaling relation (Figure 1) [39]. Furthermore, the diffusion coefficient D of hPB/alkane solutions was also measured by forward recoil spectrometry. For samples with the data were in accordance to the scaling relation [53]. The above scaling relations and experiments were included and discussed in more detail in the review by Wang [54].
Other experimental methods that have been applied to measure the diffusion of entangled linear polymers such as poly(butadiene), poly(isoprene) (PI), and poly(dimethylsiloxane) are proton multiple-quantum NMR experiments [55] in combination with time-temperature superposition. Such an experimental method can cover a large range of molecular weight (M = 10–200 kDa). In addition, such a method can measure the segmental orientation autocorrelation function which can be compared to tube model theoretical predictions [56]. The disentanglement time was evaluated as slightly below as the tube model predicts due to constraint release and contour length fluctuations [57]. The entanglement time that is consistent with rheological results [58] was evaluated by Trutschel et al. [59]. A time scaling exponent of 0.8 of the segmental orientation autocorrelation function in the Rouse regime was calculated, smaller than the theoretical unity Rouse prediction but in good agreement with Monte Carlo simulations, where the measured exponent was 0.83 [60]. It was also found that which is not in perfect accordance with the tube model predictions. In addition, in that study [59] the self-diffusion coefficients was measured by pulsed-gradient NMR [51,61] and the power law exponent was consistent with earlier studies [49,62]. Moreover, in bidisperse mixtures of 1,4 poly(butadiene), the diffusion coefficient of longer chains decreases by either increasing the chain length of short chains or reducing the weight fraction of short chains in the mixture [62]. These microscopic insights are compared in this review with results from computer simulations and theoretical approaches [59].
2.2. Nanocomposites
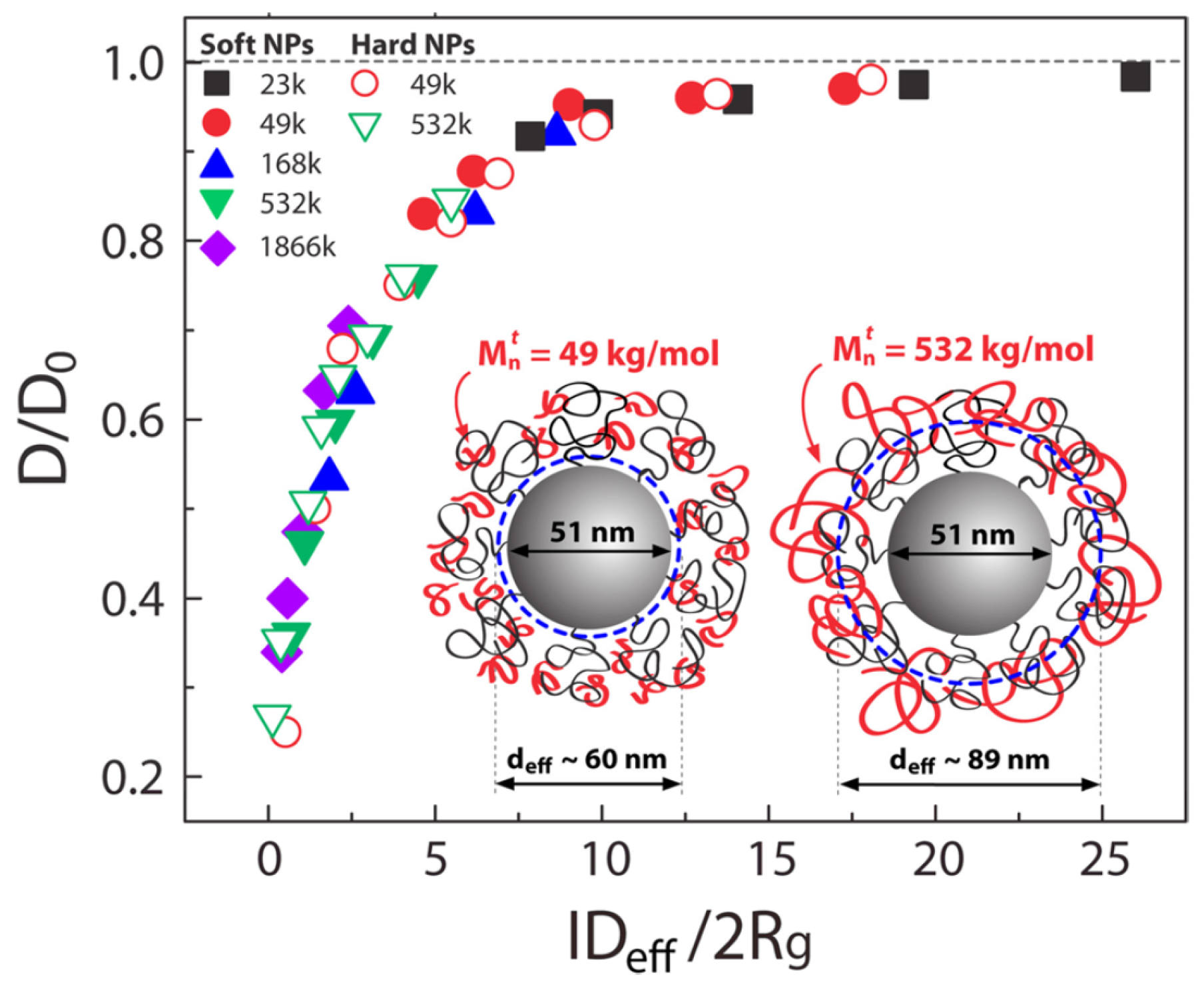
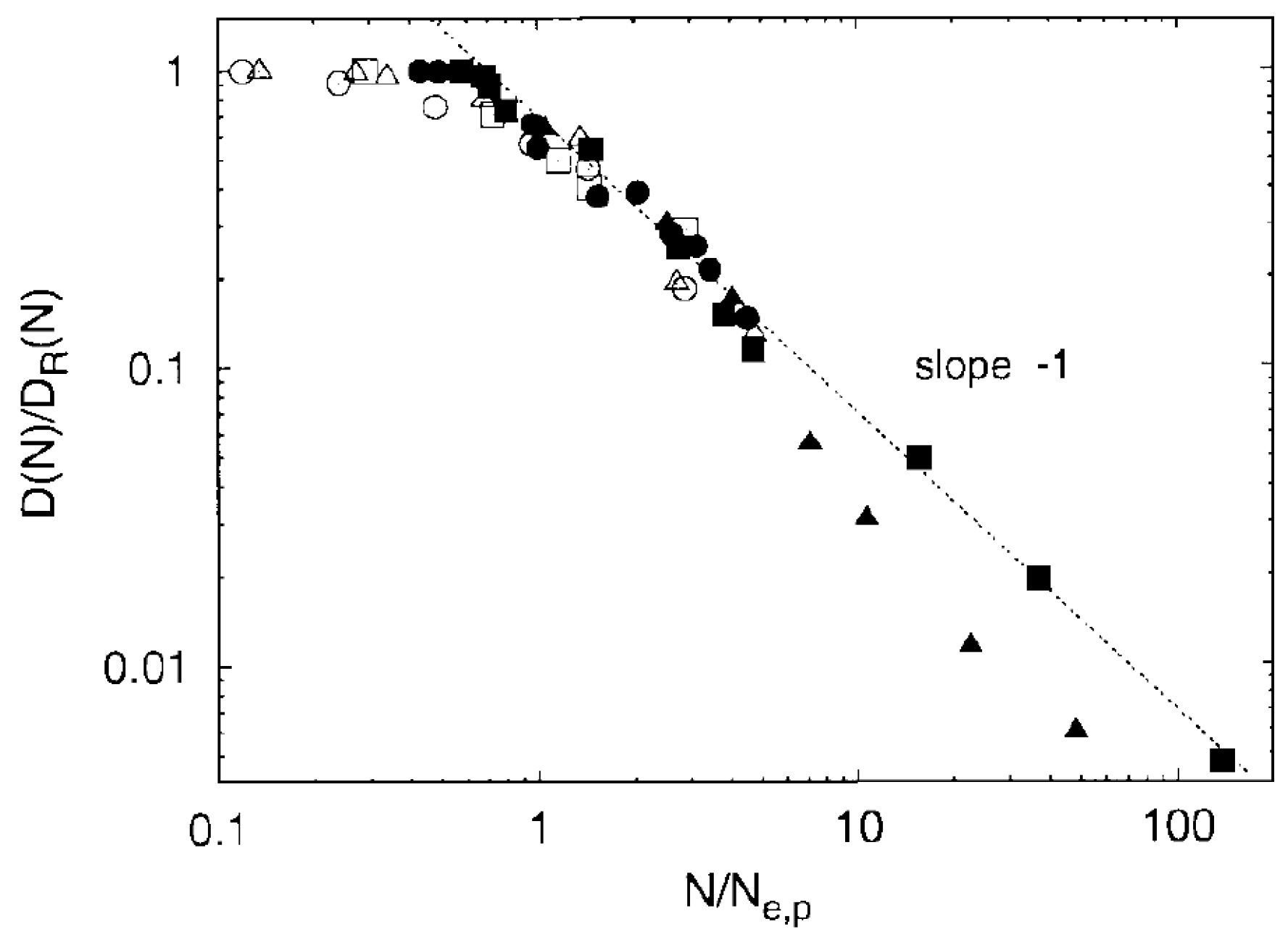
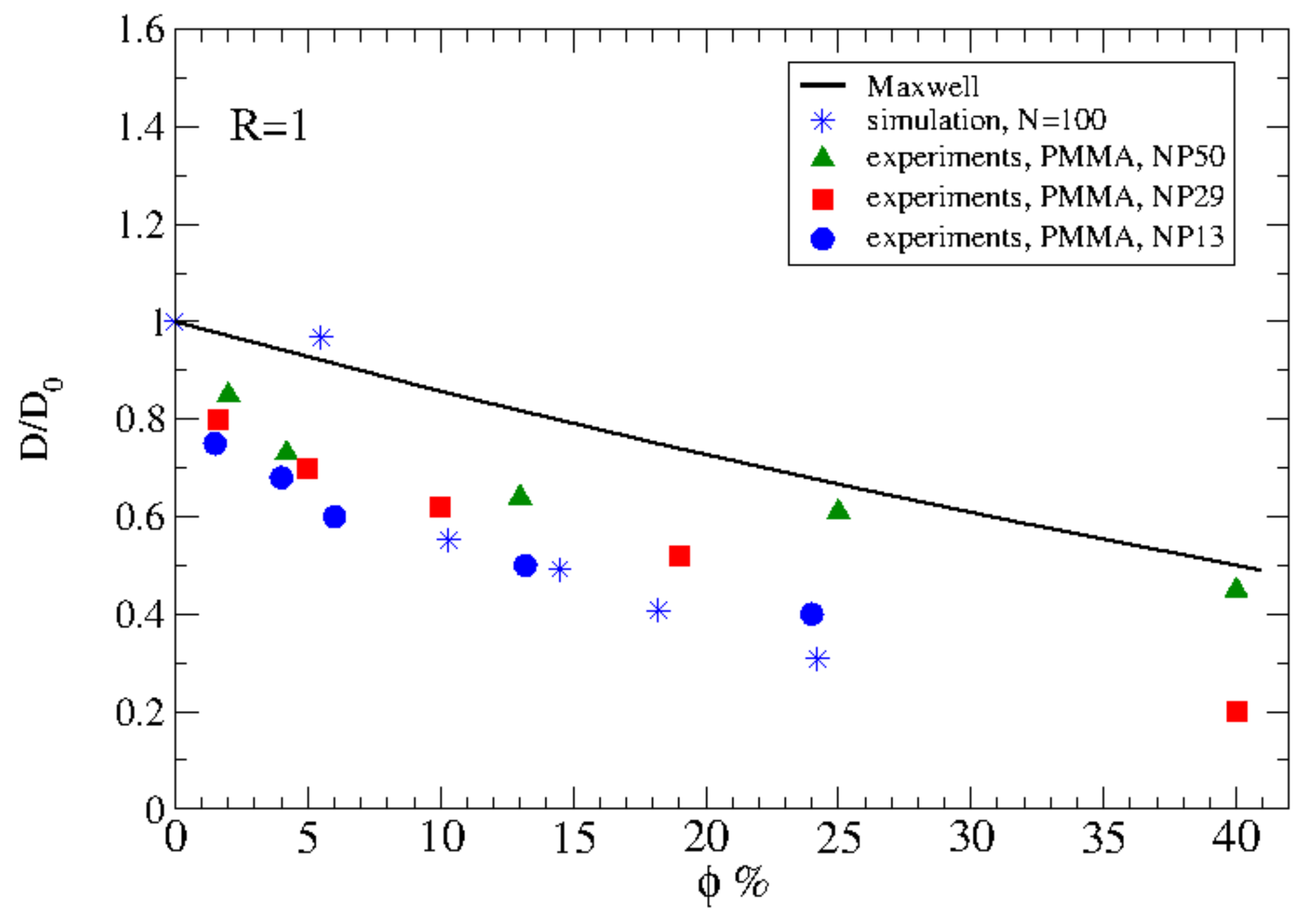
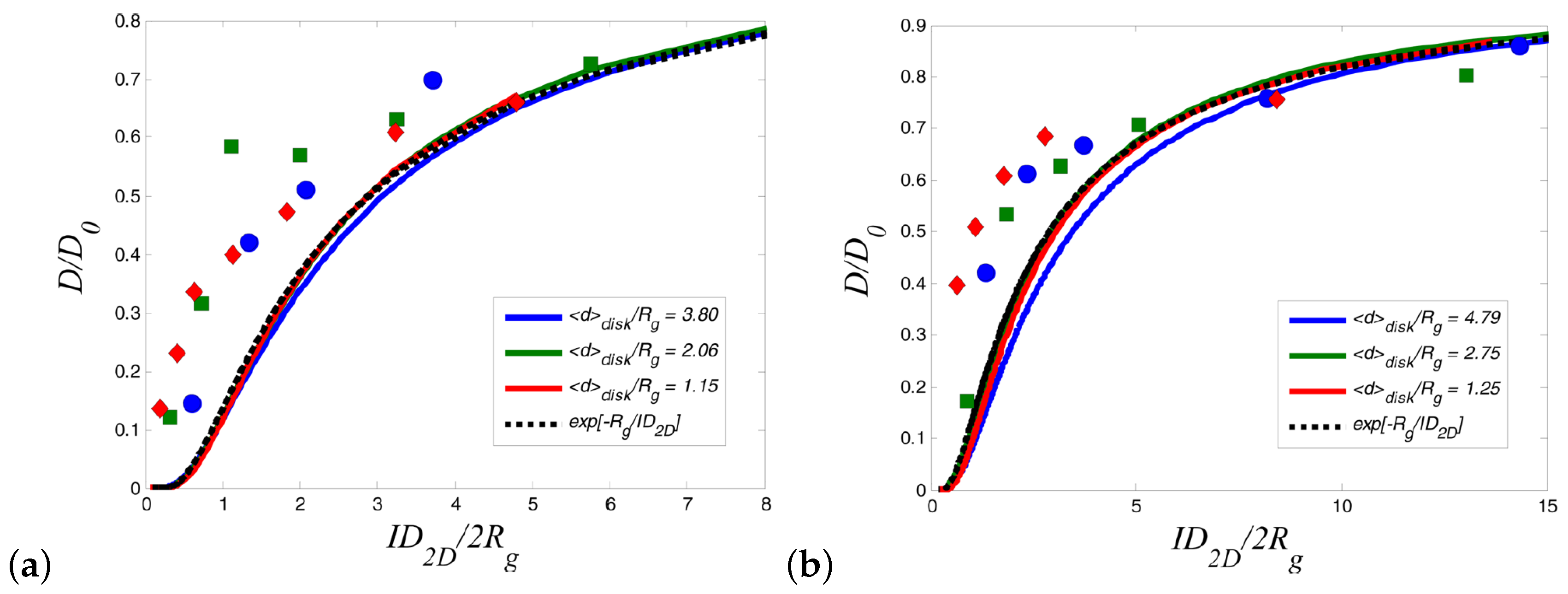
Detailed information regarding the experimental studies of polymer diffusion in nanocomposites can be found in the reviews by Lin et al. [37] and Schneider et al. [63]. In particular, we refer below the main studies regarding polymer diffusion in nanocomposites containing either spherical (such as nanosilica or gold nanospheres) or anisotropic nanoparticles (such as carbon nanotubes or TiO nanorods). The deuterated entangled poly(methyl methacrylate) (PMMA) or PS diffusion was decreased as a function of nanosilica loadings and the normalized diffusion coefficient to the bulk value as a function of the confinement parameter collapses into a master curve (Figure 2). Here, , where is the interparticle spacing, approximates the distance of closest approach for a tracer molecule in the brush and is the tracer radius of gyration. Choi et al. [64] studied the diffusion of deuterated PS in the presence of soft nanoparticles (silica nanoparticles with PS tethered chains) and produced the same curve as that of the hard nanosilicas composites [65,66] where collapsed the reduced polymer diffusion data for isothermal measurements which was considered evidence of a mechanism related to entropy. However, this collapse of breaks down as a function of temperature, and the data does not reduce as well as seen in Figure 2 explaining that mechanism of diffusion is different. In particular, for constant , D is more perturbed from bulk at higher temperatures [67].
For small and mobile NPs in an attractive polymer melt [68], was more perturbed than in nanocomposites with larger and immobile NPs. Furthermore, the mechanism for slowing the tracer polymer diffusion was related to slowed segmental dynamics (friction) rather than excluded volume or entropy as shown by Tung et al. [67]. In addition, Mu et al. [69] also investigated the diffusion of deuterated PS in PS/single-walled carbon nanotubes (SWCNT) composites and found approximately an order of magnitude decrease with SWCNT below percolation concentration. Beyond percolation the deuterated PS diffusivity increased and then recovered its bulk value. A similar behavior of the deuterated PS diffusion had also been observed for the case of PS–fullerene nanocomposites [70].
In graphene nanocomposites, the diffusion of poly(methyl methacrylate) (PMMA) confined between graphene oxide (GO) sheets was measured by the neutron reflectivity (NR) technique [71,72]. It was shown that the confined PMMA diffusivity was reduced by more than an order of magnitude if the thickness of the film was less than (where is the polymer radius of gyration in bulk), whereas the diffusivity of confined poly(styrene) (PS) was reduced by 3 times in comparison to bulk diffusivity, as the film thickness decreased from to . That discrepancy was due to PMMA-GO interaction [71]. Furthermore, the self-diffusion of confined poly(butadiene) chains in alumina channels (100 µm long, 20 and 60 nm wide) was investigated [73] by proton pulsed-gradient NMR. A reduction of the diffusivity appeared depending on the pore diameter but not on the molecular weight for short polymers (molecular weight between 2 and 24 kDa). This behavior was rationalized in terms of the monomeric friction coefficient, which suggested a 10-times enhanced friction due to the surface in a single molecular layer [73]. Moreover, by using elastic recoil detection, PS tracer diffusivity along the cylindrical aluminim oxide membranes was measured and an increase in comparison to the bulk value was observed as the confinement increases, in agreement with other studies [74].
3. Atomistic Simulations
3.1. Polymer Melts
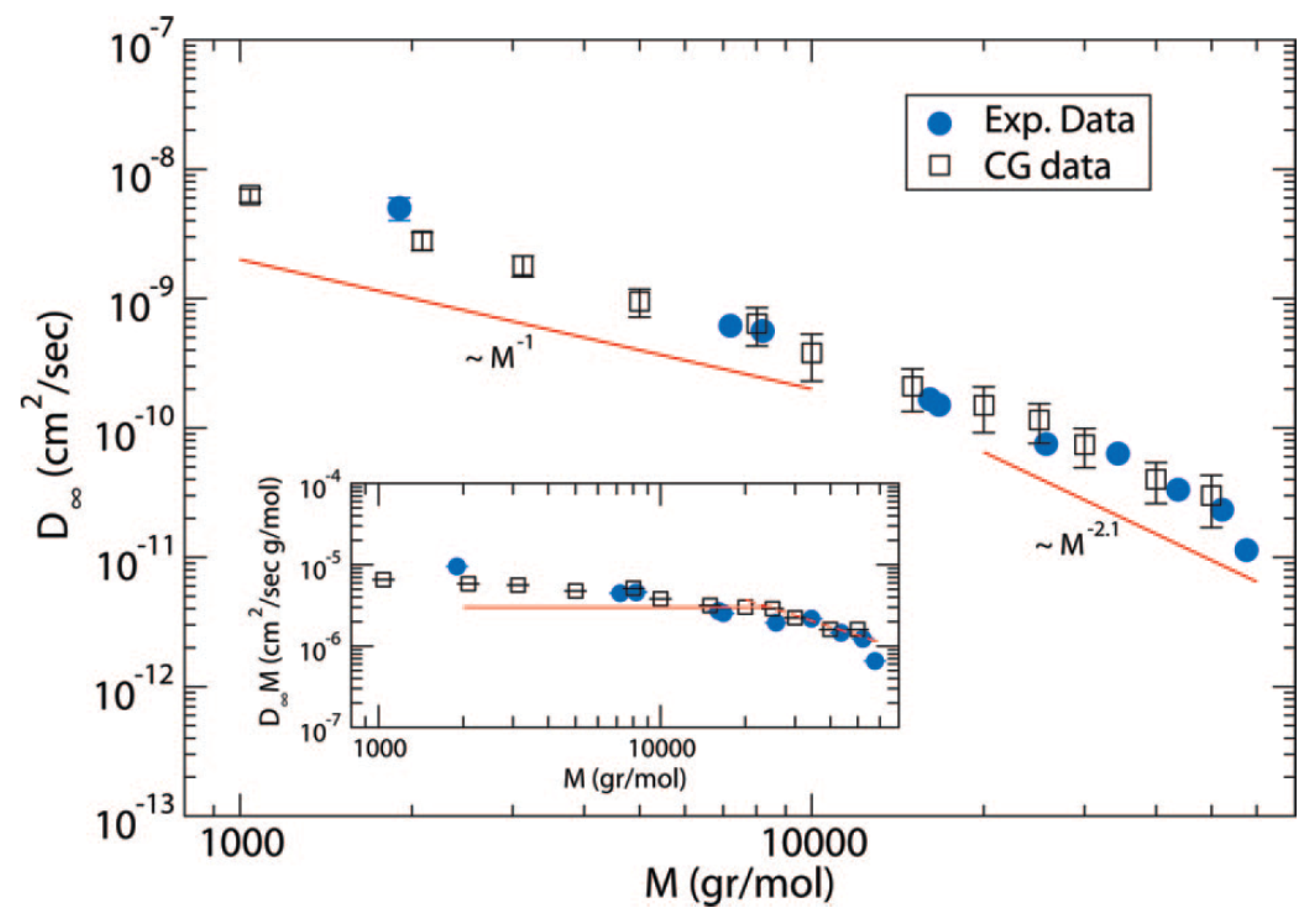
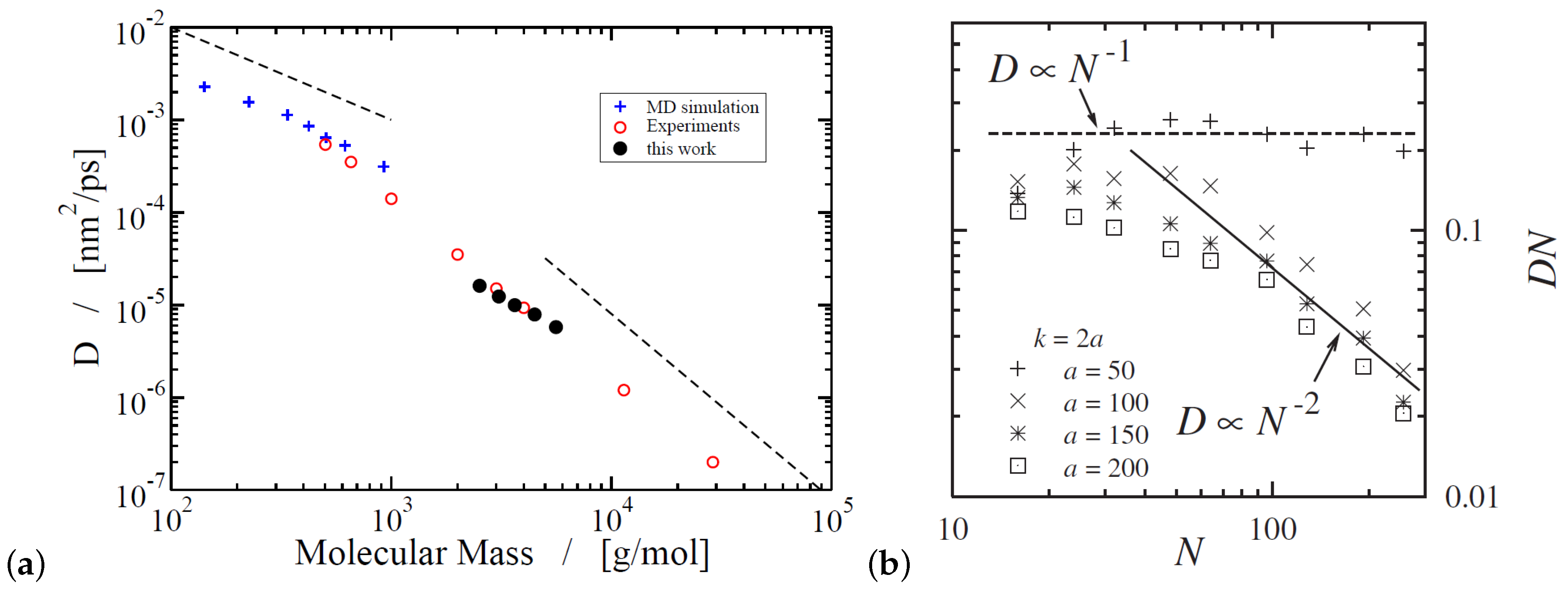
Atomistic simulations of polymer diffusion remain very computationally demanding. However, one of the very few efforts is the long (ms time scale simulations) atomistic molecular dynamics (MD) simulations of monodisperse linear poly(ethylene oxide) (PEO) melts, of molecular weight up to 20 kDa. The self-diffusion coefficient obtained, by using a united atom force field, was compared quantitatively with experimental data from neutron spin echo and pulse-field gradient NMR [75]. It was shown that the PEO diffusion coefficient (with kDa) scales as , the same as for poly(ethylene) (PE) melts [76] for chains up to monomers in which the dynamic structure factors from simulation data agreed well with neutron scattering experiments [76].
Regarding the PE melt, atomistic MD simulations were performed with a number of monomers from to . Above , the mean square displacement (MSD) and dynamics structure factor of inner segments showed a change from a Rouse towards a reptation-type behavior [78,79,80,81] with a predicted scaling relation of different than the one reported by Hur et al. [76].
A similar effort for PE with larger number of monomers () was performed by Ramos et al. [82]. The results from simulations regarding the friction coefficient , the zero-shear rate viscosity and the self-diffusivity were in agreement with experiments [27]. Another MD study of a united atom force field for PE was applied by Takahashi et al. [83]. The scaling laws for representative polymer properties, such as diffusion, viscosity, relaxation time were compared to theoretical predictions [83] as can be seen in Figure 3. Atomistic simulations of cis-1,4-poly(butadiene) (PB) melts, in high molecular length of monomers, by implementing a united atom force field, were performed for times up to 600 ns [84]. Dynamic properties, such as segmental and terminal relaxation, self-diffusivity and the single-chain dynamic structure factor, , at atmospheric pressure in a temperature range of 298–430 K were calculated [84]. The data showed that, around monomers, D changed to a reptation-like (where ) behavior at 413 K.
3.2. Polymers Confined between Surfaces
In nanocomposites there is a very limited number of polymer diffusion studies by using atomistic simulation (through implementation of a united atom force field) due to the computational demand. In particular, dynamics of confined PE chains between graphite walls was studied by Kritikos et al. [85]. The molecular weight of the monodisperse PE chains was high enough up to the entanglement regime. Three cases of distances between the walls, (2, 3, ), of PE chains were studied. By conducting MD simulations (at temperature K)—after equilibration by Monte Carlo (MC) algorithms—the relaxation time distribution was a function of distance from the graphite walls. The diffusion of PE chains in the middle layer was not inhibited by the presence of the adsorbed PE layers [85].
4. Coarse-Grained Simulations
4.1. Molecular Dynamics
4.1.1. Polymer Melts
One of the first studies in microscale simulations by MD was implemented by the simple off-lattice polymer model (freely-jointed tangent hard-sphere chain) in order to perform long time simulations to probe the entangled polymer dynamics. The highest chain lengths that were studied was segments for a volume fraction between = 0.3–0.45 [86,87,88]. The inner segments of the mean square displacement (for longest chains) followed the three different scaling regimes of the tube model. In addition, an anomalous diffusive behavior in the atomic MSD of the inner segments was observed as the inner segments cross over into the free diffusion limit [86,87]. Shanbhag [89] studied monodisperse polymer melts via the bond fluctuation model () and evaluated the diffusion coefficient D of chains using the so-called BOOTDIFF method [90], more generally useful for systems with nondiffusive short-time behavior. They obtained diffusion coefficients for , 75, 150 however, they had too few data points to extract a scaling exponent.
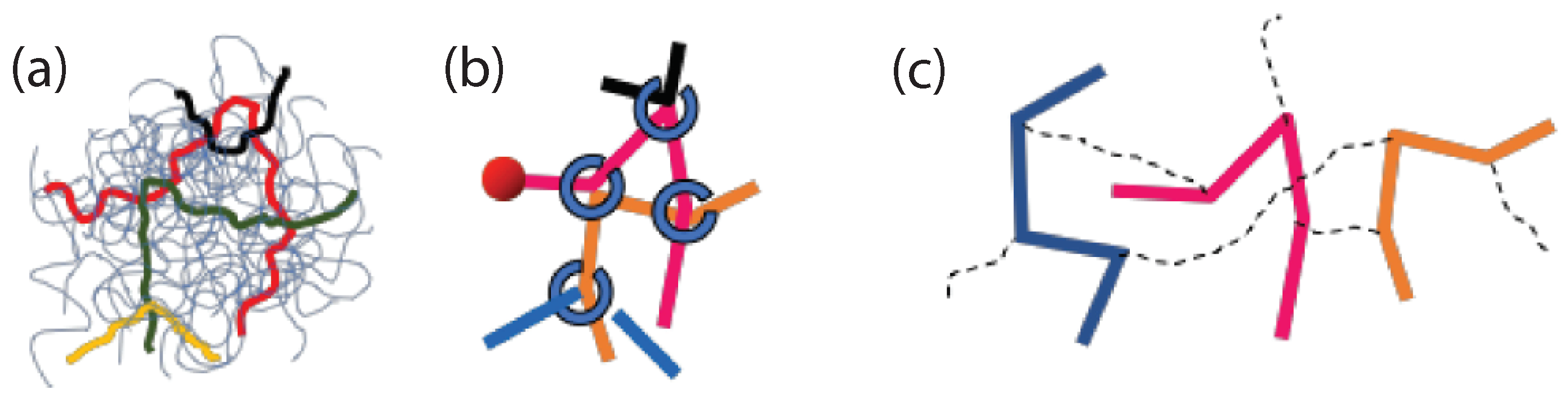
In microscale simulations there are numerous studies that use the Kremer-Grest model [26] to investigate polymer dynamics and eventually diffusion. The inclusion of the attractive part of the non bonded Lennard Jones (LJ) potential, between monomers of the chains, had insignificant effect on the chain mobility for a temperature larger than twice the glass transition temperature [94]. However, for lower temperatures the attractive part of the LJ potential reduced polymer diffusion [94] in comparison to that predicted by the Kremer-Grest model (which contains only the repulsive part of the LJ potential) [26]. The search for the reptation power laws [35] as predicted by reptation tube theory was rather difficult [83,94,95,96,97,98,99,100]. In particular it was shown that, by using the Kremer-Grest [26] model, it was difficult to identify any clear power-laws, at least for mildly entangled systems [96] in contrast to the more entangled polymers () studied by Pütz et al. [92] (Figure 4). The predictions of Doi and Edwards, regarding the four different power laws [101] present in mean square chain of mass displaments, tube diameter, and the time scales could be determined from the intersections of the power law fits.
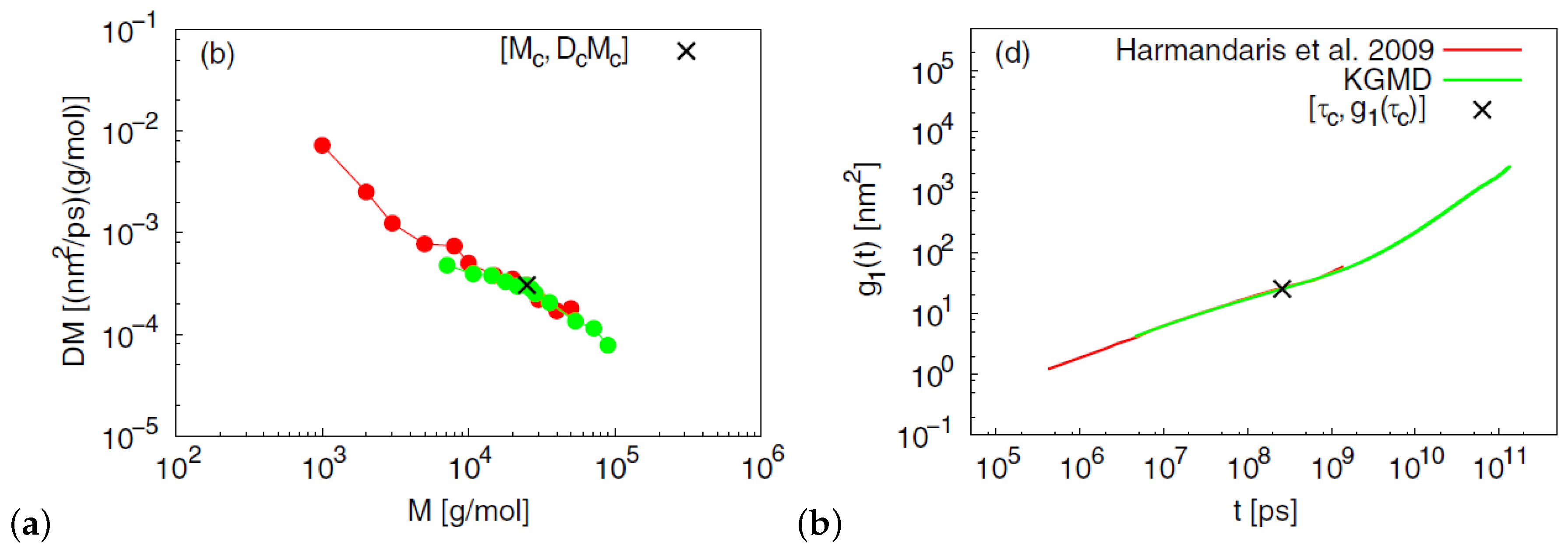
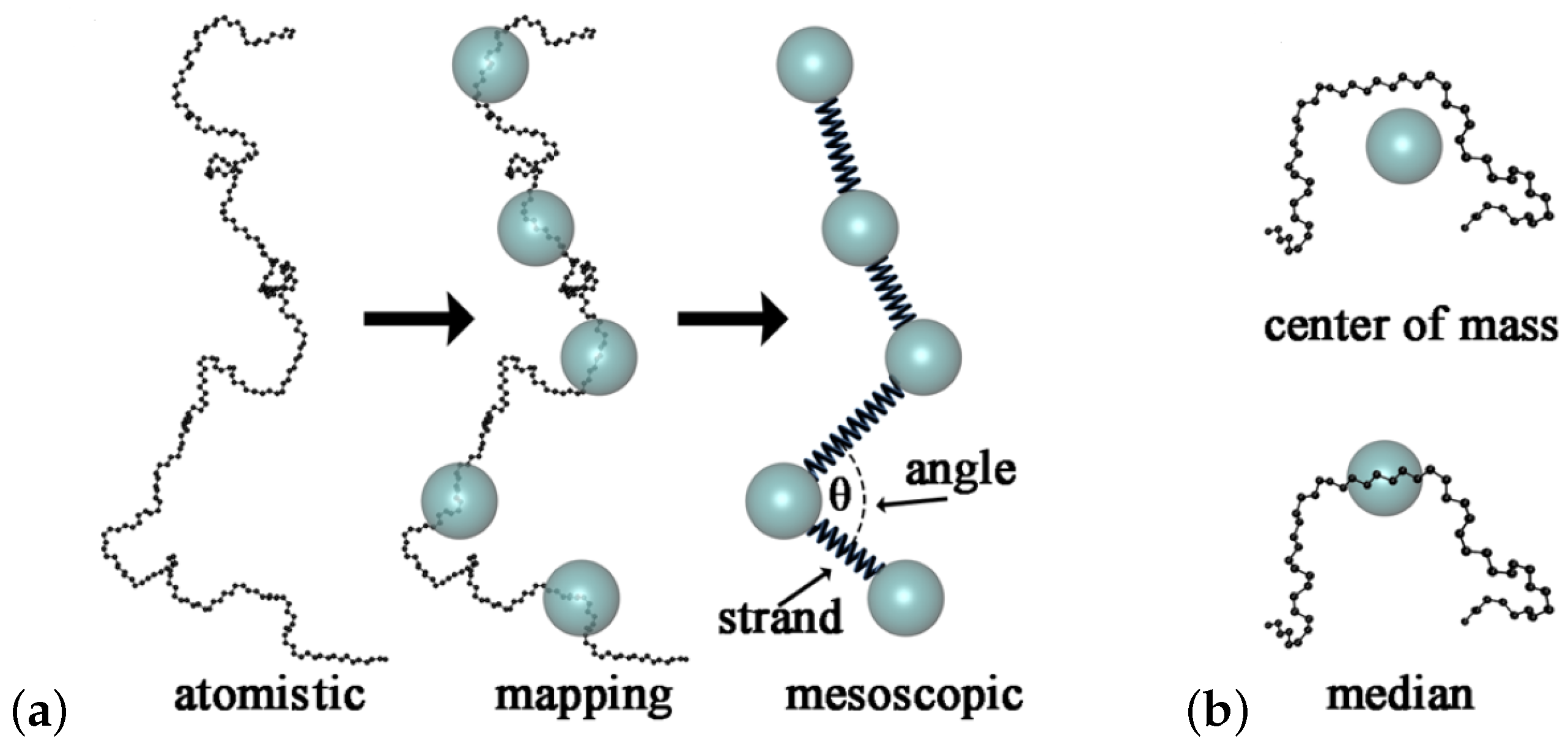
A hierarchical approach that combined both atomistic and coarse-grained dynamic simulations of entangled PS melts was studied by Harmandaris and Kremer [93] in order to capture dynamical and rheological properties. The time mapping constant was determined by comparing atomistic and coarse-grained simulations for oligomers. The chain self-diffusivity in reptation regime, after correcting for the chain end free volume, were predicted in comparison to experimental data [93] (Figure 5). In another coarse-graining effort, a mesoscale PS model was developed using the iterative Boltzmann inversion (IBI) approach [102] in order to predict the polymer dynamics of long chains. The potential was optimized, according to IBI, until the radial distribution function generated from the mesoscale model was consistent with that produced by the atomistic simulation of oligomers. Mean-squared displacements measurements captured the crossover dynamics from the Rouse to reptation behavior. In addition, the entanglement length of that PS mesoscale model was around 85 monomers at K in agreement with that of the bead-spring Kremer–Grest model [103].
Furthermore, CG models of PE were developed by the IBI approach with three to six methyl groups per CG bead. It was discussed that pressure corrections were required after the IBI for the generated CG potentials to match the pressure of atomistic melt and transfer to the CG potentials for modeling different temperatures. However, mean-square displacements (MSDs) and stress autocorrelation functions for PE melts were independent of the use of pressure-corrected potentials by the IBI. The time rescaling factor to match CG and atomistic models was the same for pressure- and non-pressure-corrected CG models, but depended on temperature [104]. In another coarse graining effort of polymer melts, the time and length between CG and atomistic models, using a generic mapping scheme based on power laws, were estimated by Takahashi et al. [77]. That scheme revealed the characteristic length and time between the different scales to link the atomistic PE model and the bead-spring Kremer–Grest (KG) model. That mapping procedure between the PS CG model and the KG model enabled measurement of polymer dynamics up to the subsecond time scale [77]. Furthermore, a coarse-grained poly(vinyl alcohol) (PVA) model with a triple-well bending potential has been developed to predict polymer dynamics, structure and crystallization during cooling [105]. However, it is an open question if such developed CG models are applicable at other temperatures. An interesting method to develop a transferrable coarse grained potential is the energy renormalization method, that has been implemented for PS and PB melts [106,107].
Polymer Polydispersity
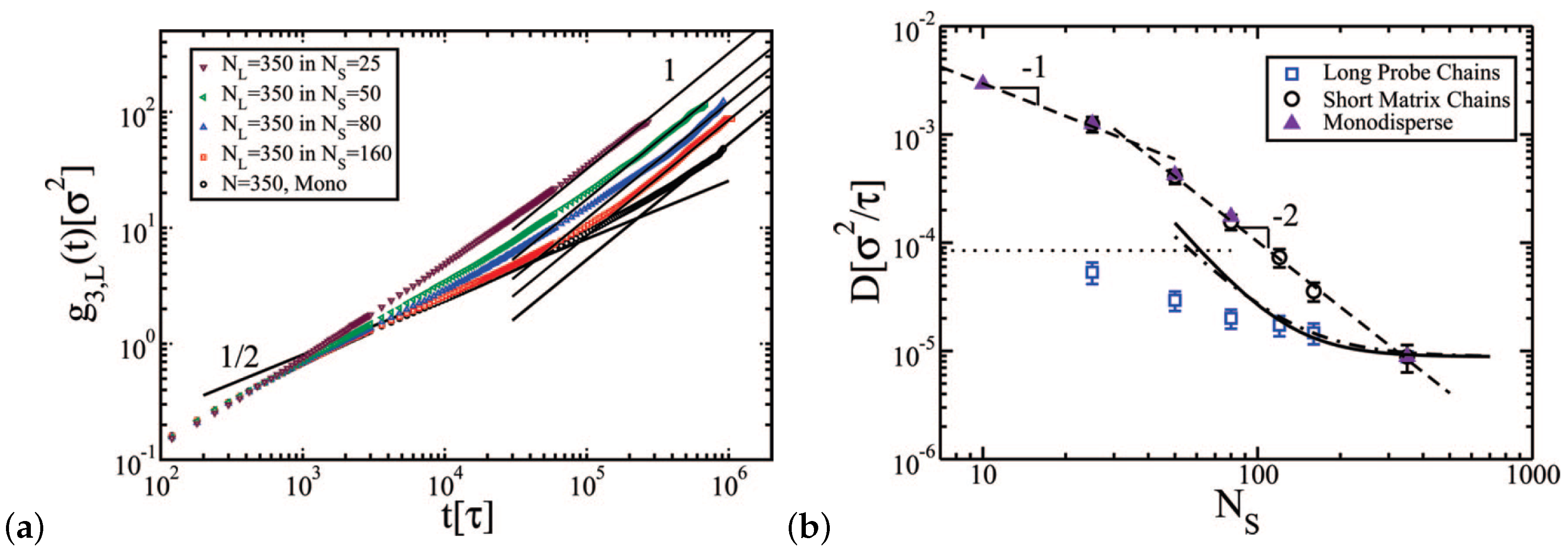
Most computer simulation studies of entangled polymers had been performed on monodisperse samples. However, polymer synthesis always results in a distribution of molecular weights with PDI = –. The effects of polydispersity on entangled PE dynamics melts were studied recently, using a coarse-grained model by Peters et al. [104]. Entangled PE melts with chain lengths of average kDa with PDI = 1.0–1.16, were studied for long scale times (600–800 ms) using MD simulations. It was found that polydispersity in that range did not alter the entanglement time or tube diameter [108]. There was a negligible difference in the PE dynamics for the distributions PDI and PDI , however, the shortest chains diffused faster than the longest ones due to a constraint release mechanism [108]. It is worth to note an atomistic simulation effort (using a united atom force field) of polydisperse linear poly(ethylene) melts with , showed a Rouse-scaling of for chains above and a signature for the onset of a reptation regime at [109]. In addition, a detailed study of bidisperse polymer melts had been performed by Wang and Larson [80]. The diffusion coefficient of long chains can be given from two different relations, Equations (1) and (2).
The mean square displacement and the linear diffusive regime of long polymer chains in the bidisperse melt are depicted in Figure 6a; diffusion coefficients are reported in Figure 6b. The ratio of polymer diffusion coefficient of long chains in a bidisperse sample to the diffusion coefficient of long chains in a monodisperse sample can be predicted by mesoscale simulations [110] in agreement with experiments by Wang et al. [62] for poly(butadiene) (PB) chains for volume fractions of long chains and .
Polymer Semiflexibility
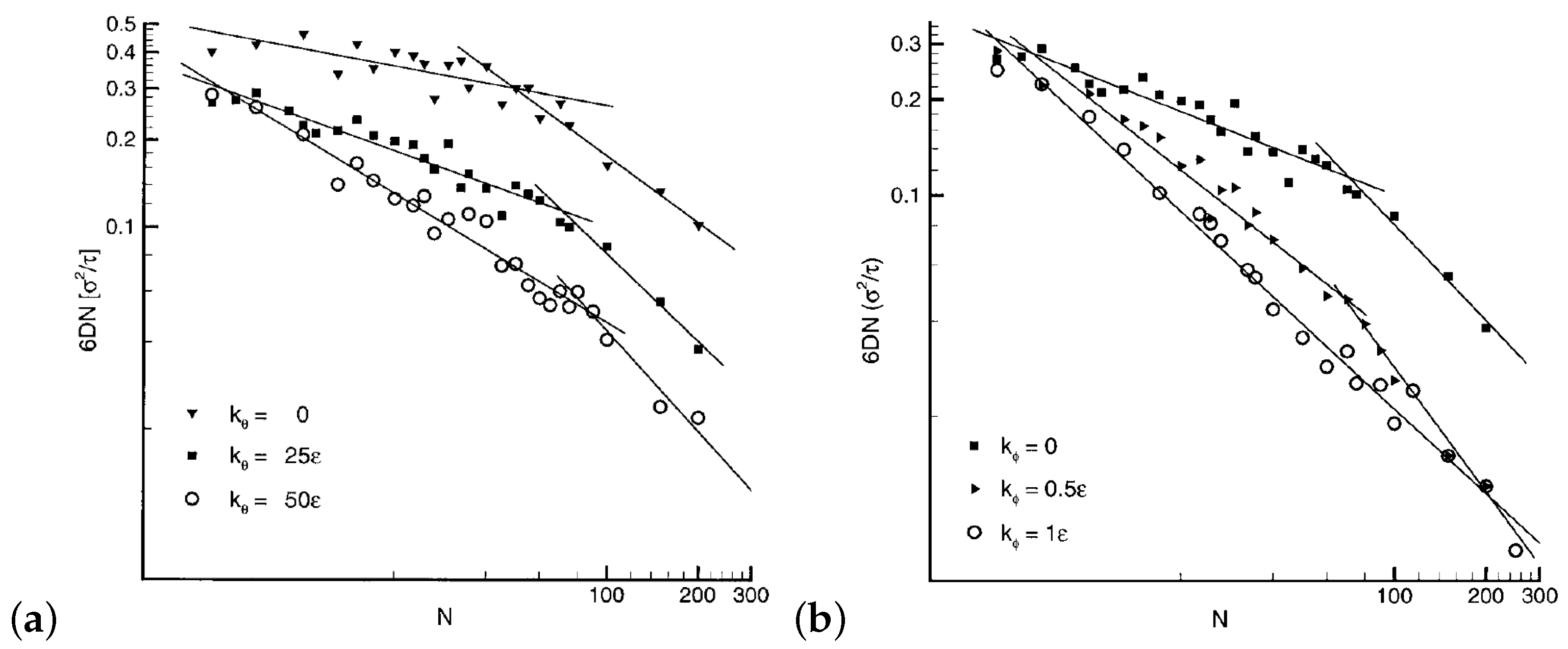
It has been shown by simulations that the semiflexibility of chains (through the insertion of a bending potential along the backbone of the polymer model) reduces polymer diffusion [111,112], since it increases the Kuhn length [113] and packing length which unavoidably decrease the entanglement length [11,114]. The polymer self-diffusivity decreases when the bending and torsion potentials increase (thus the semiflexibility increases) as can be seen in Figure 7. In addition, semiflexibility moves the transition from Rouse to reptation regimes [81,115] to longer chains. In the reptation regime the power law scaling exponent, of diffusion coefficient, becomes for the cases , or , (whereas the exponent becomes for , ) [111] in agreement with experimental [39,53], theoretical [116] and simulation predictions [117].
In a recent study, the dynamic scaling power laws of long semiflexible polymers are investigated [98]. The relaxation times in the Rouse and reptation regime is extracted by the mean square displacements, , , in quantitative agreement with theoretical predictions. The MSD data for long semiflexible chains follows exactly the reptation theory [98]. As can be seen from Figure 7, the exponent of the diffusion molecular weight dependence, in the reptation regime, is influenced by the semiflexibility of the chains and reaches the value of −2.2 for certain stiffness [111].
A higher temperature leads to an increase of the polymer chain diffusion coefficient, independent of chain stiffness [112]. However, the chain diffusion coefficient dependence on chain length is influenced strongly by chain stiffness and temperature [112]. By increasing the stiffness, or decreasing the temperature, the exponent of the chain length dependence decreases approaching the value for unentangled and mildly entangled chains [112]. Furthermore, by adding strongly attractive nanoparticles in a mildly entangled matrix, it is shown that a semiflexible polymer may diffuse faster than a flexible one, following a mechanism of nanoparticle’s attraction weakening with chain semiflexibity [118].
4.1.2. Nanocomposites
Mildly entangled polymer diffusion in nanocomposites containing spherical nanoparticles [119,120] or nanorods [121] was studied by MD simulations recently. It was shown that the polymer diffusivity decreases due to nanoparticle loading, as can be seen in Figure 8, due to an increase of the interfacial area created by the nanoparticles [119,120]. That recent work was the only one that has been performed for mildly entangled polymers diffusion in nanocomposites by molecular dynamics, while the work of Desai et al. [122] showed a similar behavior with nanoparticle loading for polymers with number of monomers just below the entanglement length.
A few studies have been implemented regarding diffusion in polymers confined between surfaces. In particular, long polymer chains disentangle under the influence of the confinement [123]. It is shown that there is a minimum in the relaxation time (calculated by measuring the correlation function of the end-to-end vectors, [124]) of long chains when decreasing the film thickness, which partially originates by the disentanglement of chains due to the confinement [123,124]. In a recent study of polymers confined in cylindrical geometry the number of entanglements decreases with cylindrical confinement in comparison to thin films [123]. The theoretical prediction of polymer diffusivity in a cylindrical geometry is in excellent agreement with the coarse grained simulations [125,126,127] however, it overpredicts the experimental measurements of PS in alumina oxide membranes [126]. As the diameter of the cylindrical confinement decreases, the chain diffusion coefficient increases (in comparison to bulk) due to chain disentanglement, however, for small pores (<5 times of the monomer segment), diffusivity decreases due to chain segregation [127]. In another recent work with a diamond network confinement it is also shown that polymer diffusivity increases in comparison to the bulk value due to the disentanglement that occurs from the nano-confinement [128], similar to the conclusion by previous studies [123,124].
4.2. Monte Carlo
4.2.1. Polymer Melts
In addition to the molecular dynamics studies of polymer diffusion there are quite a few studies implemented by Monte Carlo simulations. Rubinstein [129] proposed a “discretized” reptation model. The tube was modeled by a one-dimensional lattice and the polymer was modeled by a cluster of walkers, called reptons. Each repton represented a part of the chain and was allowed to hop between neighboring sites, but the cluster always remained connected. The repton model predicted that diffusion coefficient scaled as and viscosity scaled as [129]. The dynamics of a polymer in a network of entanglements was also studied by Deutsch and Madden [130]. The diffusion coefficient D for chains up to 100 links, scaled as , where L was the chain length. This result was different than the scaling predictions based on the tube model in which the power law dependence follows the scaling. In that particular work [130], they calculated a three-dimensional diffusion coefficient (previous work used an one-dimensional coefficient [129]). It was noted that the tube length fluctuations in the reptation model were necessary to make a direct comparison to experimental data [130] whereas were neglibile for the one-dimensional coefficient.
The crossover region from the unentangled dynamics to reptation dynamics [131,132] that had been observed from MD, was also verified by using the bond fluctuation model [133,134] in MC simulations. In particular, a study of the cubic lattice model by MC simulations (that contained both excluded volume interactions and entanglements) for polymer volume fractions in the range and chain lengths in the range , the crossover from Rouse behavior to reptation [135] was also observed. MC simulations for monodisperse linear polymer chains in dense melts with number of monomers to were implemented and standard mean-square displacements of inner monomers and of the chain’s center of mass were calculated [133,134]. The analysis revealed that the crossover from unentangled to entangled dynamics was very lengthy [136]. Even for the largest chain length of , no clear evidence for reptational diffusion [132] was shown. The scaling relation of which was very close to the experimental prediction, was predicted over a range of melt densities [137]. A scaling relation of was calculated for PE melts [138] and the start of crossover to reptation regime was observed for monomers. A different lattice Monte Carlo algorithm for bulk polymer melts had been developed by Shaffer [139]. The topology of the polymer chain could be altered without pertubing the static properties (end-to-end distance, radius of gyration). Such an algorithm can predict the tube model scaling for the polymer diffusion coefficient for a non chain crossing condition [139].
4.2.2. Nanocomposites
Early MC and theoretical studies [140,141] had shown that polymer diffusion through heterogeneous media was decreased due to the entropy loss that was created by fixed obstacles. The spacing between the neighboring nanoparticles was the main parameter for slowing down of polymer diffusion in nanocomposites. For random walk polymers of length N which diffused in randomly distributed obstacles, the chains diffused slower than the reptation model predictions [132] following a scaling relation: [142]. Using the bond fluctuation model, of a single polymer chain in regularly distributed obstacles confined in two dimensions (2D), the mean square displacement of a center monomer exhibited four dynamics regimes from shortest to longest time (diffusion) regimes [143]. In particular, where 3/5, 3/8, 3/4, 1 [143]. The second and third regimes were described by segmental diffusion in the self avoiding tube [143]. In a more recent study on lattice MC simulations of long polymers in nanocomposites [144,145] a decrease in polymer chain diffusion was observed when the nanoparticles (of radius smaller than ) were fixed in a BCC fashion in the polymer matrix, due to confinement and enthalpic effects, as can be seen in Figure 9.
A phenomenological trap model [69,70] was developed to calculate the PS diffusion in SWCNT (or fullerene) nanocomposites [6,146]. The authors assumed that there exists a volume within which the SWCNTs exert an influence on the polymers, which might be considered to be of the order of up to a radius of gyration away from the nanotube surface [69,70]. Diffusion occurs according to the following rules: a jump between two adjacent sites on a cubic lattice occurs with equal probability if the two sites are both inside or both outside the volume influenced by the nanotube, but with a reduced probability if the two sites are in different regions. When the nanotubes are isolated (dilute SWCNT loading) then any polymers within the nanotube influenced regions effectively become trapped with a residence time that depends on the probability of crossing between regions, and long-time diffusion is reduced. Once these regions start to interconnect though, polymers can diffuse long distances regardless of which region they are in so that the influence of the reduced probability of crossing diminishes. As a consequence, the diffusion coefficient starts to increase with concentration above this percolation threshold. The model does not explicitly account for the physical obstacles presented by the SWCNTs, an affect which at the concentrations of interest, up to 5% by volume is expected to be much smaller than the observed effect. The model also does not account for any polymer specific motions, such as reptation. Each diffusing polymer is treated as a single point particle. Despite the simplicity of the model, remarkably it predicts that the lowest possible reduction in the diffusion coefficient, D (in the nanocomposite), compared to (in the bulk PS), the diffusion coefficient in the absence of SWCNTs, corresponds to , a value that was subsequently found to be in excellent agreement with the more refined temperature controlled experiments carried out by Tung et al. [147]. This quantitative agreement suggests that the model captures the correct long time diffusion behavior. It does not, however, provide a molecular explanation for what causes the effective barrier for diffusion between the two regions.
4.2.3. Polymers Confined between Surfaces
The effect of bidispersity on the dynamics of polymer films capped between two neutral walls was studied by Li et al. [148] for various compositions. More specifically, they investigated a coarse-grained bond-fluctuation model via Monte Carlo. Their results rendered the characteristic entanglement length to be an important parameter that helped to interpret the effect of bidispersity on the dynamics of their model polymer films. For chains short compared with the characteristic entanglement length, and independent on the film composition, the diffusivity of the short chains was limited, caused by an average number of near-neighboring particles that increased with the decrease of the film thickness. On the contrary, the dynamics of longer chains was determined by the film’s composition. They found that with a lower weight fraction of long chains, the self-diffusion coefficient of long chains decreased slowly with the decrease of the film thickness, which was similar to that of short chains.
5. Mesoscale Simulation Methods
5.1. Dissipative Particle Dynamics
Dissipative particle dynamics (DPD) is a mesoscopic simulation technique used to predict mainly the morphology in polymer melts, blends, composites. The main feature of DPD simulations is the nature of the conservative force (non bonded force that acts between two particles). In this case the conservative force is not a Lennard–Jones force [149,150] (as in MD) but rather a harmonic force which decreases linearly if the particle distance [151,152] increases. However, this enables chain crossing and as a consequence polymers behave as phantom chains [153,154] thus the reptation scaling laws cannot be predicted [155]. However, there are a few DPD polymer melt simulation efforts to prevent unphysical polymer chain crossing, for instance by incorporating an additional repulsion based on the distance of closest approach between two bonds [156,157,158,159,160,161,162] following the philosophy by Kumar and Larson [163] and Pan and Manke [164] in order to predict the reptation diffusion in long chains. In particular, a segmental repulsion force is introduced, to reduce the frequency of artificial chain segment crossings, that depends on the distance between the midpoints of the segments, rather than the minimum distance between segments. The scaling power law exponent for the center of mass diffusion coefficient, for number of monomers above the entanglement length, is close to the theoretical value of tube model of , but the scaling power law exponent for viscosity is close to , which is smaller than the experimental value of [164]. Without such segmental repulsion forces, chain segment crossings can occur freely [155], thus the scaling of both diffusion coefficient and viscosity follows the Rouse behavior even for high chain lengths [164].
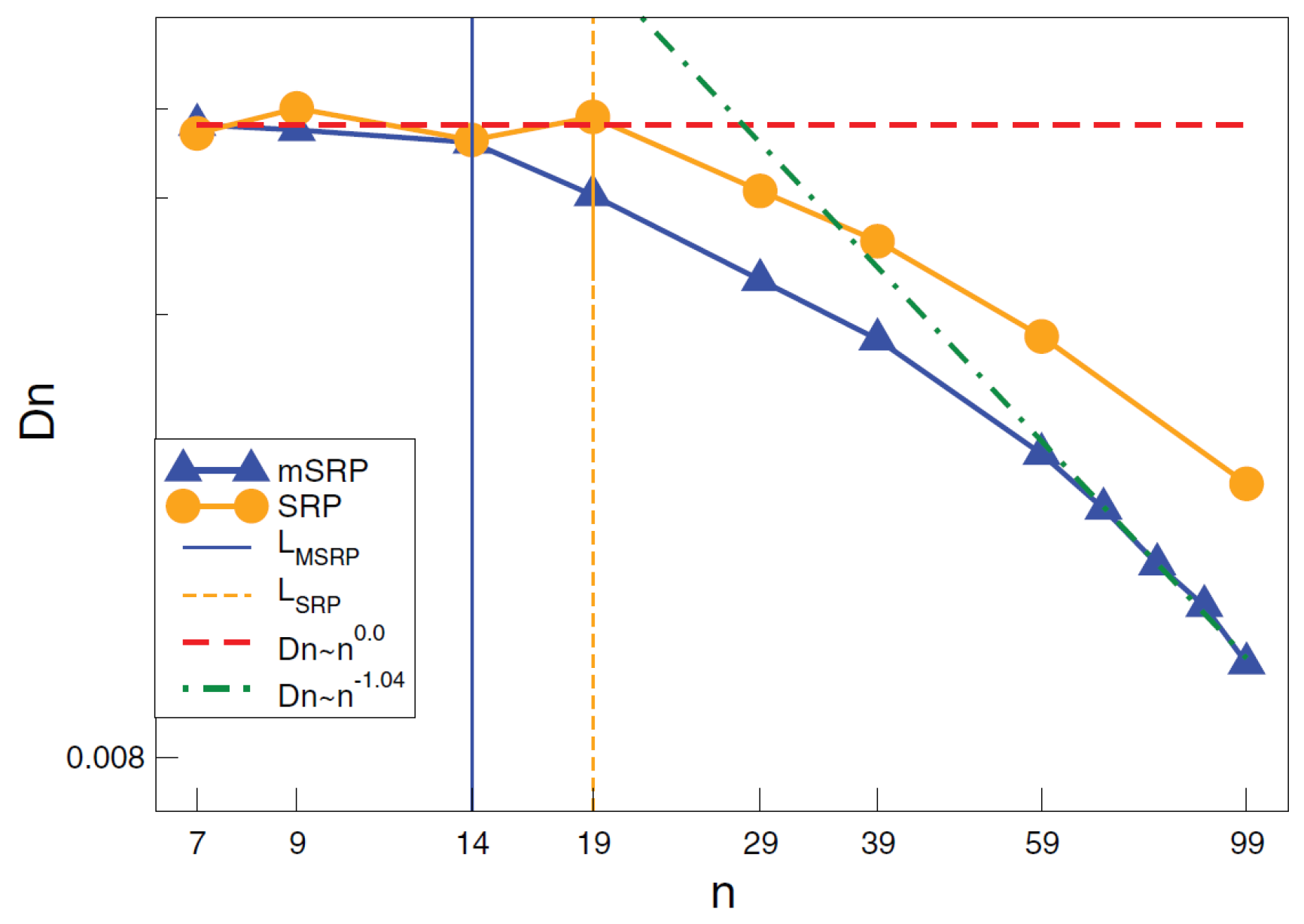
An alternative approach that was implemented is the modified segmental repulsive potential (mSRP), whose parameters were determined from topological, structural, and thermodynamic properties. The mSRP potential was able to capture entanglements and the reptational scaling law for diffusion, as shown in Figure 10, in a dense polymer melt [161].
Another approach to avoid unphysical bond crossings is either by introducing the “Twentanglement” algorithm [168,169] or by adding a rigid core around monomers [170] or by using adaptive time stepping [171]. In particular, the idea to create entanglements between coarse grained polymer chains, in the “Twentanglement” algorithm, is to view the bonds as slippery elastic bands with finite extensibility and prevents chains from crossing each other [168]. Following such methodology a chain with has a non-Rouse behavior [168] but its diffusion is higher than the experimental value of Pearson [52].
Moreover, a highly entangled coarsed grained model for PE, containing monomers, is developed that is able to form entanglements with neighboring chains and capture the diffusive linear regime at long time scales [172]. In that particular study, the entanglements are implemented as stochastic events, in which the probability of creating an entanglement, in a simulation time step, between a specific pair of coarse-grained chains depends on a weight function (quadratic function) depending the center of mass distance between these two chains [172]. These entanglements can disappear when the chains unknot, move apart or slip off [172]. This is different from the slip-spring model in which creation and annihilation of entanglements depend on the number of monomers at the chain ends [172,173,174]. Following such an approach the polymer diffusion coefficient, which is depicted in Figure 11a, presents a good agreement with the experimental data by Pearson et al. [52] but does not fit exact the power law exponent of −2, indicating that the simulated polymer chains are between Rouse and reptation regimes. Following a different approach, with a geometrical criterion for the entanglements, reptational dynamics at “high” chain length can be obtained [167] (Figure 11b). With such a geometrical criterion, no new length scales or forces need to be added [167] and entanglements are predicted in both melts and nanocomposites [175].
5.2. Slip-Springs
Another approach to simulate entangled polymer dynamics was based on the slip-link model (a number of discrete points confine the lateral motion of the polymer chain, while the chain is able to slide through them) which was first developed by Hua and Schieber [176]. This was a combination of the reptation model [21,22] and network model [177]. Based on the framework of the Hua and Schieber model, other different models [56,178,179,180,181] were proposed having different rules for the time evolution of the state variables, and these models could reproduce even quantitatively the linear viscoelastic data of monodisperse linear polymer melts. For instance, the single chain slip-link model by Likthtman [56] was based on the Rubinstein’s and Panyukov’s model [182] for polymer networks. Likthtman’s model contained contour length fluctuations, constraint release, fast Rouse and longitudinal modes, thus it was able to predict chain reptation. Based on that model, Brownian simulations were performed and compared with rheological (linear relaxation, storage and loss moduli, viscosity) and diffusion experiments. The model predictions of those properties were in agreement with experiments measurements for PS, PI, PB.
A comparison among different slip-link model simulations to account the constraint release, for bidisperse linear polymer melts, had been implemented by Masubuchi et al. [184] (Figure 12). It was found that the multichain DT model by Doi and Takimoto [179] and the multichain slip link model (primitive chain network (PCN)) by Masubuchi [178] could account for constraint release successfully and compared better to experimental modulus data, for poly(styrene) and poly(butadiene), than the model by Nair and Schieber [180]. Recently, a fast slip link model was implemented for bidisperse linear polymer melts by Shanbhag [185]. Viscosities and dynamic moduli predictions of that fast slip link model [185] were in very good agreement with experimental measurements on 1,4 poly(butadiene)s. The computational time depended on both number of slip-links, (where is the entanglement molecular weight), and number of chains, n, and scaled as . A numerical study of the slip-link model had been implemented by Biondo et al. [186]. This was used to explore both linear and non-linear rheological properties and furthermore was applied to nanocomposites (containing bare fillers) [186], which could be an alternative to studies implemented by coarse-grained simulations.
An interesting approach in DPD method [191] is to incorporate the topological constraints and, thus uncrossability of polymer chains, by creating temporary cross-links, the so-called slip-spring (variation of the slip link model) following the philosophy of Likhtman [56]. According to this, the lateral motion of the polymer chain is restricted by links connected (by virtual springs) [192] to links on other polymer chains [56,193]. These slip-springs can be created and destroyed at chain ends [56]. Following such a methodology, the polymer chains mean square displacements, diffusivities and longest relaxation time are in agreement with the results of reptation tube theory. By implementing that method, the simulation is 500 times faster than the conventional generic Kremer-Grest model [191] (where unphysical bond crossings are prevented by excluded-volume interactions) which has been used extensively in modeling polymer melts, solutions and nanocomposites by molecular dynamics. That approach of slip-spring model in DPD simulations has also been applied in bidisperse melts in comparison to the conventional KG models. A good agreement in the mean squared displacement measurements is found [110].
The effectiveness of stochastic single-chain models in describing the dynamics of entangled polymers was tested by comparing the slip-spring model to the Kremer-Grest model which was solved using stochastic MD simulations [194]. For a particular chain length in the slip-spring model, the parameters that best reproduced the mean-square displacement were determined by fitting to MD data. Such procedure involved a comparison of the time dependent stress relaxation moduli obtained by both models for a range of chain lengths. The poor agreement of the mean-square monomer displacements at short times observed was corrected by using generalized Langevin equations for the dynamics and that resulted in significantly improved agreement [194]. After identifying a limitation of the original slip-spring model in describing the static structure of the polymer chain, the model was modified so that it agreed with MD results, by introducing a pairwise repulsive potential between the monomers in the chains [194]. The single-chain slip-spring model had been extended by Khaliullin and Schieber [195] to incorporate constraint release in a mean-field way, including polymer fluctuations. The original constraint release concept by de Gennes assumed tube reorganization by Rouse motion of the tube, whose characteristic rate is proportional to the fraction of chain ends, and further determined by reptation of surrounding chains. The inclusion of the additional physics of constraint release [21,196,197] improved the linear viscoelastic predictions of the model, both for monodisperse polymers and bidisperse polymer blends [180,198]. An alternative tube reorganization mechanism, tube dilation, was proposed by Marrucci [199]. Within this concept, reptation of surrounding chains led to a widening of the tube. Constraint release played an important role in the self-consistent determination of the dilated tube diameter. The molecular picture of partial dilation based on this self consistency seemed valid for linear and star chains [200].
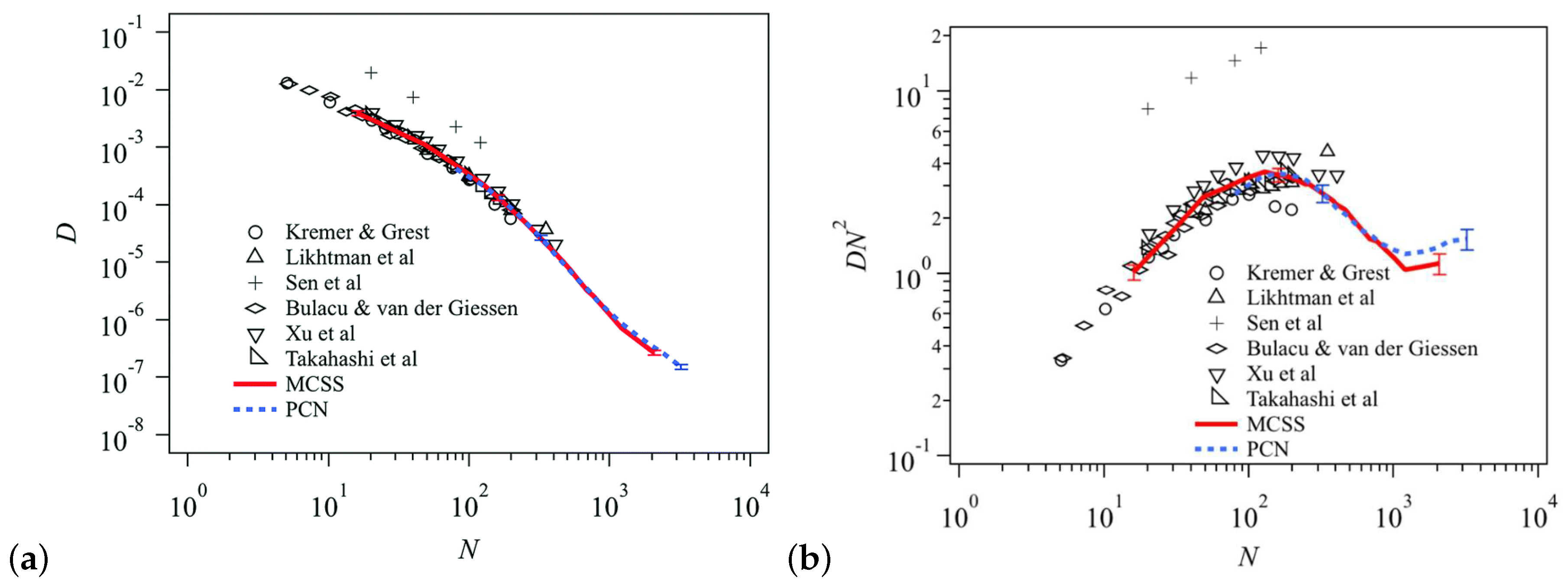
A multi-chain extension of the single-chain slip-spring model was introduced [201] that relied on a many-chain representation and introduced the topological effects that arised from the non-crossability of molecules through effective fluctuating interactions, mediated by slip-springs, between neighboring pairs of macromolecules. The total number of slip-springs was not preserved but, instead, it was controlled through a chemical potential that determined the average molecular weight between entanglements. Ramirez-Hernandez et al. [202] extended this model in a way that correctly incorporated the effects of the fluctuating environment in which polymer segments were immersed. The model was used to obtain the equation of state associated with the slip-springs, as well as linear and nonlinear rheology of entangled polymer melts. In another study of the multi-chain slip-spring model [192] the crossover regime between Rouse and entangled dynamics for the polymer chains was observed [192]. In order to test if compatibility between the different properties had been achieved; diffusion, relaxation modulus, entanglement properties were calculated for the multi-chain slip-spring model (primitive chain network model) [178,183,184,190,203] in comparison to the standard Kremer-Grest bead-spring model [26]. In particular, regarding the diffusion, three models were compatible by scaling the units of length, time and number of beads in the polymer chain (Figure 13). However, significant discrepancies were observed for the inter cross-correlations in the relaxation modulus [190].
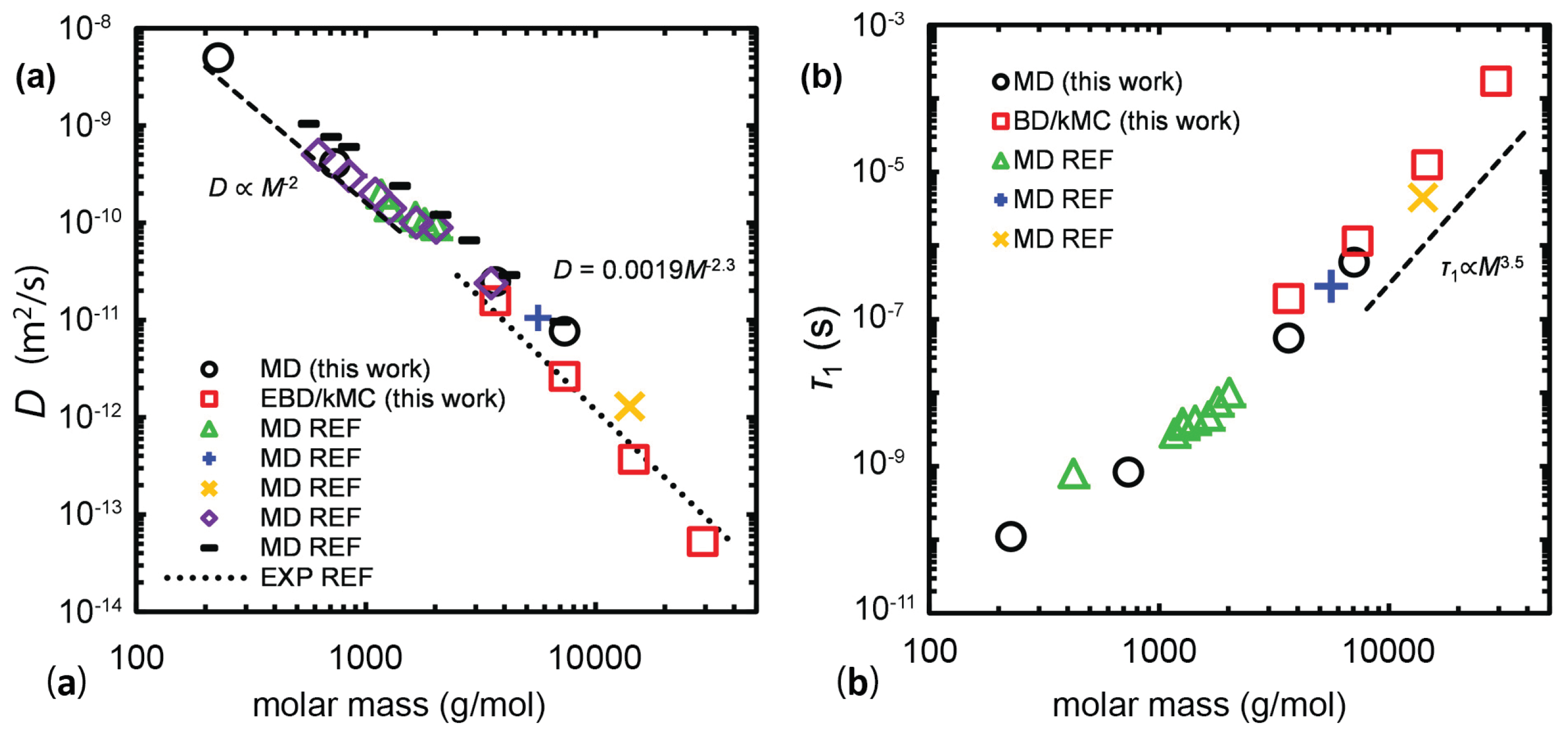
Recently, a multiscale simulation strategy that linked detailed MD simulations to slip-springs based on Brownian dynamics/kinetic Monte Carlo (BD/kMC) simulations of long PE chains ( to ) melts was implemented by Sgouros et al. [204] (Figure 14 and Figure 15) and Megariotis et al. [193]. Such (BD/kMC) methodology was based on a Helmholtz energy function that incorporated bonded, slip-spring, and nonbonded interaction terms [205]. The interesting characteristic of such development was that the nonbonded interactions in the absence of slip-springs were derived from an equation of state that was thermodynamically consistent to that obtained by detailed MD and experimental measurements.
6. Theoretical Modeling
6.1. Polymer Melts
The diffusion of a Gaussian chain in a fixed array of obstacles using the projection operator formalism by Loring [208] was investigated [209]. It was shown that the monomer friction coefficient used in the center of mass mean square displacement could be rewritten exactly in terms of the time correlation function of the total force on the chain. When the decay profile of the force correlation function had an exponential form, and its dependence on the density of obstacles written in an approximate form, the dynamics of the center of mass was found to be subdiffusive at intermediate times and diffusive at long times. Moreover, the diffusion coefficient D of the chain at long time scales and at high concentrations of small obstacles was found to vary with monomer number as , in qualitative agreement with the predictions of the reptation model [209]. It is worth noting that the self-diffusion coefficient of monodisperse linear polymer melts using an analytical theory of stress relaxation which included contour-length fluctuations predicted a scaling relation [116]. This scaling was in qualitative agreement with the polymer mode-coupling theory which predicted scaling relations , with exponent values and for linear polymer melts [210,211,212]. The polymer-mode-coupling (PMC) non reptation theory of Schweizer was able to tune the different exponents of the diffusion coefficient in the crossover to high molecular weight polymers [210,211,212,213].
6.2. Nanocomposites
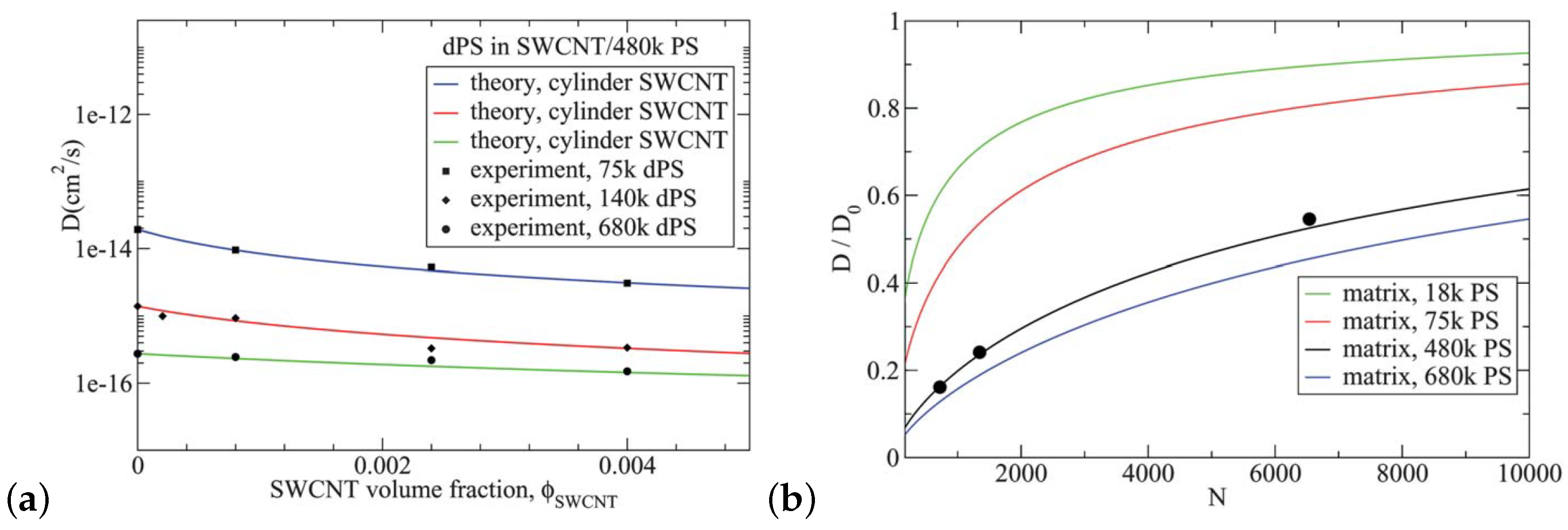
A theory based on the reptation model was developed for PS/SWCNT nanocomposites, which was modified to incorporate the effect of the surface monomer contacts. In particular, it predicted well the experimental tracer PS diffusion for low SWCNT loading below the percolation threshold as can be seen in Figure 16. In addition, the theory could predict the effects of matrix molecular weight (in agreement with experiments), nanoparticles geometry and diameter of SWCNT on the PS diffusion [214].
An excluded volume analytical model predicts the decrease of polymer diffusion in nanocomposites, in agreement to experiments at low nanoparticle loading (lower values) [215]. However, at higher loading (low values), the model predicts a slower polymer diffusion than that observed in experiments as can be seen in Figure 17. Polymer disentanglement under high loading [15] cannot be accounted for in that analytical model, thus suggesting unknown physical mechanisms are active in that regime.
7. Conclusions
We discuss the very challenging problem of reptational diffusion in melts and nanocomposites that has been studied in the last three decades. While CG models such as the Kremer-Grest model can provide us with an understanding of different dynamical regimes and agree with the predictions of the tube reptation model in polymer melts, they lack detailed chemical information. The semiflexibility can be inserted to the CG model, through a bending and a torsion potential, and has an effect on the entangled polymer diffusion and diffusion coefficient dependence on chain length. Detailed chemical information can be incorporated in fully atomistic models; however, these are limited to short chains (oligomers) due to their computational demand thus cannot capture the entangled polymer dynamics. There are different approaches to insert the chemical information, such as united atom models or the IBI method, which have been proved successful in entangled polymer dynamics modeling and diffusion coefficient dependence on chain length for mildly entangled polymer melts (such as PEO, PE, PS, PB). However, these developed models still remain computationally demanding to be applied for the diffusion in polymer nanocomposites. In particular, only the effect of graphite walls on the diffusion of mildly entangled PE chains (using a united atom force field) has been implemented. In addition, the coarse graining by the IBI method is developed at one temperature and an energy renormalization approach [106,107] should be applied to predict the polymer diffusion in different temperatures. However, this method has not still been implemented in nanocomposites.
In future, it is expected that atomistic simulations combined with models of soft potentials (the nonbonded monomers interact through a linear finite force rather than a hard core LJ force), multiblobs [218,219] or slip-springs (since well entangled polymers can be modeled in that method) will be implemented to study reptational polymer diffusion and can be useful in polymer nanocomposites, where diffusion is altered due to nanoparticle fillers size, loading, geometry and polymer-nanoparticles interaction. In general, DPD simulations are faster and can reach a larger length scale than the Kremer-Grest MD simulations. The soft LJ force is independent of the DPD thermostat, and a time step for integration between 0.01–0.02 ps can be applied [162,167] in polymer melts by still conserving the energy and momentum [162] of the system and avoid bond crossings. The slip-spring model combined with DPD simulations can improve much further the speed of polymer melt simulations, up to 500 times faster, using a time step for integration of 0.06 ps [110,191] requiring also smaller number of particles, in comparison to the Kremer-Grest model (time step 0.01 ps). In the implementation of BD combined with slip-springs, in order to model entangled polymer dynamics, a time step for integration of 0.1 ps is used [205].
Author Contributions
Conceptualization, A.K.; Methodology, A.K.; Validation, A.K., R.J.C., K.I.W., M.K., N.C.; Formal Analysis, A.K., M.K., N.C.; Investigation, A.K., R.J.C., K.I.W., M.K., N.C.; Writing—Original Draft Preparation, A.K.; Writing—Review and Editing, A.K., R.J.C., K.I.W., M.K., N.C.; Project Administration, A.K.
Funding
This work was supported by the Luxembourg Institute of Science & Technology (A.K.). N.C., A.K., K.I.W. and R.J.C. were funded through the Materials Network Program, and acknowledge financial support from the EPSRC (EP/5065373/1) and the NSF (DMR-1210379). R.J.C. acknowledges support from NSF/Polymers-DMR 1507713, NSF/PIRE-OISE 1545884 and NSF/MRSEC-DMR 1720530. Work by M.K. and A.K. related to the present review is supported by the Swiss National Science Foundation through grant 200021L_185052 and the Fonds National de la Recherche Luxembourg through grant INTER/SNF/18/13289828.
Acknowledgments
We would like to acknowledge Eric Bailey (University of Pennsylvania) for helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vacatello, M. Chain dimensions in filled polymers: An intriguing problem. Macromolecules 2002, 35, 8191–8193. [Google Scholar] [CrossRef]
- Allegra, G.; Raos, G.; Vacatello, M. Theories and simulations of polymer-based nanocomposites: From chain statistics to reinforcement. Prog. Polym. Sci. 2008, 33, 683–731. [Google Scholar] [CrossRef]
- Vacatello, M. Predicting the molecular arrangements in polymer-based nanocomposites. Macromol. Theory Simul. 2003, 12, 86–91. [Google Scholar] [CrossRef]
- Ganesan, V.; Jayaraman, A. Theory and simulation studies of effective interactions, phase behavior and morphology in polymer nanocomposites. Soft Matter 2014, 10, 13–38. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N.; Composto, R.J.; Winey, K.I. Structure, entanglements and dynamics of polymer nanocomposites containing spherical nanoparticles. IOP Conf. Ser. Mater. Sci. Eng 2014, 64, 012041. [Google Scholar] [CrossRef] [Green Version]
- Karatrantos, A.; Composto, R.J.; Winey, K.I.; Clarke, N. Primitive path network, structure and dynamics of SWCNT/polymer nanocomposites. IOP Conf. Ser. Mater. Sci. Eng. 2012, 40, 012027. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N.; Kröger, M. Modeling of polymer structure and conformations in polymer nanocomposites from atomistic to mesoscale: A Review. Polym. Rev. 2016, 56, 385–428. [Google Scholar] [CrossRef]
- Sharaf, M.A.; Mark, J.E. Monte Carlo simulations on the effects of nanoparticles on chain deformations and reinforcement in amorphous polyethylene networks. Polymer 2004, 45, 3943–3952. [Google Scholar] [CrossRef]
- Yuan, Q.W.; Kloczkowski, A.; Mark, J.E.; Sharaf, M.A. Simulations on the reinforcement of poly(dimethylsiloxane) elastomers by randomly distributed filler particles. J. Polym. Sci. Polym. Phys. Ed. 1996, 34, 1647–1657. [Google Scholar] [CrossRef]
- Liu, J.; Wu, S.; Zhang, L.; Wang, W.; Cao, D. Molecular dynamics simulation for insight into microscopic mechanism of polymer reinforcement. Phys. Chem. Chem. Phys. 2011, 13, 518–529. [Google Scholar] [CrossRef]
- Everaers, R.; Sukumaran, S.K.; Grest, G.S.; Svaneborg, C.; Sivasubramanian, A.; Kremer, K. Rheology and microscopic topology of entangled polymeric liquids. Science 2004, 303, 823–826. [Google Scholar] [CrossRef]
- Foteinopoulou, K.; Karayiannis, N.C.; Laso, M.; Kröger, M.; Mansfield, M.L. Universal scaling, entanglements, and knots of model chain molecules. Phys. Rev. Lett. 2008, 101, 265702. [Google Scholar] [CrossRef]
- Hoy, R.S.; Grest, G.S. Entanglements of an End-Grafted Polymer brush in a polymeric matrix. Macromolecules 2007, 40, 8389–8395. [Google Scholar] [CrossRef]
- Li, Y.; Kröger, M.; Liu, W.K. Nanoparticle effect on the dynamics of polymer chains and their entanglement network. Phys. Rev. Lett. 2012, 109, 118001. [Google Scholar] [CrossRef] [PubMed]
- Karatrantos, A.; Clarke, N.; Composto, R.J.; Winey, K.I. Entanglements in polymer nanocomposites containing spherical nanoparticles. Soft Matter 2016, 12, 2567–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.K.; Krishnamoorti, R. Nanocomposites: Structure, phase behavior, and properties. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Kalathi, J.T.; Grest, G.S.; Kumar, S.K. Universal viscosity behavior of polymer nanocomposites. Phys. Rev. Lett. 2012, 109, 198301. [Google Scholar] [CrossRef]
- Li, Y.; Kröger, M.; Liu, W.K. Nanoparticle geometrical effect on structure, dynamics and anisotropic viscosities of polyethylene nanocomposites. Macromolecules 2012, 45, 2099–2112. [Google Scholar] [CrossRef]
- Crosby, A.J.; Lee, J.Y. Polymer nanocomposites: The nano effect on mechanical properties. Polym. Rev. 2007, 47, 217–229. [Google Scholar] [CrossRef]
- Li, Y.; Abberton, B.C.; Kröger, M.; Liu, W.K. Challenges in multiscale modeling of polymer dynamics. Polymers 2013, 5, 751–832. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1989. [Google Scholar]
- Doi, M.; Edwards, S.F. Dynamics of concentrated polymer systems. Part 1—Brownian motion in the equilibrium state. J. Chem. Soc. Faraday Trans. 2 1978, 74, 1789–1801. [Google Scholar] [CrossRef]
- Klein, J. Evidence for reptation in an entangled polymer melt. Nature 1978, 271, 143–145. [Google Scholar] [CrossRef]
- Leger, L.; Hervet, H.; Auroy, P.; Boucher, E.; Massey, G. The reptation model: Tests through diffusion measurements in linear polymer melts. Rheol. Ser. 1996, 5, 1–16. [Google Scholar]
- Viovy, J.L.; Rubinstein, M.; Colby, R.H. Constraint release in polymer melts: Tube reorganization versus tube dilation. Macromolecules 1991, 24, 3587–3596. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Mavrantzas, V.G.; Theodorou, D.N.; Kröger, M.; Ramirez, J.; Ottinger, H.C.; Vlassopoulos, D. Crossover from the Rouse to the entangled polymer melt regime: Signals from long, detailed atomistic molecular dynamics simulations, supported by rheological experiments. Macromolecules 2003, 36, 1376–1387. [Google Scholar] [CrossRef]
- Kröger, M.; Loose, W.; Hess, S. Structural changes and rheology of polymer melts via nonequilibrium molecular dynamics. J. Rheol. 1993, 37, 1057–1079. [Google Scholar] [CrossRef]
- Kröger, M.; Hess, S. Rheological evidence for a dynamical crossover in polymer melts via nonequilibrium molecular dynamics. Phys. Rev. Lett. 2000, 85, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Stephanou, P.S.; Schweizer, T.; Kröger, M. Communication: Appearance of undershoots in start-up shear: Experimental findings captured by tumbling-snake dynamics. J. Chem. Phys. 2017, 146, 161101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Jeong, S.; Kim, J.M.; Baig, C. Molecular dynamics for linear polymer melts in bulk and confined systems under shear flow. Sci. Rep. 2017, 7, 9004. [Google Scholar] [CrossRef] [Green Version]
- Schleger, P.; Farago, B.; Lartigue, C.; Kollmar, A.; Richter, D. Clear evidence of reptation in polyethylene from neutron spin-echo spectroscopy. Phys. Rev. Lett. 1998, 81, 124–127. [Google Scholar] [CrossRef]
- Richter, D.; Willner, L.; Zirkel, A.; Farago, B.; Fetters, L.; Huang, J. Polymer motion at the crossover from Rouse to reptation dynamics. Macromolecules 1994, 27, 7437–7446. [Google Scholar] [CrossRef]
- Gold, B.; Pyckhout-Hintzen, W.; Wischnewski, A.; Radulescu, A.; Monkenbusch, M.; Allgaier, J.; Hoffmann, I.; Parisi, D.; Vlassopoulos, D.; Richter, D. Direct assessment of tube dilation in entangled polymers. Phys. Rev. Lett. 2019, 122, 088001. [Google Scholar] [CrossRef]
- De Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Read, D. Calculation of scattering from stretched copolymers using the tube model: Incorporation of the effect of elastic inhomogeneities. Macromolecules 2004, 37, 5065–5092. [Google Scholar] [CrossRef]
- Lin, C.C.; Parrish, E.; Composto, R.J. Macromolecule and particle dynamics in confined media. Macromolecules 2016, 49, 5755–5772. [Google Scholar] [CrossRef]
- Mills, P.J.; Green, P.F.; Palmstrom, C.J.; Mayer, J.W.; Kramer, E.J. Analysis of diffusion in polymers by forward recoil spectrometry. Appl. Phys. Lett. 1984, 45, 957. [Google Scholar] [CrossRef]
- Lodge, T.P. Reconciliation of the molecular weight dependence of diffusion and viscosity in entangled polymers. Phys. Rev. Lett. 1999, 83, 3218–3221. [Google Scholar] [CrossRef]
- Green, P.F.; Palmstrom, C.J.; Mayer, J.W.; Kramer, E.J. Limits of reptation in polymer melts. Phys. Rev. Lett. 1984, 53, 2145. [Google Scholar] [CrossRef]
- Mills, P.J.; Green, P.F.; Palmstrom, C.J.; Mayer, J.W.; Kramer, E.J. Polydispersity effects on diffusion in polymers: Concentration profiles of d-polystyrene measured by forward recoil spectrometry. J. Polym. Sci. Polym. Phys. Ed. 1986, 24, 1–9. [Google Scholar] [CrossRef]
- Green, P.F.; Palmstrom, C.J.; Mayer, J.W.; Kramer, E.J. Marker displacement measurements of polymer-polymer interdiffusion. Macromolecules 1985, 18, 501–507. [Google Scholar] [CrossRef]
- Green, P.F.; Kramer, E.J. Matrix effects on the diffusion of long polymer chains. Macromolecules 1986, 19, 1108. [Google Scholar] [CrossRef]
- Green, P.F.; Kramer, E.J. Temperature dependence of tracer diffusion coefficients in polystyrene. MRS 1986, 1, 202–204. [Google Scholar] [CrossRef]
- Antonietti, M.; Coutandin, J.; Sillescu, H. Critical chain lengths in polystyrene bulk diffusion. Macromolecules 1986, 19, 793. [Google Scholar] [CrossRef]
- Antonietti, M.; Fölsch, K.; Sillescu, H. Critical chain lengths in polystyrene bulk diffusion. Makromol. Chem. 1987, 188, 2317–2324. [Google Scholar] [CrossRef]
- Tead, S.F.; Kramer, E.J.; Hadziioannou, G.; Antonietti, M.; Sillescu, H.; Lutz, P.; Strazielle, C. Polymer Topology and Diffusion: A Comparison of Diffusion in Linear and Cyclic Macromolecules. Macromolecules 1992, 25, 3942–3947. [Google Scholar] [CrossRef]
- Antonietti, M.; Coutandin, J.; Sillescu, H. Chainlength and temperature-dependence of self-diffusion coefficients in polystyrene. Macromol. Chem. Rapid Commun. 1984, 5, 525–528. [Google Scholar] [CrossRef]
- Fleischer, G.; Appel, M. Chain length and temperature dependence of the self-diffusion of polyisoprene and polybutadiene in the melt. Macromolecules 1995, 28, 7281–7283. [Google Scholar] [CrossRef]
- Appel, M.; Fleischer, G. Investigation of the chain length dependence of self-diffusion of poly(dimethylsiloxane) and poly(ethylene oxide) in the melt with pulsed field gradient NMR. Macromolecules 1993, 26, 5520–5525. [Google Scholar] [CrossRef]
- Ozisik, R.; Zheng, J.; Dionne, P.J.; Picu, C.R.; Von Meerwall, D.E. NMR relaxation and pulsed-gradient diffusion study of polyethylene nanocomposites. J. Chem. Phys. 2005, 123, 134901. [Google Scholar] [CrossRef]
- Pearson, D.; Ver Strate, G.; von Meerwall, E.; Schilling, F. Viscosity and self-diffusion coefficient of linear polyethylene. Macromolecules 1987, 20, 1133–1141. [Google Scholar] [CrossRef]
- Tao, H.; Lodge, T.P.; von Meerwall, E.D. Diffusivity and viscosity of concentrated hydrogenated polybutadiene solutions. Macromolecules 2000, 33, 1747–1758. [Google Scholar] [CrossRef]
- Wang, S.Q. Chain dynamics in entangled polymers: Diffusion versus rheology and their comparison. J. Polym. Sci. B 2003, 41, 1589–1604. [Google Scholar] [CrossRef] [Green Version]
- Spiess, H.W. 50th anniversary perspective: The importance of NMR spectroscopy to macromolecular science. Macromolecules 2017, 50, 1761–1777. [Google Scholar] [CrossRef]
- Likhtman, A.E. Single-chain slip-link model of entangled polymers: Simultaneous description of neutron spin-echo, rheology, and diffusion. Macromolecules 2005, 38, 6128–6139. [Google Scholar] [CrossRef]
- Chavez, F.V.; Saalwachter, K. Time-Domain NMR observation of entangled polymer dynamics: Universal behavior of flexible homopolymers and applicability of the tube model. Macromolecules 2011, 44, 1549–1559. [Google Scholar] [CrossRef]
- Park, S.J.; Desai, P.S.; Chen, X.; Larson, R.G. Universal relaxation behavior of entangled 1,4-polybutadiene melts in the transition frequency region. Macromolecules 2015, 48, 4122–4131. [Google Scholar] [CrossRef]
- Trutschel, M.L.; Mordvinkin, A.; Furtado, F.; Willner, L.; Saalwächter, K. Time-domain NMR observation of entangled polymer dynamics: Focus on all tube-model regimes, chain center, and matrix effects. Macromolecules 2018, 51, 4108–4117. [Google Scholar] [CrossRef]
- Furtado, F.; Damron, J.; Trutschel, M.L.; Franz, C.; Schröter, K.; Ball, R.C.; Saalwächter, K.; Panja, D. NMR observations of entangled polymer dynamics: Focus on tagged chain rotational dynamics and confirmation from a simulation model. Macromolecules 2014, 47, 256–268. [Google Scholar] [CrossRef]
- Gordon, G.V.; Schmidt, R.G.; Quintero, M.; Benton, N.J.; Cosgrove, T.; Krukonis, V.J.; Williams, K.; Wetmore, P.M. Impact of polymer molecular weight on the dynamics of poly(dimethylsiloxane)-polysilicate nanocomposites. Macromolecules 2010, 43, 10132–10142. [Google Scholar] [CrossRef]
- Wang, S.; von Meerwall, E.D.; Wang, S.Q.; Halasa, A.; Hsu, W.L.; Zhou, J.P.; Quirk, R.P. Diffusion and rheology of binary polymer mixtures. Macromolecules 2004, 37, 1641–1651. [Google Scholar] [CrossRef]
- Schneider, G.J.; Nusser, K.; Willner, L.; Falus, P.; Richter, D. Dynamics of nanocomposites. Curr. Opin. Chem. Eng. 2017, 16, 65–77. [Google Scholar] [CrossRef]
- Choi, J.; Cargnello, M.; Murrey, C.B.; Clarke, N.; Winey, K.I.; Composto, R.J. Universal scaling of polymer diffusion in nanocomposites. ACS Macro Lett. 2013, 2, 485–490. [Google Scholar] [CrossRef]
- Gam, S.; Meth, J.S.; Zane, S.G.; Chi, C.; Wood, B.A.; Seitz, M.E.; Winey, K.I.; Clarke, N.; Composto, R.J. Macromolecules diffusion in a crowded polymer nanocomposite. Macromolecules 2011, 44, 3494. [Google Scholar] [CrossRef]
- Gam, S.; Meth, J.S.; Zane, S.G.; Chi, C.; Wood, B.A.; Winey, K.I.; Clarke, N.; Composto, R.J. Polymer Diffusion in a polymer nanocomposite: Effect of nanoparticle size and polydispersity. Soft Matter 2012, 8, 6512–6520. [Google Scholar] [CrossRef]
- Tung, W.S.; Griffin, P.J.; Meth, J.S.; Clarke, N.; Composto, R.J.; Winey, K.I. Temperature-dependent suppression of polymer diffusion in polymer nanocomposites. ACS Macro Lett. 2016, 5, 735–739. [Google Scholar] [CrossRef]
- Bailey, E.J.; Griffin, P.J.; Composto, R.J.; Winey, K.I. Multiscale dynamics of small, attractive nanoparticles and entangled polymers in polymer nanocomposites. Macromolecules 2019, 52, 2181–2188. [Google Scholar] [CrossRef]
- Mu, M.; Clarke, N.; Composto, R.J.; Winey, K.I. Polymer diffusion exhibits a minimum with increasing single-walled carbon nanotube concentration. Macromolecules 2009, 42, 7091–7097. [Google Scholar] [CrossRef]
- Mu, M.; Seitz, M.E.; Clarke, N.; Composto, R.J.; Winey, K. Polymer tracer diffusion exhibits a minimum in nanocomposites containing spherical nanoparticles. Macromolecules 2011, 44, 191–193. [Google Scholar] [CrossRef]
- Choi, K.I.; Kim, T.H.; Yuan, G.; Satija, S.K.; Koo, J. Dynamics of entangled polymers confined between graphene oxide sheets as studied by neutron reflectivity. ACS Macro Lett. 2017, 6, 819–823. [Google Scholar] [CrossRef]
- Miller, B.; Imel, A.D.; Holley, W.; Baskaran, D.; Dadmun, M.D. The Role of nanoparticle rigidity on the diffusion of linear polystyrene in a polymer nanocomposite. Macromolecules 2015, 48, 8369–8375. [Google Scholar] [CrossRef]
- Lange, F.; Judeinstein, P.; Franz, C.; Hartmann-Azanza, B.; Ok, S.; Steinhart, M.; Saalwachter, K. Large-scale diffusion of entangled polymers along nanochannels. Macromolecules 2015, 4, 561–565. [Google Scholar] [CrossRef]
- Shin, K.; Obukhov, S.; Chen, J.T.; Huh, J.; Hwang, Y.; Mok, S.; Dobriyal, P.; Thiyagarajan, P.; Russell, T.P. Enhanced mobility of confined polymers. Nat. Mater. 2007, 6, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Tsalikis, D.G.; Koukoulas, T.; Mavrantzas, V.G.; Pasquino, R.; Vlassopoulos, D.; Pyckhout-Hintzen, W.; Wischnewski, A.; Monkenbusch, M.; Richter, D. Microscopic structure, conformation, and dynamics of ring and linear poly(ethylene oxide) melts from detailed atomistic molecular dynamics simulations: Dependence on chain length and direct comparison with experimental data. Macromolecules 2017, 50, 2565–2584. [Google Scholar] [CrossRef]
- Hur, K.; Jeong, C.; Winkler, R.G.; Lacevic, N.; Gee, R.H.; Yoon, D.Y. Chain dynamics of ring and linear polyethylene melts from molecular dynamics simulations. Macromolecules 2011, 44, 2311–2315. [Google Scholar] [CrossRef]
- Takahashi, K.Z.; Nishimura, R.; Yamato, N.; Yasuoka, K.; Masubuchi, Y. Onset of static and dynamic universality among molecular models of polymers. Sci. Rep. 2017, 7, 12379. [Google Scholar] [CrossRef] [Green Version]
- Kremer, K.; Grest, G.; Carmesin, I. Crossover from Rouse to reptation dynamics: A molecular-dynamics simulation. Phys. Rev. Lett. 1988, 61, 566–569. [Google Scholar] [CrossRef]
- Zhou, Q.; Larson, R.G. Direct calculation of the tube potential confining entangled polymers. Macromolecules 2006, 39, 6737–6743. [Google Scholar] [CrossRef]
- Wang, Z.; Larson, R.G. Constraint release in entangled binary blends of linear polymers: A molecular dynamics study. Macromolecules 2008, 41, 4945–4960. [Google Scholar] [CrossRef]
- Steinhauser, M.O.; Schneider, J.; Blumen, A. Simulating dynamic crossover behavior of semiflexible linear polymers in solution and in the melt. J. Chem. Phys. 2009, 130, 164902. [Google Scholar] [CrossRef]
- Ramos, J.; Vega, J.F.; Martinez-Salazaar, J. Assessment of entanglement features and dynamics from atomistic simulations and experiments in linear and short chain branched polyolefins. Soft Matter 2012, 8, 6256. [Google Scholar] [CrossRef]
- Takahashi, K.Z.; Nishimura, R.; Yasuoka, K.; Masubuchi, Y. Molecular Dynamics simulations for resolving scaling laws of polyethylene melts. Polymers 2017, 9, 24. [Google Scholar] [CrossRef]
- Tsolou, G.; Mavrantzas, V.G.; Theodorou, D.N. Detailed atomistic molecular dynamics simulation of cis-1,4-poly(butadiene). Macromolecules 2005, 38, 1478–1492. [Google Scholar] [CrossRef]
- Kritikos, G.; Sgouros, A.; Vogiatzis, G.G.; Theodorou, D.N. Molecular dynamics study of polyethylene under confinement. J. Phys. Conf. Ser. 2016, 738, 012012. [Google Scholar] [CrossRef]
- Smith, S.W.; Hall, C.K.; Freeman, B.D. Large-scale molecular dynamics study of entangled hard-chain fluids. Phys. Rev. Lett. 1995, 75, 1316. [Google Scholar] [CrossRef]
- Smith, S.W.; Hall, C.K.; Freeman, B.D. Molecular dynamics study of entangled hard-chain fluids. J. Chem. Phys. 1996, 104, 5616–5637. [Google Scholar] [CrossRef]
- Smith, S.W.; Hall, C.K.; Freeman, B.D.; McCormick, J.A. Self Diffusion Coefficients and Atomic Mean-Squared Displacements in Entangled Hard Chain Fluids. In Numerical Methods for Polymeric Systems; The IMA Volumes in Mathematics and Its Applications; Whittington, S.G., Ed.; Springer: New York, NY, USA, 1998; Volume 102, p. 203. [Google Scholar]
- Shanbhag, S. Estimating self-diffusion in polymer melts: How long is a long enough molecular simulation? Molec. Simul. 2016, 42, 162–172. [Google Scholar] [CrossRef]
- Shanbhag, S. Extraction of self-diffusivity in systems with nondiffusive short-time behavior. Phys. Rev. E 2013, 88, 042816. [Google Scholar] [CrossRef]
- Paul, W.; Binder, K.; Heermann, D.W.; Kremer, K. Dynamics of polymer solutions and melts. Reptation predictions and scaling of relaxation times. J. Chem. Phys. 1991, 95, 7726. [Google Scholar] [CrossRef]
- Pütz, M.; Kremer, K.; Grest, G.S. What is the entanglement length in a polymer melt? Europhys. Lett. 2000, 49, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Harmandaris, V.A.; Kremer, K. Dynamics of polystyrene mmelts through hierarchical multiscale simulations. Macromolecules 2009, 42, 791–802. [Google Scholar] [CrossRef]
- Grest, G.S. Communication: Polymer entanglement dynamics: Role of attractive interactions. J. Chem. Phys. 2016, 145, 141101. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, A.; Ebert, U.; Schafer, L. Segment motion in the reptation model of polymer dynamics. II. Simulations. J. Stat. Phys. 1998, 50, 1375. [Google Scholar] [CrossRef]
- Likhtman, A.E.; Sukumaran, S.K.; Ramirez, J. Linear viscoelasticity from molecular dynamics simulation of entangled polymers. Macromolecules 2007, 40, 6748–6757. [Google Scholar] [CrossRef]
- Pestryaev, E.M. Comparison of various correlation times in polymer melts by molecular dynamics simulation. J. Phys. Conf. Ser. 2011, 324, 012031. [Google Scholar] [CrossRef]
- Hsu, H.P.; Kremer, K. Static and dynamic properties of large polymer melts in equilibrium. J. Chem. Phys. 2016, 144, 154907. [Google Scholar] [CrossRef] [Green Version]
- Salerno, K.M.; Agrawal, A.; Perahia, D.; Grest, G.S. Resolving dynamic properties of polymers through coarse-grained computational studies. Macromolecules 2016, 116, 058302. [Google Scholar] [CrossRef]
- Guenza, M.G. Localization of chain dynamics in entangled polymer melts. Phys. Rev. E 2014, 89, 052603. [Google Scholar] [CrossRef]
- Ebert, U.; Baumgartner, A.; Schafer, L. Short time behavior in de Gennes’ reptation model. Phys. Rev. Lett. 1997, 78, 1592–1595. [Google Scholar] [CrossRef]
- Sun, Q.; Faller, R. Crossover from unentangled to entangled dynamics in a systematically coarse-grained polystyrene melt. Macromolecules 2006, 39, 812–820. [Google Scholar] [CrossRef]
- Hou, J.X.; Svaneborg, C.; Everaers, R.; Grest, G.S. Stress relaxation in entangled polymer melts. Phys. Rev. Lett. 2010, 105, 068301. [Google Scholar] [CrossRef]
- Peters, B.L.; Salerno, K.M.; Agrawal, A.; Perahia, D.; Grest, G.S. Coarse-grained modeling of polyethylene melts: Effect on dynamics. J. Chem. Theory Comput. 2017, 13, 2890–2896. [Google Scholar] [CrossRef]
- Jabbari-Farouji, S. Static and dynamic scaling behavior of a polymer melt model with triple-well bending potential. J. Polym. Sci. B 2018, 56, 1376–1392. [Google Scholar] [CrossRef]
- Xia, W.; Song, J.; Jeong, C.; Hsu, D.D.; Phelan, F.R.; Douglas, J.F.; Keten, S. Energy-Renormalization for Achieving Temperature Transferable Coarse-Graining of Polymer Dynamics. Macromolecules 2017, 50, 8787–8796. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Hansoge, N.K.; Xu, W.S.; Phelan, F.R.; Keten, S.; Douglas, J.F. Energy renormalization for coarse-graining polymers having different segmental structures. Sci. Adv. 2019, 5, eaav4683. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.L.; Salerno, K.M.; Ge, T.; Perahia, D.; Grest, G.S. Effect of chain length dispersity on the mobility of entangled polymers. Phys. Rev. Lett. 2018, 121, 057802. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Mavrantzas, V.G.; Theodorou, D.N. Atomistic molecular dynamics simulation of polydisperse linear polyethylene melts. Macromolecules 1998, 31, 7934–7943. [Google Scholar] [CrossRef]
- Langeloth, M.; Masubuchi, Y.; Bohm, M.C.; Müller-Plathe, F. Reptation and constraint release dynamics in bidisperse polymer melts. J. Chem. Phys. 2014, 141, 194904. [Google Scholar] [CrossRef]
- Bulacu, M.; van der Giessen, E. Effect of bending and torsion rigidity on self-diffusion in polymer melts: A molecular-dynamics study. J. Chem. Phys. 2005, 123, 114901. [Google Scholar] [CrossRef] [Green Version]
- Bulacu, M.; van der Giessen, E. Molecular-dynamics simulation study of the glass transition in amorphous polymers with controlled chain stiffness. Phys. Rev. E 2007, 76, 011807. [Google Scholar] [CrossRef]
- Svaneborg, C.; Everaers, R. Characteristic time and length scales in Kremer-Grest bead-spring polymer melts as a function of chain stiffness. arXiv 2018, arXiv:1808.03503. [Google Scholar]
- Uchida, N.; Grest, G.S.; Everaers, R. Viscoelasticity and primitive path analysis of entangled polymer liquids: From F-actin to polyethylene. J. Chem. Phys. 2008, 128, 044902. [Google Scholar] [CrossRef] [Green Version]
- Groot, R.D. Mesoscale simulation of semiflexible chains. I. Endpoint distribution and chain dynamics. J. Chem. Phys. 2013, 138, 224903. [Google Scholar] [CrossRef] [PubMed]
- Frischknecht, A.L.; Milner, S.T. Diffusion with contour length fluctuations in linear polymer melts. Macromolecules 2000, 33, 5273–5277. [Google Scholar] [CrossRef]
- Padding, T.; Briels, W.J. Time and length scales of polymer melts studied by coarse-grained molecular dynamics simulations. J. Chem. Phys. 2002, 117, 925–943. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Zhang, H.; Huang, X.W.; Huang, H.H.; Luo, M.B. Monte Carlo simulation on the dynamics of a semi-flexible polymer in the presence of nanoparticles. Phys. Chem. Chem. Phys. 2018, 20, 26333–26343. [Google Scholar] [CrossRef] [PubMed]
- Karatrantos, A.; Composto, R.J.; Winey, K.I.; Clarke, N. Polymer and spherical nanoparticle diffusion in nanocomposites. J. Chem. Phys. 2017, 146, 203331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picu, R.C.; Rakshit, A. Dynamics of free chains in polymer nanocomposites. J. Chem. Phys. 2007, 126, 144909. [Google Scholar] [CrossRef]
- Li, S.J.; Qian, H.J.; Lu, Z.Y. Translational and rotational dynamics of an ultra-thin nanorod probe particle in linear polymer melts. Phys. Chem. Chem. Phys. 2018, 20, 20996–21007. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.; Keblinski, P.; Kumar, S.K. Molecular dynamics simulations of polymer transport in nanocomposites. J. Chem. Phys. 2005, 122, 134910. [Google Scholar] [CrossRef]
- Meyer, H.; Kreer, T.; Cavallo, A.; Wittmer, J.P.; Baschnagel, J. On the dynamics and disentanglement in thin and two-dimensional polymer films. Eur. Phys. J. Spec. Top. 2007, 141, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wei, D.; Han, C.C.; Liao, Q. Dynamics of polymer melts confined by smooth walls: Crossover from nonentangled region to entangled region. J. Chem. Phys. 2007, 126, 204907. [Google Scholar] [CrossRef] [PubMed]
- Sussman, D.; Tung, W.S.; Winey, K.I.; Schweizer, K.S.; Riggleman, R.A. Entanglement reduction and anisotropic chain and primitive path conformations in polymer melts under thin film and cylindrical confinement. Macromolecules 2014, 47, 6462. [Google Scholar] [CrossRef]
- Tung, W.S.; Composto, R.J.; Riggleman, R.A.; Winey, K.I. Local polymer dynamics and diffusion in cylindrical nanoconfinement. Macromolecules 2015, 48, 2324–2332. [Google Scholar] [CrossRef]
- Pressly, J.F.; Riggleman, R.A.; Winey, K.I. Polymer diffusion is fastest at intermediate levels of cylindrical confinement. Macromolecules 2018, 51, 9789–9797. [Google Scholar] [CrossRef]
- Zhang, T.; Riggleman, R.A.; Winey, K.I. Polymer conformations and dynamics under confinement with two length scales. Macromolecules 2019, 52, 217–226. [Google Scholar] [CrossRef]
- Rubinstein, M. Discretized model of entangled-polymer dynamics. Phys. Rev. Lett. 1987, 59, 1946. [Google Scholar] [CrossRef]
- Deutsch, J.; Madden, T.L. The diffusion coefficient of a reptating polymer. J. Chem. Phys. 1989, 91, 3252. [Google Scholar] [CrossRef]
- Wittmer, J.; Paul, W.; Binder, K. Rouse and reptation dynamics at finite temperature: A Monte Carlo Simulation. Macromolecules 1992, 25, 7211–7219. [Google Scholar] [CrossRef]
- Kreer, T.; Baschnagel, J.; Müller, M.; Binder, K. Monte Carlo simulation of long chain polymer melts: Crossover from Rouse and reptation dynamics. Macromolecules 2001, 34, 1105–1117. [Google Scholar] [CrossRef]
- Carmesin, I.; Kremer, K. The bond fluctuation method: A new effective algorithm for the dynamics of polymers in all spatial dimensions. Macromolecules 1988, 21, 2819–2823. [Google Scholar] [CrossRef]
- Kremer, K.; Binder, K. Monte Carlo simulation of lattice models for macromolecules. Comput. Phys. Rep. 1988, 7, 259–310. [Google Scholar] [CrossRef]
- Paul, W.; Binder, K.; Heermann, D.W.; Kremer, K. Crossover scaling in semidilute polymer solutions: A Monte Carlo test. J. Phys. II 1991, 1, 37–60. [Google Scholar] [CrossRef]
- Kolinski, A.; Skolnick, J.; Yaris, R. Monte Carlo studies on the long time dynamic properties of dense cubic lattice multichain systems. I. The homopolymeric melt. J. Chem. Phys. 1987, 86, 7164–7173. [Google Scholar] [CrossRef]
- Kolinski, A.; Skolnick, J.; Yaris, R. Does reptation describe the dynamics of entangled, finite length polymer systems? A model simulation. J. Chem. Phys. 1987, 86, 1567–1585. [Google Scholar] [CrossRef]
- Lin, H.; Mattice, W.L.; Von Meerwall, E.D. Dynamics of polyethylene melts studied by Monte Carlo simulations on a high coordination lattice. J. Polym. Sci. B 2006, 44, 2556. [Google Scholar] [CrossRef]
- Shaffer, J.S. Effects of chain topology on polymer dynamics: Bulk melts. J. Chem. Phys. 1994, 101, 4205. [Google Scholar] [CrossRef]
- Muthukumar, M.; Baumgartner, A. Effects of entropic barriers on polymer dynamics. Macromolecules 1989, 22, 1937–1941. [Google Scholar] [CrossRef]
- Muthukumar, M.; Baumgartner, A. Diffusion of a polymer chain in random media. Macromolecules 1989, 22, 1941–1946. [Google Scholar] [CrossRef]
- Reiter, J. A random walk chain reptating in a network of obstacles: Monte Carlo study of diffusion and decay of correlations and a comparison with the Rouse and reptation models. J. Chem. Phys. 1991, 94, 3222. [Google Scholar] [CrossRef]
- Azuma, R.; Takayama, H. Diffusion of single long polymers in fixed and low density matrix of obstacles confined to two dimensions. J. Chem. Phys. 1999, 111, 8666–8671. [Google Scholar] [CrossRef] [Green Version]
- Dionne, P.J.; Osizik, R.; Picu, C.R. Structure and dynamics of polyethylene nanocomposites. Macromolecules 2005, 38, 9351–9358. [Google Scholar] [CrossRef]
- Termonia, Y. Chain confinement in polymer nanocomposites and its effect on polymer bulk properties. J. Polym. Sci. B 2010, 48, 687–692. [Google Scholar] [CrossRef]
- Karatrantos, A.; Composto, R.J.; Winey, K.I.; Kröger, M.; Clarke, N. Entanglements and dynamics of polymer melts near a SWCNT. Macromolecules 2012, 45, 7274. [Google Scholar] [CrossRef]
- Tung, W.S.; Clarke, N.; Composto, R.J.; Winey, K.I. Temperature dependence of polymer diffusion in MWCNT/PS nanocomposites. Macromolecules 2013, 46, 2317–2322. [Google Scholar] [CrossRef]
- Li, S.J.; Chen, Q.Y.; Ding, M.M.; Shi, T.F. Effect of bidispersity on dynamics of confined polymer films. Polymers 2018, 10, 1327. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Hoogerbrugge, P.J.; Koelman, J.M.V.A. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys. Lett. 1992, 19, 155. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Vacatello, M. Phantom chain simulations of polymer-nanofiller systems. Macromolecules 2003, 36, 3411–3416. [Google Scholar] [CrossRef]
- Vacatello, M. Phantom chain simulations of realisticaly sized polymer-based nanocomposites. Macromol. Theory Simul. 2006, 15, 303–310. [Google Scholar] [CrossRef]
- Spenley, N.A. Scaling laws for polymers in dissipative particle dynamics. Europhys. Lett. 2000, 49, 534. [Google Scholar] [CrossRef]
- Lahmar, F.; Tzoumanekas, C.; Theodorou, D.N.; Rousseau, B. Onset of entanglements revisited. Dynamical analysis. Macromolecules 2009, 42, 7485–7494. [Google Scholar] [CrossRef]
- Holleran, S.P.; Larson, R.G. Using spring repulsions to model entanglement interactions in Brownian dynamics simulations of bead-spring chains. Rheol. Acta 2008, 47, 3–17. [Google Scholar] [CrossRef]
- Goujon, F.; Malfreyt, P.; Tildesley, D.J. Mesoscopic simulation of entanglements using dissipative particle dynamics: Application to polymer brushes. J. Chem. Phys. 2008, 129, 034902. [Google Scholar] [CrossRef]
- Goujon, F.; Malfreyt, P.; Tildesley, D.J. Mesoscopic simulation of entangled polymer brushes under shear: Compression and rheological properties. Macromolecules 2009, 42, 4310. [Google Scholar] [CrossRef]
- Yamanoi, M.; Pozo, O.; Maia, J.M. Linear and non-linear dynamics of entangled linear polymer melts by modified tunable coarse-grained level Dissipative Particle Dynamics. J. Chem. Phys. 2011, 135, 044904. [Google Scholar] [CrossRef]
- Sirk, T.W.; Sliozberg, Y.R.; Brennan, J.K.; Lisal, M.; Andzelm, J.W. An enhanced entangled polymer model for dissipative particle dynamics. J. Chem. Phys. 2012, 136, 134903. [Google Scholar] [CrossRef]
- Khai, S.; Yamanoi, M.; Maia, J. The Lowe-Andersen thermostat as an alternative to the dissipative particle dynamics in the mesoscopic simulation of entangled polymers. J. Chem. Phys. 2013, 138, 174903. [Google Scholar]
- Kumar, S.; Larson, R.G. Brownian dynamics simulations of flexible polymers with spring–spring repulsions. J. Chem. Phys. 2001, 114, 6937. [Google Scholar] [CrossRef]
- Pan, G.; Manke, C.W. Developments toward simulation of entangled polymer melts by dissipative particle dynamics. Int. J. Mod. Phys. B 2003, 17, 231. [Google Scholar] [CrossRef]
- Fan, L.L.; Liu, L. Coarse grain simulations of linear polymer melts for chain lengths near the reptation transition. IOP Conf. Ser. Mater. Sci. Eng. 2018, 369, 012017. [Google Scholar] [CrossRef] [Green Version]
- Mondello, M.; Grest, G.S. Viscosity calculations of n-alkanes by equilibrium molecular dynamics. J. Chem. Phys. 1997, 106, 9327–9336. [Google Scholar] [CrossRef]
- Nikunen, P.; Karttunen, M.; Vattulainen, I. Reptational dynamics in dissipative particle dynamics simulations of polymer melts. Phys. Rev. E 2007, 75, 036713. [Google Scholar] [CrossRef] [PubMed]
- Padding, T.; Briels, W.J. Uncrossability constraints in mesoscopic polymer melt simulations: Non-Rouse behavior of C120H242. J. Chem. Phys. 2001, 115, 2846–2859. [Google Scholar] [CrossRef]
- Padding, J.T.; Briels, W.J. Coarse-grained molecular dynamics simulations of polymer melts in transient and steady shear flow. J. Chem. Phys. 2003, 118, 10276–10286. [Google Scholar] [CrossRef]
- Liu, H.; Xue, Y.H.; Qian, H.J.; Lu, Z.Y.; Sun, C.C. A practical method to avoid bond crossing in two-dimensional dissipative particle dynamics simulations. J. Chem. Phys. 2008, 129, 024902. [Google Scholar] [CrossRef] [PubMed]
- Hoda, N.; Larson, R.G. Brownian dynamics simulations of single polymer chains with and without self-entanglements in theta and good solvents under imposed flow fields. J. Rheol. 2010, 54, 1061. [Google Scholar] [CrossRef]
- Zhu, Y.L.; Liu, H.; Lu, Z.Y. A highly coarse-grained model to simulate entangled polymer melts. J. Chem. Phys. 2012, 136, 144903. [Google Scholar] [CrossRef]
- Edwards, D.; Vilgis, T. The effect of entanglements in rubber elasticity. Polymer 1986, 27, 483–492. [Google Scholar] [CrossRef]
- Schieber, J.D.; Andreev, M. Entangled polymer dynamics in equilibrium and flow modelled through slip links. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 367–381. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N.; Composto, R.J.; Winey, K.I. Topological entanglement length in polymer melts and nanocomposites by a DPD polymer model. Soft Matter 2013, 9, 3877–3884. [Google Scholar] [CrossRef]
- Hua, C.C.; Schieber, J.D. Segment connectivity, chain-length breathing, segmental stretch, and constraint release in reptation models. I. Theory and single-step strain predictions. J. Chem. Phys. 1998, 109, 10018. [Google Scholar] [CrossRef]
- Green, M.S.; Tobolsky, A.V. A New Approach to the Theory of Relaxing Polymeric Media. J. Chem. Phys. 1946, 14, 80. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Takimoto, J.I.; Koyama, K.; Ianniruberto, G.; Marrucci, G.; Grego, F. Brownian simulations of a network of reptating primitive chains. J. Chem. Phys. 2001, 115, 4387. [Google Scholar] [CrossRef]
- Doi, M.; Takimoto, J. Molecular modelling of entanglement. Phys. Trans. R. Soc. Lond. A 2003, 361, 641–652. [Google Scholar] [CrossRef]
- Nair, D.M.; Schieber, J.D. Linear viscoelastic predictions of a consistently unconstrained Brownian slip-link model. Macromolecules 2006, 39, 3386–3397. [Google Scholar] [CrossRef]
- Schieber, J.D.; Neergaard, J.; Gupta, S. A full-chain, temporary network model with sliplinks, chain-length fluctuations, chain connectivity and chain stretching. J. Rheol. 2003, 47, 213–233. [Google Scholar] [CrossRef]
- Rubinstein, M.; Panyukov, S. Elasticity of Polymer Networks. Macromolecules 2002, 35, 6670. [Google Scholar] [CrossRef]
- Masubuchi, Y. Contraction of Entangled Polymers After Large Step Shear Deformations in Slip-Link Simulations. Polymers 2019, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Watanabe, H.; Ianniruberto, G.; Grego, F.; Marrucci, G. Comparison among Slip-Link Simulations of Bidisperse LinearPolymer Melts. Macromolecules 2008, 41, 8275. [Google Scholar] [CrossRef]
- Shanbhag, S. Fast Slip Link Model for Bidisperse Linear Polymer Melts. Macromolecules 2019, 52, 3092–3103. [Google Scholar] [CrossRef]
- Biondo, D.D.; Masnada, E.M.; Merabia, S.; Couty, M.; Barrat, J.L. Numerical study of a slip-link model for polymer melts and nanocomposites. J. Chem. Phys. 2013, 138, 194902. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Kumar, S.K.; Keblinski, P. Viscoelastic properties of polymer melts from equilibrium molecular dynamics simulations. Macromolecules 2005, 38, 650–653. [Google Scholar] [CrossRef]
- Takahashi, K.Z.; Nishimura, R.; Yamato, N.; Yasuoka, K.; Masubuchi, Y. Critical test of bead-spring model to resolve the scaling laws of polymer melts: A molecular dynamics study. Mol. Simul. 2017, 43, 1196–1201. [Google Scholar] [CrossRef]
- Xu, X.; Chen, J.; An, L. Simulation studies on architecture dependence of unentangled polymer melts: A molecular dynamics study. J. Chem. Phys. 2015, 142, 074903. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Uneyama, T. Comparison among multi-chain models for entangled polymer dynamics. Soft Matter 2018, 14, 5986–5994. [Google Scholar] [CrossRef]
- Langeloth, M.; Masubuchi, Y.; Bohm, M.C.; Müller-Plathe, F. Recovering the reptation dynamics of polymer melts in dissipative particle dynamics via slip-springs. J. Chem. Phys. 2013, 138, 104907. [Google Scholar] [CrossRef] [PubMed]
- Uneyama, T.; Masubuchi, Y. Multi-chain slip-spring model for entangled polymer dynamics. J. Chem. Phys. 2012, 137, 154902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megariotis, G.; Vogiatzis, G.G.; Sgouros, A.P.; Theodorou, D.N. Slip spring-based mesoscopic simulations of polymer networks: Methodology and the corresponding computational code. Polymers 2018, 10, 1156. [Google Scholar] [CrossRef]
- Sukumaran, S.K.; Likhtman, A.E. Modeling entangled dynamics: Comparison between stochastic single chain and multichain models. Macromolecules 2009, 42, 4300–4309. [Google Scholar] [CrossRef]
- Khaliullin, R.N.; Schieber, J.D. Self-consistent modeling of constraint release in a single-chain mean-field slip-link model to bidisperse systems. Macromolecules 2009, 42, 7504–7517. [Google Scholar] [CrossRef]
- McLeish, T.C.B. Tube theory of entangled polymer dynamics. Adv. Phys. 2002, 61, 1379–1527. [Google Scholar] [CrossRef]
- Graham, R.S.; Likhtman, A.E.; McLeish, T.C.B. Microscopic theory of linear, entangled polymer chains under rapid deformation including chain stretch and convective constraint release. J. Rheol. 2003, 47, 1171–1200. [Google Scholar] [CrossRef]
- Katzarova, M.; Kashyap, T.; Schieber, J.D.; Venerus, D.C. Linear viscoelastic behavior of bidisperse polystyrene blends: Experiments and slip-link predictions. Rheologica Acta 2018, 57, 327–338. [Google Scholar] [CrossRef]
- Marrucci, G. Relaxation by reptation and tube enlargement—A model for polydisperse polymers. J. Polym. Sci. B 1985, 23, 159–177. [Google Scholar] [CrossRef]
- Qiao, X.; Sawada, T.; Matsumiya, Y.; Watanabe, H. Constraint release in moderately entangled monodisperse star polyisoprene systems. Macromolecules 2006, 39, 7333–7341. [Google Scholar] [CrossRef]
- Ramirez-Hernández, A.; Peters, B.L.; Andreev, M.; Schieber, J.D.; de Pablo, J.J. A multichain polymer slip-spring model with fluctuating number of entanglements for linear and nonlinear rheology. J. Chem. Phys. 2015, 143, 243147. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Hernandez, A.; Peters, B.L.; Schneider, L.; Andreev, M.; Schieber, J.D.; Müller, M.; de Pablo, J.J. A multi-chain polymer slip-spring model with fluctuating number of entanglements: Density fluctuations, confinement, and phase separation. J. Chem. Phys. 2017, 146, 014903. [Google Scholar] [CrossRef] [Green Version]
- Masubuchi, Y. Multichain slip-spring simulations for branch polymers. Macromolecules 2019, 51, 10184–10193. [Google Scholar] [CrossRef]
- Sgouros, A.P.; Megariotis, G.; Theodorou, D.N. Slip-spring model for the linear and nonlinear viscoelastic properties of molten polyethylene derived from atomistic simulations. Macromolecules 2017, 50, 4524–4541. [Google Scholar] [CrossRef]
- Vogiatzis, G.G.; Megariotis, G.; Theodorou, D.N. Equation of state based slip spring model for entangled polymer dynamics. Macromolecules 2017, 50, 3004–3029. [Google Scholar] [CrossRef]
- Nafar Sefiddashti, M.H.; Edwards, M.G.; Khomami, B. Individual chain dynamics of a polyethylene melt undergoing steady shear flow. J. Rheol. 2015, 59, 119–153. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Mavrantzas, V.G.; Theodorou, D.N. Segmental dynamics in polyethylene melts through atomistic molecular dynamics simulations. In Recent Research Topics and Developments in Chemical Physics: From Quantum Scale to Macroscale; Transworld Research Network: Kerala, India, 2008; Volume 661, pp. 179–196. [Google Scholar]
- Loring, R.F. Diffusion of a flexible polymer chain in a disordered medium. J. Chem. Phys. 1988, 88, 6631–6640. [Google Scholar] [CrossRef]
- Dua, A.; Cherayil, B.J. The anomalous diffusion of polymers in random media. J. Chem. Phys. 2000, 112, 421–427. [Google Scholar] [CrossRef]
- Fuchs, M.; Schweizer, K.S. Polymer-mode-coupling theory of finite-size-fluctuation effects in entangled solutions, melts, and gels. 1. General formulation and predictions. Macromolecules 1997, 30, 5133–5155. [Google Scholar] [CrossRef]
- Schweizer, K.S. Microscopic theory of the dynamics of polymeric liquids: General formulation of a mode–mode-coupling approach. J. Chem. Phys. 1989, 91, 5802. [Google Scholar] [CrossRef]
- Schweizer, K.S. Mode-coupling theory of the dynamics of polymer liquids: Qualitative predictions for flexible chain and ring melts. J. Chem. Phys. 1989, 91, 5822–5839. [Google Scholar] [CrossRef]
- Schweizer, K.S.; Szamel, G. Mode-coupling theory ofentangled polymer fluids. Transp. Theory Stat. Phys. 1995, 24, 947–977. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N. A theoretical model for the prediction of diffusion in polymer/SWCNT nanocomposites. Soft Matter 2011, 7, 7334–7341. [Google Scholar] [CrossRef]
- Meth, J.S.; Gam, S.; Choi, J.; Lin, C.C.; Composto, R.J. Excluded volume model for the reduction fo polymer diffusion into nanocomposites. J. Phys. Chem. B 2013, 117, 15675–15683. [Google Scholar]
- Martin, J.; Krutyeva, M.; Monkenbusch, M.; Arbe, A.; Allgaier, J.; Radulescu, A.; Falus, P.; Maiz, J.; Mijangos, C.; Colmenero, J.; et al. Direct observation of confined single chain dynamics by neutron scattering. Phys. Rev. Lett. 2010, 104, 197801. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Gam, S.; Meth, J.S.; Clarke, N.; Winey, K.I. Do attractive polymer-nanoparticle interactions retard polymer diffusion in nanocomposites. Macromolecules 2013, 46, 4502–4509. [Google Scholar] [CrossRef]
- Pierleoni, C.; Capone, B.; Hansen, J.P. A soft effective segment representation of semidilute polymer solutions. J. Chem. Phys. 2007, 127, 171102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locatelli, E.; Capone, B.; Likos, C.N. Multiblob coarse-graining for mixtures of long polymers and soft colloids. J. Chem. Phys. 2016, 145, 174901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Melt self-diffusion data for hydrogenated (or deuterated) poly(butadiene) samples (hPB and dPB, respectively) adjusted to 175 °C, as a function of molecular weight and data for six other polymers from the literature, with the global power law fit [39,42,43,45,46,48,49,50,51]. Reprinted from [39] with permission from American Institute of Physics (AIP).
Figure 1.
Melt self-diffusion data for hydrogenated (or deuterated) poly(butadiene) samples (hPB and dPB, respectively) adjusted to 175 °C, as a function of molecular weight and data for six other polymers from the literature, with the global power law fit [39,42,43,45,46,48,49,50,51]. Reprinted from [39] with permission from American Institute of Physics (AIP).

Figure 2.
Effect of confinement parameter, defined as the interparticle distance relative to the tracer polymer size, on diffusion. The reduced diffusion coefficient of deuterated PS (dPS) with molecular weights M = 23, 49, 168, 532, and 1866 kDa as a function of the confinement parameter yields a master curve. Closed and open symbols represent dPS diffusion in nanocomposites with soft (SiO–PS87k) and hard (SiO–I) nanoparticles, respectively. [64]. Reprinted from [64] with permission from the American Chemical Society (ACS).
Figure 2.
Effect of confinement parameter, defined as the interparticle distance relative to the tracer polymer size, on diffusion. The reduced diffusion coefficient of deuterated PS (dPS) with molecular weights M = 23, 49, 168, 532, and 1866 kDa as a function of the confinement parameter yields a master curve. Closed and open symbols represent dPS diffusion in nanocomposites with soft (SiO–PS87k) and hard (SiO–I) nanoparticles, respectively. [64]. Reprinted from [64] with permission from the American Chemical Society (ACS).

Figure 3.
Results of mapping between multiscale MD of PS and Kremer-Grest MD (KGMD). (a) Diffusion coefficient times M versus molecular weight M, (b) mean square displacement, , of central monomers for 50 kDa PS [77]. Reprinted from [77] with permission from Springer Nature.

Figure 4.
Scaled diffusion coefficient vs. scaled chain length for poly(styrene) (, 485 K, (filled circles) [46], poly(ethylene) (, K, filled squares) [52], PEB2 (, K, filled triangles) [49], bead-spring Kremer-Grest model (, open triangles), the bond-fluctuation model for , (, open squares) [91], and tangent hard spheres at , open circles) [87]. All data are scaled with from the plateau modulus or with 2.25 from . Reprinted from [92] with permission from IOPscience.
Figure 4.
Scaled diffusion coefficient vs. scaled chain length for poly(styrene) (, 485 K, (filled circles) [46], poly(ethylene) (, K, filled squares) [52], PEB2 (, K, filled triangles) [49], bead-spring Kremer-Grest model (, open triangles), the bond-fluctuation model for , (, open squares) [91], and tangent hard spheres at , open circles) [87]. All data are scaled with from the plateau modulus or with 2.25 from . Reprinted from [92] with permission from IOPscience.

Figure 5.
Self-diffusion coefficient of PS melts at K [93], after correcting for the chain length dependent friction coefficient, , as a function of the molecular weight. In the inset vs M is shown at K [93]. Experimental data are corrected for the different temperature ( K) with the temperature dependence reported in Reference [46]. Reprinted from [93] with permission from ACS.
Figure 5.
Self-diffusion coefficient of PS melts at K [93], after correcting for the chain length dependent friction coefficient, , as a function of the molecular weight. In the inset vs M is shown at K [93]. Experimental data are corrected for the different temperature ( K) with the temperature dependence reported in Reference [46]. Reprinted from [93] with permission from ACS.

Figure 6.
(a) Mean square displacement (MSD) of center of mass of the long probe chains with monomers in binary blends with various short matrix chains [80]. (b) Diffusion coefficients of the long probe chains with length and the short matrix chains, as a function of the matrix chain length of the short chains, , in binary blends with fixed long chain segment fractions . The triangles are simulation results obtained from monodisperse melts with fixed chain segment number density (Lennard-Jones units). The thick solid and dotted-dashed lines are the predictions of Equations (1) and (2), respectively, using the z and K values from the experimental work of Green and Kramer [43]. The dotted horizontal line shows the Rouse diffusivity of the probe chains, attained in the matrix offering only frictional, and not topological, resistance to the long chain motion. Reprinted from [80] with permission from ACS.
Figure 6.
(a) Mean square displacement (MSD) of center of mass of the long probe chains with monomers in binary blends with various short matrix chains [80]. (b) Diffusion coefficients of the long probe chains with length and the short matrix chains, as a function of the matrix chain length of the short chains, , in binary blends with fixed long chain segment fractions . The triangles are simulation results obtained from monodisperse melts with fixed chain segment number density (Lennard-Jones units). The thick solid and dotted-dashed lines are the predictions of Equations (1) and (2), respectively, using the z and K values from the experimental work of Green and Kramer [43]. The dotted horizontal line shows the Rouse diffusivity of the probe chains, attained in the matrix offering only frictional, and not topological, resistance to the long chain motion. Reprinted from [80] with permission from ACS.

Figure 7.
(a) The dependence of on N for different bending rigidities in the absence of torsion stiffness, . (b) The dependence of on N for different torsion rigidities, at a constant bending strength . Reprinted from [111] with permission from ACS.
Figure 7.
(a) The dependence of on N for different bending rigidities in the absence of torsion stiffness, . (b) The dependence of on N for different torsion rigidities, at a constant bending strength . Reprinted from [111] with permission from ACS.

Figure 8.
Dependence of diffusivity (normalized to its bulk value) of mildly entangled polymers () for different nanoparticle loading:(i) Maxwell prediction (black line) (ii) simulation data (stars), (iii) PMMA tracer diffusion: diameter nm (circles), (iv) nm (squares), (v) nm (triangles). Reprinted from [119] with permission from ACS.
Figure 8.
Dependence of diffusivity (normalized to its bulk value) of mildly entangled polymers () for different nanoparticle loading:(i) Maxwell prediction (black line) (ii) simulation data (stars), (iii) PMMA tracer diffusion: diameter nm (circles), (iv) nm (squares), (v) nm (triangles). Reprinted from [119] with permission from ACS.

Figure 9.
(a) Model picture of a nanocomposite system, with nanoparticles of radius (where is the length of a chain segment) in a body-centered cubic configuration with periodic boundary conditions. (b) Dependence of D on chain length N in the presence of different particles, at different nanoparticle volume fractions : of small particles of radius (upper triangles), of large particles with radius (open circles), and pure melt (filled circles). Reprinted from [145] with permission from Wiley.
Figure 9.
(a) Model picture of a nanocomposite system, with nanoparticles of radius (where is the length of a chain segment) in a body-centered cubic configuration with periodic boundary conditions. (b) Dependence of D on chain length N in the presence of different particles, at different nanoparticle volume fractions : of small particles of radius (upper triangles), of large particles with radius (open circles), and pure melt (filled circles). Reprinted from [145] with permission from Wiley.

Figure 10.
Diffusion as a function of chain length for mSRP and SRP. Rouse (dash) and reptation (dash-dot) scaling limits intersect at approximately for mSRP. The initial deviation from Rouse diffusion scaling for mSRP and SRP occurs at (solid) and (small dash), respectively. Reprinted from [161] with permission from ACS.
Figure 10.
Diffusion as a function of chain length for mSRP and SRP. Rouse (dash) and reptation (dash-dot) scaling limits intersect at approximately for mSRP. The initial deviation from Rouse diffusion scaling for mSRP and SRP occurs at (solid) and (small dash), respectively. Reprinted from [161] with permission from ACS.

Figure 11.
(a) Self-diffusion coefficients of linear polymers as a function of molecular mass. The black dots are the results of Fan and Liu [165], the filled circles represent experimental data from Pearson et al. [52], and the plus symbols show molecular dynamics simulation results obtained by Mondello and Grest [166]. The dashed lines illustrate the slopes expected for the Rouse regime (left, ) and the reptation regime (right, ). Reprinted from [165] with permission from IOPscience. (b) Proper scaling limits are reached for the diffusion coefficient D, by DPD simulations ( is the force bond constant). Reprinted from [167] with permission from the American Physical Society (APS).
Figure 11.
(a) Self-diffusion coefficients of linear polymers as a function of molecular mass. The black dots are the results of Fan and Liu [165], the filled circles represent experimental data from Pearson et al. [52], and the plus symbols show molecular dynamics simulation results obtained by Mondello and Grest [166]. The dashed lines illustrate the slopes expected for the Rouse regime (left, ) and the reptation regime (right, ). Reprinted from [165] with permission from IOPscience. (b) Proper scaling limits are reached for the diffusion coefficient D, by DPD simulations ( is the force bond constant). Reprinted from [167] with permission from the American Physical Society (APS).

Figure 12.
Schematics of slip-spring models. (a) Entangled chains colored in an entangled polymer melt, (b) corresponding slip-spring primitive chain network (PCN) model [178], and (c) a group of chains considered in the Doi and Takimoto (DT) model [179]. Reproduced from [183] with permission from Polymers.
Figure 12.
Schematics of slip-spring models. (a) Entangled chains colored in an entangled polymer melt, (b) corresponding slip-spring primitive chain network (PCN) model [178], and (c) a group of chains considered in the Doi and Takimoto (DT) model [179]. Reproduced from [183] with permission from Polymers.

Figure 13.
(a) Center-of-mass diffusion coefficient and (b) its normalized value with respect to the classical reptation behavior (bottom) plotted against the number of beads per chain, N. Red solid and blue dotted curves are the results of multi-chain slip-spring (MCSS) and primitive chain network (PCN). The MCSS and PCN data are rescaled by the scale-conversion factors. KG results obtained from the literature [26,96,111,187,188,189] are shown by symbols. Error bar shows the standard deviation for 8 independent simulation runs. Reproduced from [190] with permission from the Royal Society of Chemistry.
Figure 13.
(a) Center-of-mass diffusion coefficient and (b) its normalized value with respect to the classical reptation behavior (bottom) plotted against the number of beads per chain, N. Red solid and blue dotted curves are the results of multi-chain slip-spring (MCSS) and primitive chain network (PCN). The MCSS and PCN data are rescaled by the scale-conversion factors. KG results obtained from the literature [26,96,111,187,188,189] are shown by symbols. Error bar shows the standard deviation for 8 independent simulation runs. Reproduced from [190] with permission from the Royal Society of Chemistry.

Figure 14.
From the atomistic to the mesoscopic representation. (a) Mapping of a real polymer chain with 260 carbon atoms onto a sequence of five coarse-grained beads (). (b) Two approaches for mapping atomistic polymer segments onto beads. A bead can be assigned either to the center of mass of a segment or to the coordinates of the central atom(s) of a segment. Reprinted from [204] with permission from ACS.
Figure 14.
From the atomistic to the mesoscopic representation. (a) Mapping of a real polymer chain with 260 carbon atoms onto a sequence of five coarse-grained beads (). (b) Two approaches for mapping atomistic polymer segments onto beads. A bead can be assigned either to the center of mass of a segment or to the coordinates of the central atom(s) of a segment. Reprinted from [204] with permission from ACS.

Figure 15.
(a) Dependence of the self-diffusion coefficient D and the longest relaxation time (obtained from the autocorrelation function of the end-to-end vector) on molar mass, from this work (MD, circles; BD/kMC, red squares) [204], as well as (b) from various simulation works in the literature (green triangles [109], blue crosses [206], orange X-markers [207], black dashes [76], purple diamonds [27]). The dotted line is a fit on the experimental data performed by Lodge performed by Lodge [39] while the dashed lines are guides to the eye. Reprinted from [204] with permission from ACS.
Figure 15.
(a) Dependence of the self-diffusion coefficient D and the longest relaxation time (obtained from the autocorrelation function of the end-to-end vector) on molar mass, from this work (MD, circles; BD/kMC, red squares) [204], as well as (b) from various simulation works in the literature (green triangles [109], blue crosses [206], orange X-markers [207], black dashes [76], purple diamonds [27]). The dotted line is a fit on the experimental data performed by Lodge performed by Lodge [39] while the dashed lines are guides to the eye. Reprinted from [204] with permission from ACS.

Figure 16.
(a) Diffusivity of tracer of different molecular weights in an entangled matrix (480 kDa PS) as a function of the SWCNT volume fraction [214] in a PS/SWCNT nanocomposite system: (i) theoretical prediction for 75 kDa dPS (blue line), (ii) 140 kDa dPS (red line), (iii) 680 kDa dPS (green line line) (iv) experimental results [69] for 75 kDa dPS (squares), (v) experimental results for 140 kDa dPS (diamonds), and (vi) experimental results for 680 kDa dPS (circles). (b) Normalized diffusivity of tracer in an entangled matrix as a function of the number of monomers of the entangled polymer chains at the SWCNT percolation threshold (): (i) theoretical prediction for 18 kDa PS matrix (green line) (ii) 75 kDa PS matrix (red line) (iii) 480 kDa PS matrix (black line) (iv) 680 kDa PS matrix (blue line) (v) experimental results for PS/SWCNT system in 480 kDa PS matrix at (symbols) [69]. Reproduced from [214] with permission from the Royal Society of Chemistry.
Figure 16.
(a) Diffusivity of tracer of different molecular weights in an entangled matrix (480 kDa PS) as a function of the SWCNT volume fraction [214] in a PS/SWCNT nanocomposite system: (i) theoretical prediction for 75 kDa dPS (blue line), (ii) 140 kDa dPS (red line), (iii) 680 kDa dPS (green line line) (iv) experimental results [69] for 75 kDa dPS (squares), (v) experimental results for 140 kDa dPS (diamonds), and (vi) experimental results for 680 kDa dPS (circles). (b) Normalized diffusivity of tracer in an entangled matrix as a function of the number of monomers of the entangled polymer chains at the SWCNT percolation threshold (): (i) theoretical prediction for 18 kDa PS matrix (green line) (ii) 75 kDa PS matrix (red line) (iii) 480 kDa PS matrix (black line) (iv) 680 kDa PS matrix (blue line) (v) experimental results for PS/SWCNT system in 480 kDa PS matrix at (symbols) [69]. Reproduced from [214] with permission from the Royal Society of Chemistry.

Figure 17.
(a) Plot of reduced diffusion coefficient [215] versus the confinement parameter for the poly(styrene) data from Martin et al. [216]. (b) Reduced diffusion coefficient versus 2D confinement parameter for the deuterated PMMA/PMMA couple from Lin et al. [217]. Reproduced from [215] with permission from AIP.
Figure 17.
(a) Plot of reduced diffusion coefficient [215] versus the confinement parameter for the poly(styrene) data from Martin et al. [216]. (b) Reduced diffusion coefficient versus 2D confinement parameter for the deuterated PMMA/PMMA couple from Lin et al. [217]. Reproduced from [215] with permission from AIP.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Karatrantos, A.; Composto, R.J.; Winey, K.I.; Kröger, M.; Clarke, N. Modeling of Entangled Polymer Diffusion in Melts and Nanocomposites: A Review. Polymers 2019, 11, 876. https://doi.org/10.3390/polym11050876
AMA Style
Karatrantos A, Composto RJ, Winey KI, Kröger M, Clarke N. Modeling of Entangled Polymer Diffusion in Melts and Nanocomposites: A Review. Polymers. 2019; 11(5):876. https://doi.org/10.3390/polym11050876
Chicago/Turabian StyleKaratrantos, Argyrios, Russell J. Composto, Karen I. Winey, Martin Kröger, and Nigel Clarke. 2019. "Modeling of Entangled Polymer Diffusion in Melts and Nanocomposites: A Review" Polymers 11, no. 5: 876. https://doi.org/10.3390/polym11050876
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.