
Searching for a Needle in a Haystack: Cas9-Targeted Nanopore Sequencing and DNA Methylation Profiling of Full-Length Glutenin Genes in a Big Cereal Genome

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
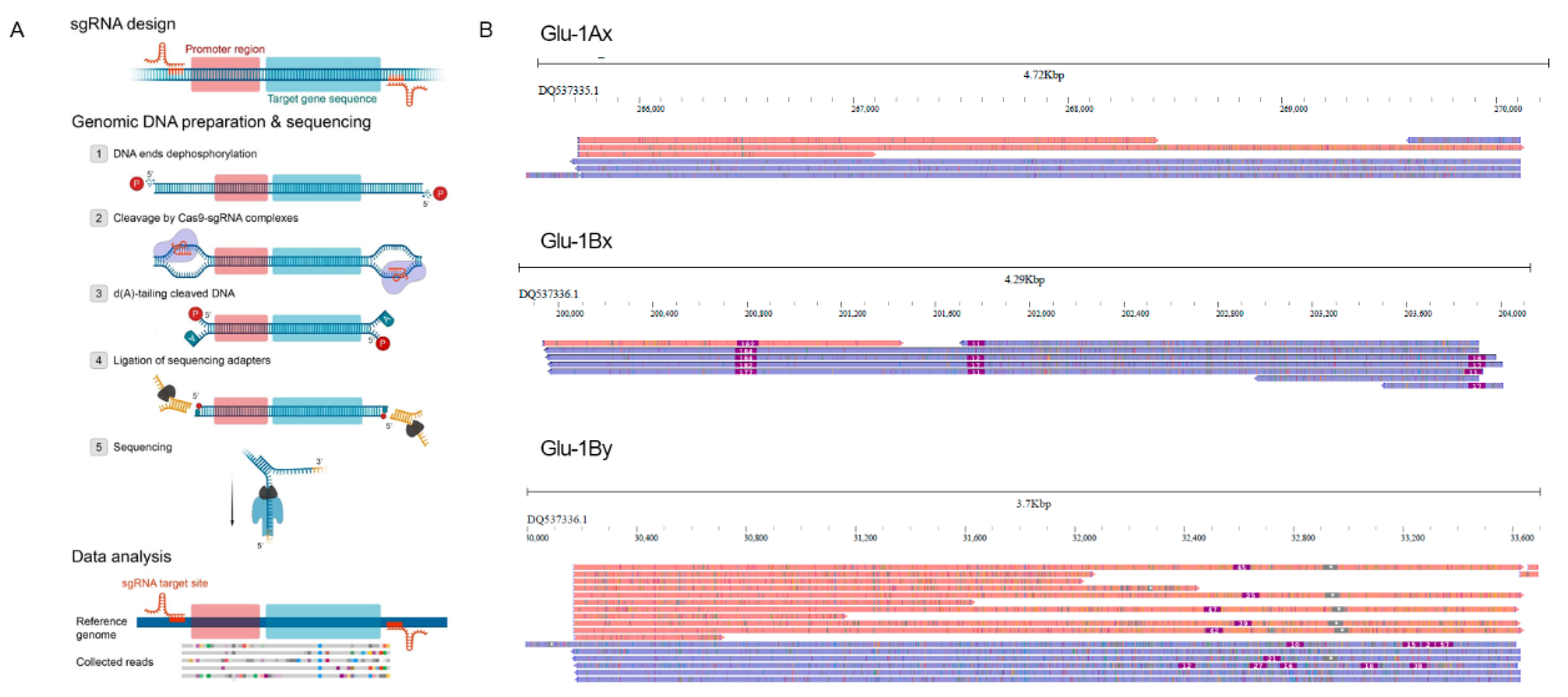
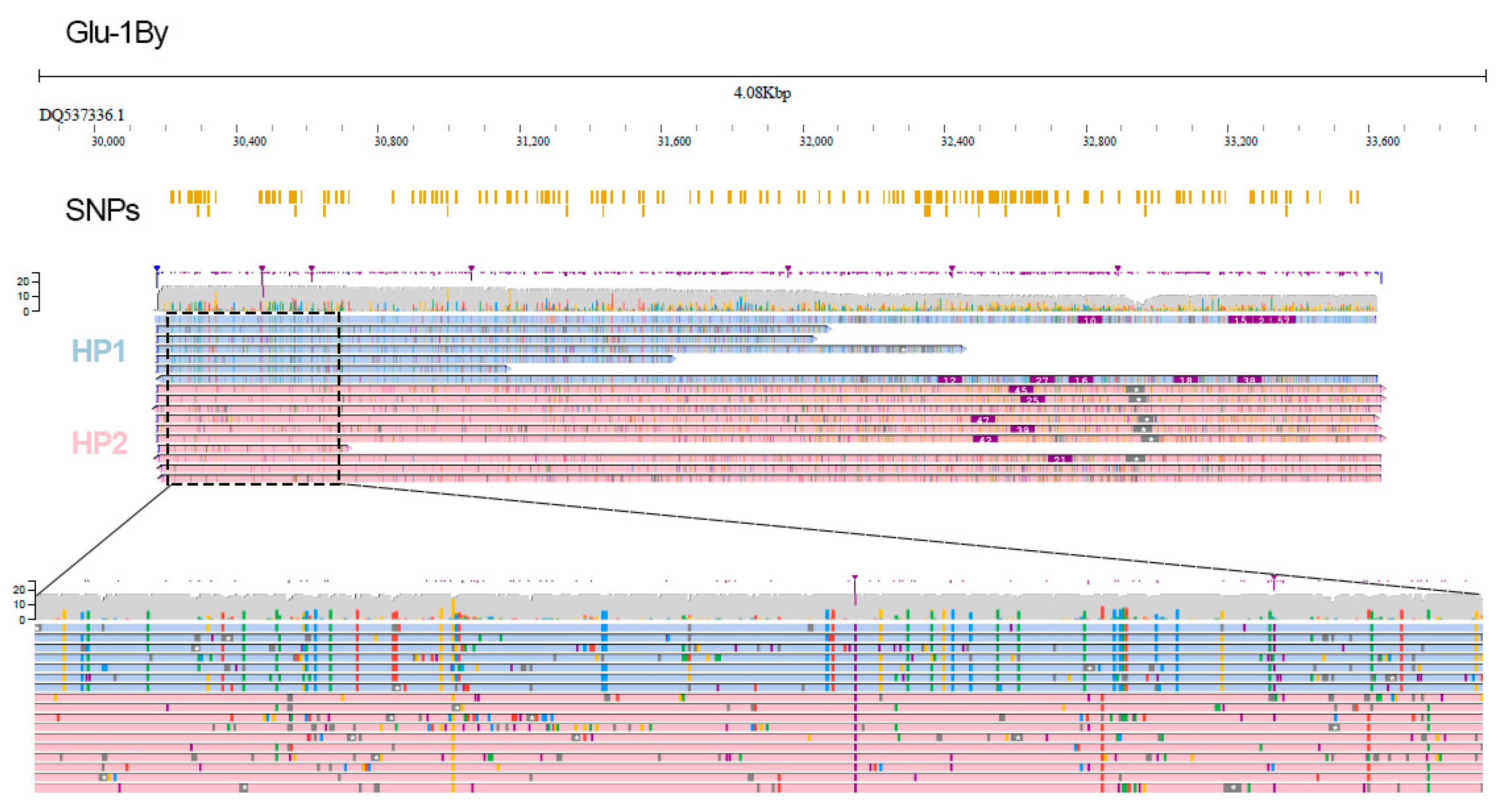
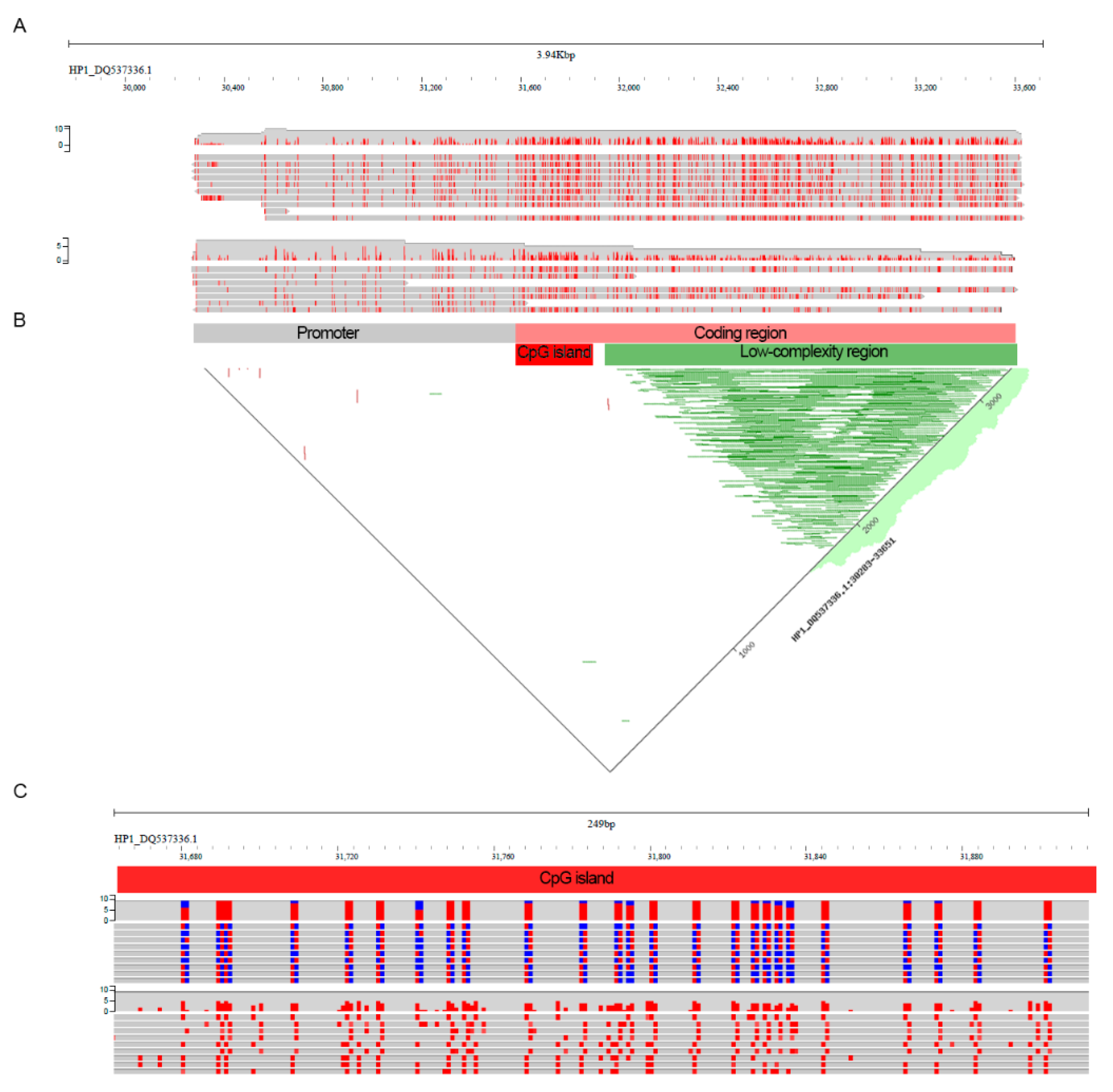
2. Results
3. Discussion
4. Materials and Methods
4.1. Plant Material and DNA Isolation
4.2. gRNA Design and In Vitro Transcription
4.3. nCATS Library Preparation
4.4. Nanopore Sequencing and Basecalling
4.5. SNP Calling and Phasing
4.6. Methylation Calling and Visualization
4.7. PCR Validation of the Insertion in Glu-1Bx Gene
4.8. Phylogenetic Tree Construction
4.9. Visualization and Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kozarewa, I.; Armisen, J.; Gardner, A.F.; Slatko, B.E.; Hendrickson, C.L. Overview of target enrichment strategies. Curr. Protoc. Mol. Biol. 2015, 112, 7.21.21–27.21.23. [Google Scholar] [CrossRef]
- Madsen, E.B.; Höijer, I.; Kvist, T.; Ameur, A.; Mikkelsen, M.J. Xdrop: Targeted sequencing of long DNA molecules from low input samples using droplet sorting. Hum. Mutat. 2020, 41, 1671–1679. [Google Scholar] [CrossRef]
- Gabrieli, T.; Sharim, H.; Fridman, D.; Arbib, N.; Michaeli, Y.; Ebenstein, Y. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Acids Res. 2018, 46, e87. [Google Scholar] [CrossRef] [Green Version]
- Bennett-Baker, P.E.; Mueller, J.L. CRISPR-mediated isolation of specific megabase segments of genomic DNA. Nucleic Acids Res. 2017, 45, e165. [Google Scholar] [CrossRef] [Green Version]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef]
- Yuen, Z.W.-S.; Srivastava, A.; Daniel, R.; McNevin, D.; Jack, C.; Eyras, E. Systematic benchmarking of tools for CpG methylation detection from nanopore sequencing. Nat. Commun. 2021, 12, 3438. [Google Scholar] [CrossRef] [PubMed]
- Giesselmann, P.; Brändl, B.; Raimondeau, E.; Bowen, R.; Rohrandt, C.; Tandon, R.; Kretzmer, H.; Assum, G.; Galonska, C.; Siebert, R.; et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat. Biotechnol. 2019, 37, 1478–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, P.; Huang, N.; Nie, F.; Zhang, J.; Zhang, Z.; Wu, B.; Bai, L.; Liu, W.; Xiao, C.-L.; Luo, F.; et al. Genome-wide detection of cytosine methylations in plant from Nanopore data using deep learning. Nat. Commun. 2021, 12, 5976. [Google Scholar] [CrossRef] [PubMed]
- López-Girona, E.; Davy, M.W.; Albert, N.W.; Hilario, E.; Smart, M.E.M.; Kirk, C.; Thomson, S.J.; Chagné, D. CRISPR-Cas9 enrichment and long read sequencing for fine mapping in plants. Plant Methods 2020, 16, 121. [Google Scholar] [CrossRef]
- Kron, P.; Husband, B.C.H.C. Hybridization and the reproductive pathways mediating gene flow between native Malus coronaria and domestic apple, M. domestica. Botany 2009, 87, 864–874. [Google Scholar] [CrossRef]
- Bennett, M.D.; Smith, J.B.; Riley, R. Nuclear DNA amounts in angiosperms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1976, 274, 227–274. [Google Scholar] [CrossRef]
- Hülgenhof, E.; Weidhase, R.A.; Schlegel, R.; Tewes, A. Flow cytometric determination of DNA content in isolated nuclei of cereals. Genome 1988, 30, 565–569. [Google Scholar] [CrossRef]
- Mergoum, M.; Singh, P.K.; Peña, R.J.; Lozano-del Río, A.J.; Cooper, K.V.; Salmon, D.F.; Gómez Macpherson, H. Triticale: A “New” crop with old challenges. In Cereals; Carena, M.J., Ed.; Springer US: New York, NY, USA, 2009; pp. 267–287. [Google Scholar]
- Clavijo, B.J.; Venturini, L.; Schudoma, C.; Accinelli, G.G.; Kaithakottil, G.; Wright, J.; Borrill, P.; Kettleborough, G.; Heavens, D.; Chapman, H.J.G.r. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017, 27, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Kirov, I.; Merkulov, P.; Gvaramiya, S.; Komakhin, R.; Omarov, M.; Dudnikov, M.; Kocheshkova, A.; Soloviev, A.; Karlov, G.; Divashuk, M. Illuminating the transposon insertion landscape in plants using Cas9-targeted Nanopore sequencing and a novel pipeline. bioRxiv 2021, 10, 1101. [Google Scholar] [CrossRef]
- Bennet, M.D.; Leitch, I.J.; Price, H.J.; Johnston, J.S. Comparisons with Caenorhabditis (∼100 Mb) and Drosophila (∼175 Mb) Using Flow Cytometry Show Genome Size in Arabidopsis to be ∼157 Mb and thus ∼25% Larger than the Arabidopsis Genome Initiative Estimate of ∼125 Mb. Ann. Bot. 2003, 91, 547–557. [Google Scholar] [CrossRef]
- Shewry, P.R.; Halford, N.G. Cereal seed storage proteins: Structures, properties and role in grain utilization. J. Exp. Bot. 2002, 53, 947–958. [Google Scholar] [CrossRef] [Green Version]
- Ravel, C.; Fiquet, S.; Boudet, J.; Dardevet, M.; Vincent, J.; Merlino, M.; Michard, R.; Martre, P. Conserved cis-regulatory modules in promoters of genes encoding wheat high-molecular-weight glutenin subunits. Front. Plant Sci. 2014, 5, 621. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Fang, L.; Yu, J.; Zhao, Y.; Chen, F.; Xia, G. 5-Azacytidine treatment and TaPBF-D over-expression increases glutenin accumulation within the wheat grain by hypomethylating the Glu-1 promoters. Theor. Appl. Genet. 2018, 131, 735–746. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, C.; Qin, M.; Li, W.; Hou, J.; Shi, X.; Dai, Z.; Yao, W.; Tian, B.; Lei, Z.; et al. Promoter DNA hypermethylation of TaGli-γ-2.1 positively regulates gluten strength in bread wheat. J. Adv. Res. 2021. in press. [Google Scholar] [CrossRef]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Hino, K.; Bono, H.; Ui-Tei, K. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 2014, 31, 1120–1123. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Shendure, J. FlashFry: A fast and flexible tool for large-scale CRISPR target design. BMC Biol. 2018, 16, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahsan, M.U.; Liu, Q.; Fang, L.; Wang, K. NanoCaller for accurate detection of SNPs and indels in difficult-to-map regions from long-read sequencing by haplotype-aware deep neural networks. Genome Biol. 2021, 22, 261. [Google Scholar] [CrossRef]
- Danecek, P.; McCarthy, S.A. BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics 2017, 33, 2037–2039. [Google Scholar] [CrossRef] [PubMed]
- WhatsHap: Weighted haplotype assembly for future-generation sequencing reads. J. Comput. Biol. 2015, 22, 498–509. [CrossRef]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. JBrowse: A dynamic web platform for genome visualization and analysis. Genome Biol. 2016, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, F.; Correia, D.; Lefort, V.; Doppelt-Azeroual, O.; Mareuil, F.; Cohen-Boulakia, S.; Gascuel, O. NGPhylogeny.fr: New generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019, 47, W260–W265. [Google Scholar] [CrossRef] [Green Version]
- Noé, L.; Kucherov, G. YASS: Enhancing the sensitivity of DNA similarity search. Nucleic Acids Res. 2005, 33, W540–W543. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Locus | Number of On Target Reads | Enrichment Rate |
|---|---|---|
| Glu-1Bx DQ537336.1:199,854..204,146 | 8 | ~200× |
| Glu-1By DQ537336.1:29,265..35,054 | 17 | ~645× |
| Glu-1Ax DQ537335.1:265,694..270,243 | 7 | ~200× |
| Locus | gRNA Sequence |
|---|---|
| Glu-1Bx DQ537336.1:199,854..204,146 | F1: AAAACGTCCATGCATAAGTA; F2: ATTACATGTAGCCACCGACA; R1: TCACGTTTATTGTATAGCTA; R2: CAGAGAGTTCTATCACTGCC |
| Glu-1By DQ537336.1:29,265..35,054 | F1: GGGCCCTGTGCGGTTCGCAC; F2: CCTGGATTATGTTGGACGAT; R1: CCCTCCATCCGACACATTAT; R2: TGCTCTGTGTTAACATGGTA |
| Glu-1Ax DQ537335.1:265,694..270,243 | F1: GCAACGATTATGGGGCTGCA; F2: CTCCCTCATGAGTTGTATGC; R1: ATGCGTCGCCGCCCTCTAGC; R2: TGCTCCGCGCTAACATGGTA |
| Primer Id | Primer Sequences | Insertion Name |
|---|---|---|
| Glu_x_prom F | caaccatgcatagaagaaagctc | Insertion 180 |
| Glu_x_prom R | ccttcttggggtttggcaga | |
| BxUnique1_350F | ccctgctgcgaagaagttac | Insertion 12 |
| BxUnique1_350R | tggcctggatagtatgacccctg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirov, I.; Polkhovskaya, E.; Dudnikov, M.; Merkulov, P.; Vlasova, A.; Karlov, G.; Soloviev, A. Searching for a Needle in a Haystack: Cas9-Targeted Nanopore Sequencing and DNA Methylation Profiling of Full-Length Glutenin Genes in a Big Cereal Genome. Plants 2022, 11, 5. https://doi.org/10.3390/plants11010005
Kirov I, Polkhovskaya E, Dudnikov M, Merkulov P, Vlasova A, Karlov G, Soloviev A. Searching for a Needle in a Haystack: Cas9-Targeted Nanopore Sequencing and DNA Methylation Profiling of Full-Length Glutenin Genes in a Big Cereal Genome. Plants. 2022; 11(1):5. https://doi.org/10.3390/plants11010005
Chicago/Turabian StyleKirov, Ilya, Ekaterina Polkhovskaya, Maxim Dudnikov, Pavel Merkulov, Anastasia Vlasova, Gennady Karlov, and Alexander Soloviev. 2022. "Searching for a Needle in a Haystack: Cas9-Targeted Nanopore Sequencing and DNA Methylation Profiling of Full-Length Glutenin Genes in a Big Cereal Genome" Plants 11, no. 1: 5. https://doi.org/10.3390/plants11010005