3.1. Phase Morphology of PCL/PBS Blends and PCL/PBS/(PC/MWCNTs) Blend Nanocomposites
Phase morphology plays an important role in the mechanical behavior of polymer blends. The type of morphology and the sizes of dispersed phases in the polymer blends are important factors that determine the physical properties of these blends. In order to evaluate the morphology of the PCL/PBS blends and its filled nanocomposites, scanning electron microscopic (SEM) and atomic force microscopic (AFM) analyses were conducted.
Figure 1 shows the SEM micrographs of the PCL/PBS blends and the PCL/PBS/(PC/MWCNTs) blend nanocomposites at different blend and masterbatch ratios.
The SEM images in
Figure 1a,f show a sea-island morphology with discrete droplets of the minor phase in the matrix of the major phase, typical of immiscible polymer blends. The approximate diameters of the dispersed minor phases are shown in
Table 2. It can be seen that the size of the minor phase for the 30/70 PCL/PBS blend (
Figure 1a) is smaller than that of the 70/30 PCL/PBS blend (
Figure 1f). There are several factors that determine the final particle size of the dispersed phases in polymer blends, such as surface energy, interfacial tension, polar character, blend composition, molar mass, viscosity ratio, and differences between the degree of crystallinity of the components in the blend, as well as time, shear stress, and temperature of mixing [
21]. Amongst the mentioned factors, viscosity ratio plays a major role in the sizes of the dispersed phases [
22].
In the case of the blends filled with the (PC/MWCNTs) masterbatch (
Figure 1b–e), the SEM images show three phases: (i) The matrix (either PCL or PBS rich phase depending on the composition), (ii) dispersed polymer droplets of small size (either PCL or PBS rich phase depending on the composition), typically 3 μm or less, and (iii) dispersed phases of tens of micron sizes containing large concentrations of MWCNTs, which clearly is the (PC/MWCNTs) masterbatch, and which are indicated in
Figure 1 with arrows. However, although most MWCNTs are located in the PC dispersed phases (
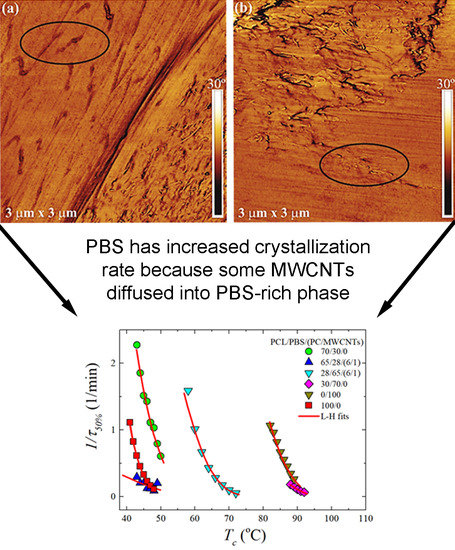
Figure 1d,e), some of the MWCNTs (indicated with a black circle in the AFM images in
Figure 2) migrated from the dispersed PC phase to the polymer matrix outside the dispersed phase. This is due to the partial miscibility and intimate contact at the interfaces between the PC-rich and PCL-rich phases, or the PC-rich and PBS-rich phases, as previously reported [
23,
24,
25,
26].
It is worth noting that when the PCL/PBS blends were loaded with 1 wt% MWCNTs, the sizes of the dispersed polymer droplets for the 65/28/(6/1) w/w PCL/PBS/(PC/MWCNTs) nanocomposite were smaller than those of the 28/65/(6/1) w/w PCL/PBS/(PC/MWCNTs) nanocomposite. When the PCL/PBS blends were loaded with 4 wt% MWCNTs, the sizes of the dispersed polymer droplets were smaller in the 22/51/(23/4) w/w PCL/PBS/(PC/MWCNTs) nanocomposite than those in the 51/22/(23/4) w/w PCL/PBS/(PC/MWCNTs) blend nanocomposite (
Table 2). The smaller droplets indicate that there is compatibilization between the component phases. However, amongst the nanocomposites investigated in this paper, the 22/51/(23/4) PCL/PBS/(PC/MWCNTs) nanocomposite is the one giving the smallest particle sizes of the dispersed phase. This is due to the lower interfacial tension values between PBS and the PC/MWCNTs masterbatch compared to those between PCL and the PC/MWCNTs masterbatch (see
Table 3). At equilibrium the particles will likely disperse in the phase where the affinity between the polymer and the nanoparticles is high.
The contact angles, total surface energies, as well as their dispersive and polar surface components, were calculated using the Owens-Wendt method [
12,
27,
28] (Equations (1) and (2)).
where
θ is the contact angle,
γ is the surface energy, the subscripts ‘
s’ and ‘
l’ respectively indicate solid and liquid, while ‘
d’ and ‘
p’ respectively indicate the dispersive and polar components. If the contact angle of at least two liquids, usually polar and nonpolar liquids with known
and
values, are measured on a solid surface, the
and
and the total surface energy (
) of the solid can be calculated by using Equations (2) and (3) [
29]. The interfacial tensions between the components in a blend were calculated from the surface energy measurement results using the geometric mean equation (Equation (3)) [
12], and the wetting coefficient (Equation (4)) from the interfacial tensions.
where
= interfacial tension between components 1 and 2 in the blend,
and
are the total surface energies of components 1 and 2,
and
are the dispersive surface energies of components 1 and 2, and
and
are the polar surface energies of the components in the nanocomposites. The wetting coefficient, w
α, is calculated by using Equation (4).
where γ
polymerB-Filler is the interfacial tension between polymer B and the filler, γ
polymerA-Filler the interfacial tension between polymer A and the filler, and γ
polymerA-polymerB the interfacial tension between polymers A and B. The value of the wetting coefficient is normally used to determine where the filler is likely expected to disperse. If ω
α < −1, the particles are predicted to be localised in polymer B, if ω
α > 1, they are dispersed in polymer A, and if the value of ω
α is between −1 and 1, the nanoparticles are likely dispersed on the interface between the two polymers in the blend. In rare cases where the particles are dispersed in both the interface and one of the phases, the third condition does not apply, so that a negative ω
α indicates dispersion of the particles in polymer B as well as the interface, and a positive ω
α indicates dispersion of the particles in polymer A and on the interface [
30,
31]. The results are summarised in
Table 3 and
Table 4.
Table 3 shows that the polar component of the surface energy, γ
p, for the masterbatch is closer to that of PBS. In terms of the interfacial tension values reported in
Table 4, it can be seen that the interfacial tension between PCL and (PC/MWCNTs) (0.97 mN m
−1) is larger than that between PBS and (PC/MWCNTs) (0.36 mN m
−1). These results and the −0.26 mN m
−1 wetting coefficient value suggest that the nanotubes would preferably disperse better in the PBS-rich phase.
Similar results were reported by He et al. [
11], who introduced MWCNTs to double crystalline PBS/PCL blends using solution mixing and solvent casting processes. The MWCNTs exhibited a much finer dispersion morphology in the PBS/MWCNT nanocomposite than in the PCL/MWCNT nanocomposite. This was attributed to the wetting coefficient data for MWCNTs introduced into the binary copolymer, which was calculated as 1.73 (harmonic-mean equation) or −3.43 (geometric-mean equation). According to the authors, this meant that the MWCNTs were selectively distributed in the PBS phase.
Taking into account the results presented so far, we can conclude that a fair number of MWCNTs diffused from the PC-rich phase into the PCL-rich and PBS-rich phases, although one would expect, from the interfacial tension results, to find more MWCNTs in the PBS-rich phase. The majority of the MWCNTs were, however, still confined to the PC-rich droplets, despite the partial miscibility of the blends, and the intimate phase boundaries between the different components in the blends.
The ternary morphology obtained for the blends with a PC/MWCNT masterbatch, and especially the large sizes of the PC/MWCNTs phases, is obviously not the best as stress transfer may not be ideal, even when the phase boundaries obtained are small. Tuning of the morphology to obtain better mixing will have to be studied by varying extrusion conditions, screw configurations and processing variables in general. However, such a study was outside the scope of the present work.
3.3. Non-Isothermal DSC
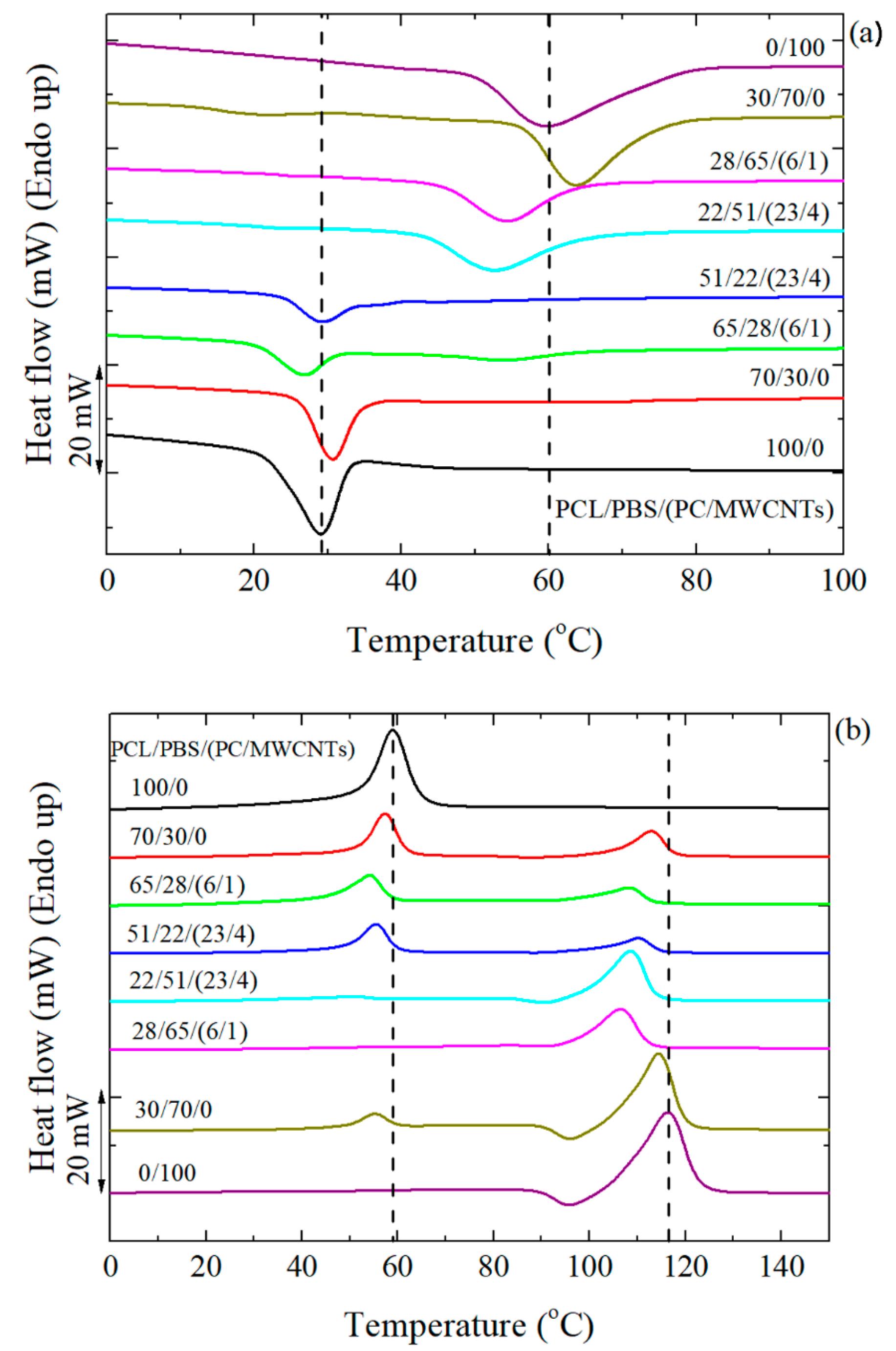
Figure 3a shows the DSC cooling scans after erasing the thermal history, and
Figure 3b the subsequent heating scans performed at 20 °C min
−1 for the different investigated samples. Since PBS crystallizes first, its crystallization peak appears at about 60 °C, followed by the crystallization peak of the PCL component at 30 °C.
When 30 wt% PCL was added to 70 wt% PBS, there was a shift in the crystallization temperature (
Tc) of PBS to higher temperatures. This indicates a nucleation effect of the PBS component in the blend, which can only be produced by a transfer of impurities from the PCL to the PBS component, as the PCL is molten at the relevant temperatures for PBS crystallization [
32]. In the case of adding 30 wt% PBS to 70 wt% PCL, there was no significant shift in the
Tc value of the PCL component. This is because the PCL/PBS blends are immiscible as evidenced by the composition independent glass transition temperature and the biphasic melt [
7]. Other authors studied the miscibility behavior of a related poly(ε-caprolactone)/poly(propylene succinate) (PCL/PPSu) blend. The PCL/PPSu blends were found to show a very limited miscibility in the molten state, since the polymer-polymer interaction parameter (χ
12) was −0.11 [
33].
When the PCL/PBS blends were loaded with different amounts of PC/MWCNTs masterbatch, the Tc of the PBS-rich nanocomposites shifted to lower temperatures than those of the neat PBS and the blend with PBS as the major phase. The same is true for the PCL-rich nanocomposites, although the change was less significant. This is due to the competition between two effects: (1) The partial miscibility of the PC-rich, the PCL-rich, and the PBS-rich phases, and (2) the nucleation effect of the MWCNTs. Miscibility between the different components in the blend tends to decrease the values of Tc (as high Tg PC chains are being solubilized in the PBS-rich phase and to a lower extent in the PCL-rich phase) while the nucleation effect tends to increase the Tc values. In this case, since the MWCNTs and PC were simultaneously added, the miscibility effect dominates because of the decrease in the Tc values of the nanocomposites.
In the subsequent heating scans shown in
Figure 3b, the melting temperatures (
Tm) of the 70/30 and 30/70 PCL/PBS blends show very little variation compared to the homopolymers. Normally, when the blends are immiscible, the
Tm is not affected. In the case of the filled nanocomposites, the
Tm values decreased (more especially in the PBS-rich nanocomposites) compared to the neat materials and the blends. The decrease in the melting temperature values is due to miscibility effects between the PC-rich and the PCL-rich or PBS-rich phases. The PCL melting peak in the 28/65/(6/1) PCL/PBS/(PC/MWCNTs) nanocomposite is almost invisible. This is probably due to the PCL partial miscibility with the more rigid PC chains, making it difficult for PCL to crystallize, therefore forming fewer crystals.
To further examine the results presented in
Figure 3, the
Tc and
Tm values were plotted as a function of sample composition in
Figure 4.
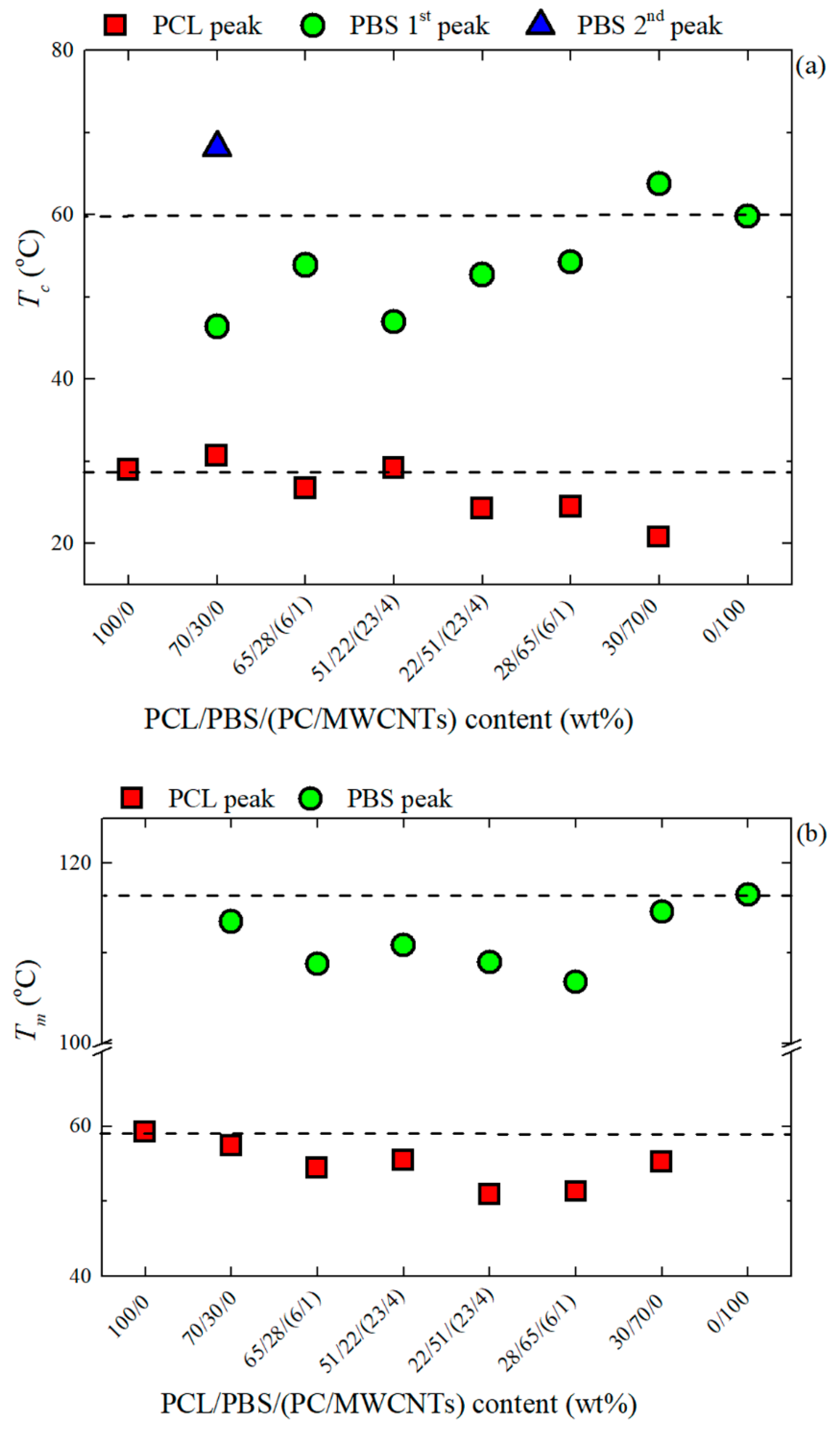
Figure 4a shows that the components in the 30/70 PCL/PBS blend are slightly more nucleated than the components in the 70/30 PCL/PBS blend. This is because of the smaller particle size of the PCL phase observed in the 30/70 blend (d
n = 0.4 microns) compared to that of the PBS component in the 70/30 blend (d
n = 1.3 microns), giving rise to a better dispersion and enhanced nucleation. The decrease in the
Tc values for the nanocomposites is attributed to the competition between partial miscibility with the PC-rich phase and the nucleation effect of the MWCNTs. It is clear that miscibility is the dominating factor.
In
Figure 4b, the changes in
Tm are most significant when the masterbatch is added, because of the partial miscibility between the PCL-rich and PC-rich phases, as well as the PBS-rich and PC-rich phases. In the PBS-rich phase nanocomposites, the decrease in
Tm is more significant than in the PCL-rich nanocomposites. This is an indication that the PBS-rich phase is more compatible with the PC-rich phase than the PCL-rich phase, as indicated by the interfacial tension values in
Table 4.
Figure 5 shows the normalized crystallization and melting enthalpy values for all the samples. The normalized enthalpies for the PCL/PBS blends are almost the same as those of the homopolymers. This indicates that the total crystallinities of each of the two polymers are not significantly influenced by the presence of the other polymer in the blend. However, in the presence of the masterbatch, the normalized crystallization and melting enthalpy values for both polymers are lower than those of the neat polymers. This is due to the PC addition. When PC is added to the complex ternary blend, the resulting
Tg of the PCL-rich phase or the PBS-rich phase will be higher, as long as there is some partial solubility of PC chains in these respective phases. An effectively higher
Tg will make chain diffusion to the crystallization front more difficult. As previously discussed above, the DMTA results do show a significant increase of the
Tg for the PBS-rich phase when the PC/Masterbatch is added. In the case of the PCL-phase, the trend is not very regular with composition, but some slightly higher values are observed upon PC/Masterbatch addition depending on which criterion variable is used to determine the
Tg values.
The general trends observed in
Figure 4 and
Figure 5 indicate that the PBS-rich thermal properties are more affected by the addition of PCL and PC/Masterbatch than in the case of the PCL-rich phase (with the addition of PBS and PC/Masterbatch). This is consistent with the already mentioned notion that the miscibility between PBS and PC seems to be higher than that of PC and PCL, for the polymers employed in this work. It must be remembered that miscibility is a strong function of molecular weight.
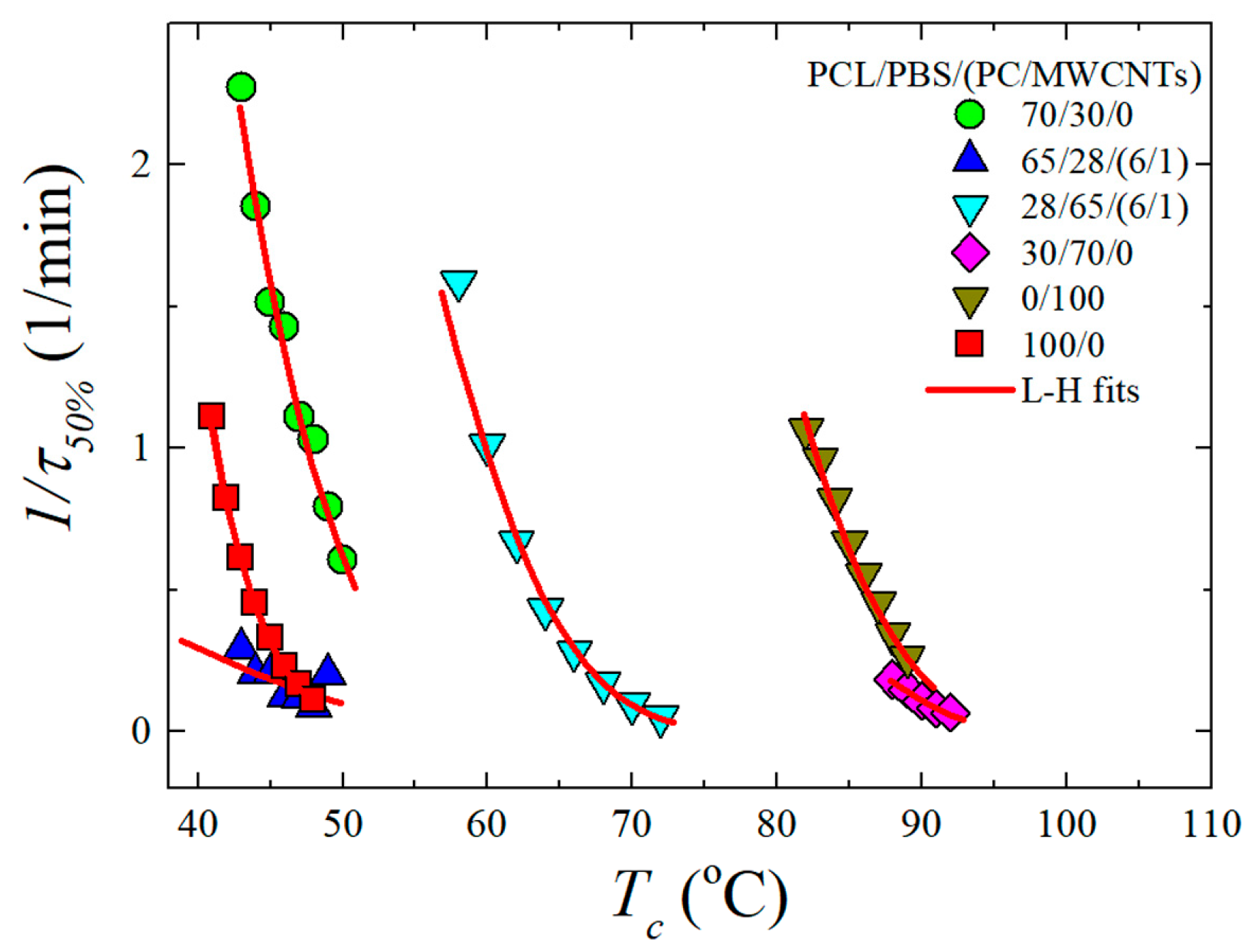
3.4. Overall Isothermal Crystallization Studied by DSC
The blend consists of two crystallisable components, i.e., PCL and PBS. Therefore, the influence of the PC/MWCNTs masterbatch on the isothermal crystallization kinetics of PCL (for temperatures below 60 °C) and PBS (for temperatures above 60 °C) is presented in this section. The inverse of the half crystallization time (1/
t50%) as a function of the isothermal crystallization temperature (
Tc) for some of the investigated samples is shown in
Figure 6. PBS clearly crystallizes at a higher temperature than PCL. The experimental data was fitted with the Lauritzen and Hoffman (LH) theory (see the
Supplementary Information).
The PBS component in the neat 30/70 PCL/PBS blend shows a small change in the overall crystallization kinetics in comparison to the neat PBS. This is because the 30/70 PCL/PBS blend is immiscible as observed in the SEM images (
Section 3.1). However, since the 30/70 blend shows nucleation effects, one would expect to see some increase in the overall crystallization rate, which includes both nucleation and growth components. But apparently, under isothermal conditions, the moderate nucleation effects observed in non-isothermal conditions (i.e., cooling from the melt), are not relevant.
When the PC/MWCNTs masterbatch is added to the 30/70 PCL/PBS blend, the crystallization rate of the PBS rich component in the blend is substantially lower than that of the neat PBS and the blend. This is because of the partial miscibility between the PC-rich and the PBS-rich phases. Miscible PC chains (that are more rigid than PBS chains) within the PBS-rich phases can decrease the rate of crystallization.
The PCL component in the 70/30 blend crystallizes faster than neat PCL. This is attributed to a nucleation effect of the previously crystallized PBS-rich phase. In order to perform the crystallization kinetics of the 70/30 blend, the sample is cooled from the melt and PBS can crystallize during such cooling. When the low Tc temperatures needed to crystallize the PCL component are reached, the PBS has already finished crystallizing. Then the equipment is switched to the isothermal mode and the crystallization of the PCL component is measured. The blend with the masterbatch shows a slightly lower crystallization rate. This is due to the competition between the nucleation of the MWCNTs and the partial miscibility with PC. However, the miscibility effect obviously dominated, giving rise to a lower crystallization rate.
It is worth noting that the difference between the crystallization rate of neat PBS and the PBS-rich blend nanocomposite is much larger than that between the neat PCL and the PCL-rich blend nanocomposite. This suggests that the PBS-rich phase is more miscible with PC in comparison to the PCL-rich phase. A result consistent with the non-isothermal crystallization data presented above.
Fitting DSC Isothermal Data to the Avrami Model
The data obtained during the isothermal crystallization experiments were analysed using the Avrami equation (Equation (5)) [
34].
where
t is the experimental time,
t0 is the induction time,
Vc is the relative volumetric transformed fraction,
n is the Avrami index, and
K is the overall crystallization rate constant. The procedure used to perform the fittings to the data was developed by Lorenzo et al. [
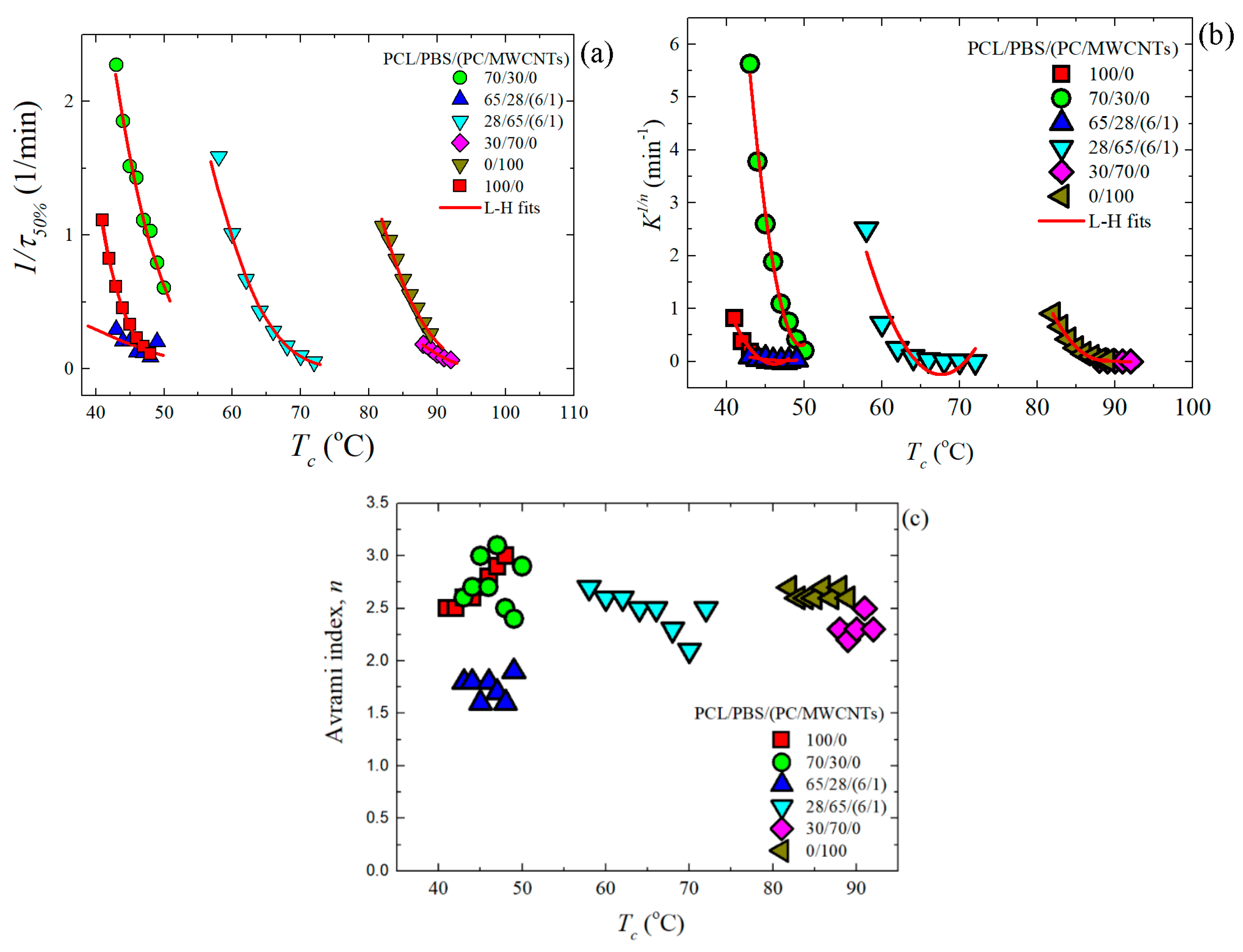
19]. The kinetic parameters for all the investigated samples are plotted in
Figure 7 and tabulated in
Table S1.
Figure 7a shows 1/
t50%-values as a function of
Tc, the trend of which was explained earlier in the discussion (
Section 3.3). A similar trend was obtained with the
K1/n-values of the Avrami model (see
Figure 7b), since this constant is also related to the overall crystallization rate.
Figure 7c shows the Avrami index values,
n, for all the samples. In the case of the PCL-rich samples, neat PCL and the 70/30 blend have
n-values between 2.5 and 3.0. This is consistent with instantaneously nucleated spherulites. However, for its filled nanocomposite (65/28/(6/1) PCL/PBS/(PC/MWCNTs)), the value of
n is between 1.5 and 2.0, which is approximately 2.0, indicating instantaneous axialites. It is known that the addition of a nucleating agent can cause the Avrami index values to change from 3.0 to 2.0 (as the dimensionality of growth can switch from 3D to 2D when the nucleation density is greatly enhanced) [
19,
34,
35]. It was demonstrated in our previous study that the MWCNTs nucleate the PCL-rich phase [
23]. The carbon nanotubes therefore affect the nucleation and the resultant morphology, while the crystallization rate is determined by the miscibility between the components.
The Avrami index values for neat PBS are within the range of 2.5–2.7, which is close to 3.0 indicating spherulitic morphology with instantaneous nucleation. For the blend and its nanocomposite, the n-values can be averaged to 2.3. This is because of a larger number of nucleation sites, which caused the formation of more crystals with an accompanying decrease in the probability of developing 3-D spherulites. 2D axialites instantaneously nucleated are probably formed in this case.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}